
Abstract
Sixty years after Watson and Crick published the double helix model of DNA's structure, thirteen members of Genome Biology's Editorial Board select key advances in the field of genome biology subsequent to that discovery.
Similar content being viewed by others


April 25th 2013 is the sixtieth anniversary of the infamous Watson and Crick Nature paper describing a model for the structure of DNA, published 25 April 1953: the now infamous 'double helix' [1]. Two accompanying papers from Rosalind Franklin, Maurice Wilkins and colleagues leant experimental support to the proposed structure in the form of X-ray diffraction data [2, 3], as described elsewhere in this issue of Genome Biology [4]. The model was a landmark discovery in the history of modern science, and was notable for its cross-disciplinary importance: the question addressed was of immense biological importance, but it was physicists and chemists whose expertise and techniques were needed in order to arrive at an answer. One of these physicists, Ray Gosling, describes the unveiling of Watson and Crick's double helix structure as a 'eureka' moment [4]: its simplicity and elegance were striking, and not only explained the X-ray diffraction data but also the mode of replication of life itself. It is rare for a scientific discovery to achieve such an iconic status, to pervade popular culture and the public consciousness, as well as to become an emblem of scientific inquiry - as exemplified by Genome Biology's double helix-inspired logo. Although Avery had already shown DNA to be the genetic material [5], it took the convincing simplicity of Watson and Crick's double helix for this notion to widely take hold, in place of theories favoring proteins. The discovery, therefore, had many important implications, and set the scene for future breakthroughs in the field of genome biology.
To celebrate sixty years of such discoveries, we asked a jury composed of Genome Biology Editorial Board members to select key advances in the field since 25 April 1953. The brief was to choose a development that was either the most important or the most surprising, or that had the most personal impact, and to briefly summarize why. A number of selections focused on technological advances - from restriction mapping through microarrays and high-throughput sequencing. These technologies have clearly done much to inform our understanding of the biology of genomes. The most popular choice, however, was the discovery of introns. Much like the double helix, this discovery had something of the 'X factor' to it: biologists trained in the post-intron era may take the concept of gene fragmentation for granted, but at the time it was a truly radical and paradigm-shifting idea. The sense of surprise made a strong impression on those old enough to remember the discovery, and one of the groups involved went so far as to describe it as 'amazing' in the title of their paper [6].
'Genes in pieces': introns
Brenton Graveley
In 1977, Rich Roberts and Phil Sharp independently reported that genes can be interrupted by introns [6, 7]. In my opinion, this is the biggest and most surprising discovery in genome biology since the structure of DNA was reported by Watson and Crick on 25 April 1953 [1]. Accordingly, Roberts and Sharp were awarded the Nobel Prize in Physiology or Medicine in 1993 for this discovery. The existence of introns was entirely unexpected, and led to the subsequent discovery of the spliceosome, the macromolecular complex that removes introns. Alternative splicing, where exons can be joined together in different patterns, is known to occur for nearly all human genes and plays important roles in both increasing protein diversity and regulating biological processes. The implications of these findings are very far reaching and it is now known that many human diseases are caused by mutations in the RNA sequences that direct splicing, as well as in the proteins and RNAs that catalyze and regulate intron removal.
Jernej Ule
Walter Gilbert wrote an opinion article in 1978 beginning with the statement, 'Our picture of the organisation of genes in higher organisms has recently undergone a revolution' [8]. In 1977, the Roberts and Sharp groups reported that viral mRNA molecules were shorter than their corresponding genes [6, 7]. By 1978, a flurry of papers demonstrated that most genes in eukaryotic organisms contain regions that are removed during the production of mRNAs. In his article, Gilbert proposed that we call these regions 'introns' (for intragenic regions).
Despite knowing the sequence of only a small number of genes in 1978, Gilbert was able to correctly predict that introns account for approximately ten times more genomic sequence than exons. But what was the function of this prevalent genomic feature? Since introns are removed from the nascent RNA, they marked a departure from early studies of genetic code. Introns did not fit easily into the simple, linear transfer of genetic information from DNA to mRNA to protein. They required a different kind of thinking.
Gilbert predicted that parts of introns could in some cases become exons, a process that we now recognize as alternative splicing. Alternative splicing would increase protein diversity and, if its regulation was cell-type specific, it might even drive cellular differentiation. These hypotheses have since been validated by decades of research. Moreover, Gilbert envisioned that introns could be fertile ground for evolutionary tinkering, since mutations that modulate inclusion of exons could change the patterns of alternative splicing.
'The extra material is scattered in the genome, to be called into action at any time... Evolution can seek new solutions without destroying the old.' Here, Gilbert suggests that mutations can create novel mRNA isoforms without removing the previous isoforms. Thus, in spite of the apparent burden of extra genomic sequence, introns create a mechanism primed to explore evolutionary innovations. The idea that life has a vast in-built potential that remains to be realized had a strong impact on my thinking and research. This led us to the recent findings that the human genome contains a reservoir of transposable elements that can gradually evolve into exons by mutations that influence the competitive binding of multiple splicing factors [9]. Gilbert summarized the dual nature of introns as burden and blessing with typical elegance, 'introns are both frozen remnants of history and the sites of future evolution.'
Steven Henikoff
I was just finishing up as a graduate student in 1977 when I first learned of the amazing discovery of split genes in adenovirus [6, 7]. Shortly thereafter, rumors circulated that genes in pieces, or what Wally Gilbert referred to as exons and introns [8], are far more common than originally supposed, and in fact are the rule, rather than the exception. It was a thrill to realize that something that we all took for granted, from the dominant Escherichia coli paradigm of uninterrupted protein-coding genes, does not hold in eukaryotic genomes. It is hard to think of another example in which a concept so fundamental as split genes and post-transcriptional splicing was not suspected to exist before its discovery. Wally began the debate over 'why?' [8] and others asked 'how?' [10], but for me, the question was: what other surprises lay in store in the realm of eukaryotic gene organization? Thus began my interest in what later became referred to as genomics, and when I had my own lab, studying genes in pieces led to the surprising discovery of genes in pieces in genes in pieces [11].
Ford Doolittle
The discovery of spliceosomal introns in the genomes of eukaryotes [12–15] ranks very near the top of my list, in 'significance, surprise and (especially) personal impact'. I was at the time (1977) on sabbatical in the lab of Wally Gilbert and he, having just become aware of that amazing discovery, came up with 'exon shuffling' as an appealing raison-d'etre for introns' existence [8]. Because I also knew of Carl Woese's then very recent discovery of archaea and Carl's inference therefrom that eukaryotes and prokaryotes might have diverged separately from a more primitive common ancestor [16], and because I did not believe that early eukaryotes would have taken on the burden of introneousness just because it might help them in the distant future, I cobbled together the theory that became known as 'introns early' [17].
Almost certainly that theory is false: it seems much more likely that spliceosomal introns are Group II introns gone to seed - fallen apart into 'five easy pieces' [18] and increasingly dependent on dozens of proteins, the accretion of which may have been largely through a neutral evolutionary ratchet. But 'introns early' had a great run, in large part thanks to Wally, and stimulated many to think more deeply about the evolution and functional significance of genome architecture. In particular, it forced us to accept that Dobzhansky's rubric that 'nothing in biology makes sense except in the light of evolution' is just as valid at the molecular level as the organismal. Genomes are much more than repositories of encoded information needed to make organisms, and we can be led astray if we adopt an overly functionalist/adaptationist perspective. Genomicists and systems biologists are especially at risk of doing this, as the recent controversy surrounding the ENCODE project illustrates [19].
Genomes become tangible: restriction mapping
George Weinstock
When I started graduate school in 1970 there were no restriction enzymes and agarose gels, let alone DNA sequences. Genetics was dominated by model organisms like E. coli, phage lambda, Saccharomyces cerevisiae and Drosophila melanogaster. It was based on linkage maps and recombination frequencies, complementation tests and cistrons, abstract concepts that only a geneticist could love and that probably made the field less accessible to other biologists. There were experiments relating genetic traits to physical locations on a chromosome, but these were relatively low resolution and had only limited impact on converting our view of the genome from the abstract concepts of genetic methodology to the biochemical reality of DNA structure. Then, in the mid-1970s, restriction enzymes [20, 21] and agarose gels [22, 23] began to change all of that. Suddenly it was possible to reduce the abstractions of linkage maps to detailed restriction maps (for example, [24]) with concrete physical distances instead of operational but obscure recombination frequencies. Being able to view genome structure in these tangible terms was a paradigm shift that made for a more comfortable way of describing genome structure for both the biologist and lay community. Of course, all of that led to great downstream advances - PCR, DNA sequencing, and much more. Now most biologists only think of genomes in palpable terms, never in the abstract genetic concepts, and probably would have a hard time imagining how you could do research without this. So to me, that DNA technology of the mid-1970s was the day genetics changed and we shifted from one era to the next.
A new regulatory paradigm: microRNA
John Rinn
The laws of genetics have never depended on what genes are made of chemically, and would hold true even if they were made of green cheese.
Ed Lewis
Genetic approaches are as fundamental to biology as math is to physics. One of the most bizarre, dogma-shifting and inspiring genetic studies is exemplified in the article by Victor Ambros and colleagues entitled 'The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14' [25]. The study was published alongside related findings from Gary Ruvkun and colleagues [26].
As a testament to the strength of genetics, Ambros and colleagues went to heroic lengths to identify the function of the lin-4 gene, which Horvitz and Sulston had previously identified in a forward genetic screen for novel Caenorhabditis elegans cell-lineage mutants [27]. Dr Ambros' lab started to investigate the mechanistic role of lin-4, using classic complementation to hone in on the gene product. They narrowed the locus down to a 3.2 kbp region, and then further refined it to a 693 bp region, that could complement or recover the lin-4 mutant phenotype of delayed development. Typically, this genetic resolution would have identified a protein-coding exon, leading to a more obvious explanation of the lin-4 gene product.
The first twist in this expedition into 'noncodarnia' was that the gene mapped not to a coding region, but within an intron of a protein-coding mRNA. Dogmatically, this region was already doomed to be labeled as junk, based on being encoded within an intron. Even worse, Northern blot and RNase protection analyses identified 61 nucleotide and 22 nucleotide RNA species, which was smaller than any protein mRNA known at the time. Grounded in purely genetic observations, the authors continued to pursue the most heroic genetic exercise in this study: non-complementation. After surveying no less than 20,000 mutants of the lin-4 bearing chromosome, only one was unable to complement the original lin-4 mutant. The source was a C-to-T transition within both the 61 nucleotide and 22 nucleotide lin-4 RNA species. Thus, despite beginning with a most unorthodox genetic observation, the authors now had evidence that a mutation in this tiny RNA could indeed recapitulate the original lin-4 mutant.
Taking these observations at face value, the authors put forward a model - perhaps the only possible model - in which this deviant 22 nucleotide or 61 nucleotide RNA hybridizes to the 3' UTR of the lin-14 gene, and in doing so serves as a negative inhibitor. The inhibition of lin-14 would explain the original observation of delayed development in lin-4 mutants. The unquestionable genetics underlying the interplay of lin-4 and lin-14 led, ten years ahead of its time, to a model that has become dogma. We now know, of course, that the authors had discovered a whole new form of gene regulation, widespread in nature, mediated by what Ambros and others [28–30] eventually christened microRNAs, or miRNAs. The visionary discussion section of Ambros and colleagues' lin-4 article simply raises intellectual goosebumps.
The authors never deviated from the inherent truths of genetic observation, no matter how odd the underlying gene product. Despite having discovered this abstruse gene product, which in its day smelled something like green cheese, the genetic basis and implications were to them clear, correct and prophetic.
The original 'data explosion': microarrays
Alicia Oshlack
Ten years ago, I had no real concept of DNA. Genes, in my limited understanding, were passed down to you by your parents but I had not made any clear connection between genes and DNA. My awareness was focused on the outer reaches of the universe and contemplating how gases revolve around black holes in the centers of quasars: my PhD topic. Had it not been for the discovery of microarrays, I may never have had the opportunity to turn my scientific career on its head and begin training in bioinformatics. The first expression microarrays were published in 1995 by Pat Brown's group at Stanford [31], and by 2003 hundreds of discoveries were being made using the many different versions of this technology. The invention of microarrays produced a data explosion (although perhaps not as phenomenal as the one we are currently experiencing) and with this came a need for quantitative scientists to develop analysis methodology and interpretation of data. I was fortunate enough to have been given an opportunity to join the bioinformatics division of two pioneers in the field of microarray analysis, Gordon Smyth and Terry Speed. I consider the first couple of years of the job akin to a second PhD, in which my primary focus was methodology development for the analysis of microarray data. If it had not been for the invention of expression microarrays, my career may have ended up light years away from what I now believe to be the most interesting job in the world.
Unlocking 'genetic messages': sequencing technologies
Michael Schatz
The most significant development in genome biology since 25 April 1953 has been the rise of large-scale DNA sequencing, pioneered by Fred Sanger in 1977 [32], and refined over the last several decades in first, second, and now third generation automated sequencing technologies. Determining the structure of DNA in 1953 was incredibly significant for determining the molecular framework for so many aspects of biology, including the frameworks for replication, transcription, inheritance, mutation and evolution, to name but a few. However, determining the structure of DNA is analogous to determining the structure of a piece of paper, void of any literature or mathematics written upon it. Now, with the rise of high-throughput sequencing, we have the capability to read the genetic messages written on those molecular pages. This capability has unlocked not only the human genome but also those of thousands of other species, revealing their genes, regulatory sequences and overall structures, which has in turn led to many important advances in biology and medicine. The next frontier in genome biology is to apply these technologies, and the related molecular assays empowered by DNA sequencing, over large populations of species, individuals and cells to compare, model and predict how these systems behave.
'Sequence is power': the human and mouse genome projects
Chris Ponting
To me, one important moment in genomics was in 2002 when we could measure the human genome against an evolutionary yardstick, the mouse genome. Jim Kent of UCSC produced a wonderful visual guide (Figure 25a in [33]) to the most important features of mammalian genes. By collapsing levels of sequence conservation between thousands of mouse and human orthologs into one metagene, he showed how, from a common sequence over 90 million years ago, mutation has etched away intronic sequence whilst selection has greatly preserved the exons, particularly toward their boundaries. For me, Jim's analysis was important in illustrating so very clearly how evolutionarily dynamic are our genes and genomes.
Mark Gerstein
I think the most significant advance in genomics since the unraveling of the double helical structure in 1953 has been the sequencing of the first personal genome, that of Craig Venter. The sequencing of a personal genome, not a reference or anonymous genome, links genomics to people and transforms the field from an abstract science of molecules to something very personal and connected to humankind. When we look back on 1953 from 2053, we will realize that the personal genome is what really brought DNA to the masses.
Peter Fraser
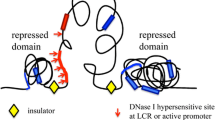
I suppose the most important 'happening' in genome biology since 1953 would have to be the human and mouse genome projects [33, 34]. Prior to the completed sequences, these genomes were a seemingly infinite mystery. Individual investigators clung onto and pored over a few hard-fought kilobases of DNA sequence around their genes of interest. Going much beyond this in a time when sequences were still determined on gels was akin to interstellar travel. The genome projects essentially turned each genome into a global village of finite proportions that is now rapidly filling with features. Though the genome projects themselves are not really a discovery, they have made possible countless other discoveries. I have decided to focus on one of these smaller discoveries, important in its time, but one of more personal significance. It serves as an excellent example of the many discoveries that the genome projects revolutionized. The discovery was of the properties of the beta-globin dominant control region, also called the locus activating region and later named by committee as the locus control region. This was a region about 50 kilobases upstream of the beta-globin gene that was known to have profound effects on globin gene expression [35]. The element was in the dark area of the genome in which no sequence existed, far away from the relative light of the gene-local kilobase or so of sequences flanking the globin genes. Instead of sequence, only painstakingly determined restriction maps existed for this distant region. After discovery of its function, the focus switched to how such a long-range element could affect gene expression over such a distance. The debates were lively, passionate, some would say fierce, and this not only energized the field but, finally, 15 years later, led to experiments that provided evidence for a physical interaction-based mechanism, via long-range folding or 'looping' of chromatin [36]. During this time, the various genome projects were gradually increasing the amount of genomic sequences available. Soon it was realized that many genes were controlled by long-range elements, some up to a megabase away, and nowadays it appears that most, if not all, genes may be controlled by many such elements [37]. But it didn't stop there; built on the foundation of the complete genome sequence and fuelled by advanced high-throughput sequencing technologies, the folding of these individual loops into chromosomes, together with the way chromosomes are arranged and interact in the nucleus, is increasingly being appreciated as an important contributing factor to the control of many genome functions [38, 39]. So, although the monumental genome projects are the antithesis of the elegance of the Watson and Crick model, they must be seen as equally significant because of their present and future impacts. Sequence may not be knowledge, but it is power.
Retelling the human story: analysis of ancient and historical DNA
Detlef Weigel
'What has been the most surprising discovery in the field of genome biology for you?' In hindsight, it appears that many applications of high-throughput sequencing, in which I include automated Sanger sequencing, have long been obvious. Certainly, being able to read entire genomes was in principle already thinkable when I was in college in the early 1980s. What I believe would have been very difficult to imagine is the ability to study genomes from organisms that have been dead for thousands or even tens of thousands of years. The vision that my Max Planck colleague Svante Pääbo has had in this regard, and the dogged manner with which he has pursued the sequencing of ancient genomes, is absolutely remarkable. The work of Svante and his students has already revolutionized our understanding of human evolution in the distant past [40–48]. I believe it has similar potential for changing our knowledge of more recent history. A taste of what I am certain is an imminent avalanche of such studies has been provided by Bos and colleagues' Nature study exploring the Black Death outbreak in medieval times [49].
The exception to the rule: lateral gene transfer
Curtis Huttenhower
I would suggest the discovery of horizontal gene transfer as one of the most remarkable in the history of genome biology. First, it arguably predates the discovery of the structure of DNA itself, having been implicated in Frederick Griffith's transformation experiment in 1928 [50]. As happens too often in biology, this simultaneously introduced both a rule (DNA as the agent of vertical genetic transmission) and its exception (lateral transfer). Second, this apparently straightforward concept was gradually shown to comprise a diversity of related biological mechanisms, perhaps culminating with Joshua Lederberg's 1951 work on phage transduction [51] and 1958 Nobel Prize for bacterial recombination. Even then, the extent and plasticity of lateral transfer were only emphasized later in the 1983 Nobel Prize to Barbara McClintock for the discovery of distinct mobile genetic elements - a discovery made decades earlier [52] but not widely accepted until the 1970s. The range of mechanisms by which transfer occurs has had implications as practical as driving the spread of antibiotic resistance and as far-flung as casting doubt on the reconstruction of a universal tree of life. Third, horizontal transfer is notable in remaining an area of active research almost a century after its discovery. Its implications in ancient evolution or in eukaryotes are not yet clear. Likewise, its prevalence, dynamics, ecology, and functional consequences in microbial communities continue to be explored. I hope that the 100th anniversary of DNA sees an even longer list of biological ramifications of horizontal gene transfer and continued discoveries in the field.
Abbreviations
- bp:
-
base pair
- ENCODE:
-
The Encyclopedia of DNA Elements
- miRNA:
-
microRNA
- PCR:
-
polymerase chain reaction
- UCSC:
-
University of California at Santa Cruz.
References
Watson JD, Crick FH: Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953, 171: 737-738. 10.1038/171737a0.
Franklin RE, Gosling RG: Molecular configuration in sodium thymonucleate. Nature. 1953, 171: 740-741. 10.1038/171740a0.
Wilkins MHF, Stokes AR, Wilson HR: Molecular structure of deoxypentose nucleic acids. Nature. 1953, 171: 738-740. 10.1038/171738a0.
Attar N: Raymond Gosling: the man who crystallized genes. Genome Biol. 2013, 14: 402-10.1186/gb-2013-14-4-402.
Avery OT, Macleod CM, McCarty M: Studies on the chemical nature of the substance inducing transformation of pneumococcal types: Induction of transformation by a desoxyribonucleic acid fraction isolated from pneumococcus type III. J Exp Med. 1944, 79: 137-158. 10.1084/jem.79.2.137.
Chow LT, Gelinas RE, Broker TR, Roberts RJ: An amazing sequence arrangement at the 5' ends of adenovirus 2 messenger RNA. Cell. 1977, 12: 1-8. 10.1016/0092-8674(77)90180-5.
Berget SM, Moore C, Sharp PA: Spliced segments at the 5' terminus of adenovirus 2 late mRNA. Proc Natl Acad Sci USA. 1977, 74: 3171-3175. 10.1073/pnas.74.8.3171.
Gilbert W: Why genes in pieces?. Nature. 1978, 271: 501-501. 10.1038/271501a0.
Zarnack K, König J, Tajnik M, Martincorena I, Eustermann S, Stévant I, Reyes A, Anders S, Luscombe NM, Ule J: Direct competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell. 2013, 152: 453-466. 10.1016/j.cell.2012.12.023.
Brody E, Abelson J: The "spliceosome": yeast pre-messenger RNA associates with a 40S complex in a splicing-dependent reaction. Science. 1985, 228: 963-967. 10.1126/science.3890181.
Henikoff S, Keene MA, Fechtel K, Fristrom JW: Gene within a gene: nested Drosophila genes encode unrelated proteins on opposite DNA strands. Cell. 1986, 44: 33-42. 10.1016/0092-8674(86)90482-4.
Jeffreys AJ, Flavell RA: The rabbit beta-globin gene contains a large large insert in the coding sequence. Cell. 1977, 12: 1097-1108. 10.1016/0092-8674(77)90172-6.
Tonegawa S, Maxam AM, Tizard R, Bernard O, Gilbert W: Sequence of a mouse germ-line gene for a variable region of an immunoglobulin light chain. Proc Natl Acad Sci USA. 1978, 75: 1485-1489. 10.1073/pnas.75.3.1485.
Glover DM, Hogness DS: A novel arrangement of the 18S and 28S sequences in a repeating unit of Drosophila melanogaster rDNA. Cell. 1977, 10: 167-176. 10.1016/0092-8674(77)90212-4.
Breathnach R, Mandel JL, Chambon P: Ovalbumin gene is split in chicken DNA. Nature. 1977, 270: 314-319. 10.1038/270314a0.
Woese CR, Fox GE: Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci USA. 1977, 74: 5088-5090. 10.1073/pnas.74.11.5088.
Doolittle WF: Genes in pieces: were they ever together?. Nature. 1978, 272: 581-582. 10.1038/272581a0.
Sharp PA: Five easy pieces. Science. 1991, 254: 663-10.1126/science.1948046.
Doolittle WF: Is junk DNA bunk? A critique of ENCODE. Proc Natl Acad Sci USA. 2013, 110: 5294-5300. 10.1073/pnas.1221376110.
Danna K, Nathans D: Specific cleavage of simian virus 40 DNA by restriction endonuclease of Hemophilus influenzae. Proc Natl Acad Sci USA. 1971, 68: 2913-2917. 10.1073/pnas.68.12.2913.
Smith HO, Wilcox KW: A restriction enzyme from Hemophilus influenzae. I. Purification and general properties. J Mol Biol. 1970, 51: 379-391. 10.1016/0022-2836(70)90149-X.
Takahashi M, Ogino T, Baba K: Estimation of relative molecular length of DNA by electrophoresis in agarose gel. Biochim Biophys Acta. 1969, 174: 183-187. 10.1016/0005-2787(69)90241-X.
Sharp PA, Sugden B, Sambrook J: Detection of two restriction endonuclease activities in Haemophilus parainfluenzae using analytical agarose--ethidium bromide electrophoresis. Biochemistry. 1973, 12: 3055-3063. 10.1021/bi00740a018.
Mulder C, Arrand JR, Delius H, Keller W, Pettersson U, Roberts RJ, Sharp PA: Cleavage maps of DNA from adenovirus types 2 and 5 by restriction endonucleases EcoRI and HpaI. Cold Spring Harb Symp Quant Biol. 1975, 39: 397-400.
Lee RC, Feinbaum RL, Ambros V: The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993, 75: 843-854. 10.1016/0092-8674(93)90529-Y.
Wightman B, Ha I, Ruvkun G: Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993, 75: 855-862. 10.1016/0092-8674(93)90530-4.
Horvitz HR, Sulston JE: Isolation and genetic characterization of cell-lineage mutants of the nematode Caenorhabditis elegans. Genetics. 1980, 96: 435-454.
Lee RC, Ambros V: An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001, 294: 862-864. 10.1126/science.1065329.
Lau NC, Lim LP, Weinstein EG, Bartel DP: An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001, 294: 858-862. 10.1126/science.1065062.
Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T: Identification of novel genes coding for small expressed RNAs. Science. 2001, 294: 853-858. 10.1126/science.1064921.
Schena M, Shalon D, Davis RW, Brown PO: Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995, 270: 467-470. 10.1126/science.270.5235.467.
Sanger F, Air GM, Barrell BG, Brown NL, Coulson AR, Fiddes JC, Hutchison CA, Slocombe PM, Smith M: Nucleotide sequence of bacteriophage |[phi]|X174 DNA. Nature. 1977, 265: 687-695. 10.1038/265687a0.
Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, et al: Initial sequencing and comparative analysis of the mouse genome. Nature. 2002, 420: 520-562. 10.1038/nature01262.
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, et al: Initial sequencing and analysis of the human genome. Nature. 2001, 409: 860-921. 10.1038/35057062.
Grosveld F, van Assendelft GB, Greaves DR, Kollias G: Position-independent, high-level expression of the human beta-globin gene in transgenic mice. Cell. 1987, 51: 975-985. 10.1016/0092-8674(87)90584-8.
Carter D, Chakalova L, Osborne CS, Dai Y, Fraser P: Long-range chromatin regulatory interactions in vivo. Nat Genet. 2002, 32: 623-626. 10.1038/ng1051.
Sanyal A, Lajoie BR, Jain G, Dekker J: The long-range interaction landscape of gene promoters. Nature. 2012, 489: 109-113. 10.1038/nature11279.
Cope NF, Fraser P, Eskiw CH: The yin and yang of chromatin spatial organization. Genome Biol. 2010, 11: 204-10.1186/gb-2010-11-3-204.
Gibcus JH, Dekker J: The hierarchy of the 3D genome. Mol Cell. 2013, 49: 773-782. 10.1016/j.molcel.2013.02.011.
Pääbo S: Molecular cloning of Ancient Egyptian mummy DNA. Nature. 1985, 314: 644-645. 10.1038/314644a0.
Krings M, Stone A, Schmitz RW, Krainitzki H, Stoneking M, Pääbo S: Neandertal DNA sequences and the origin of modern humans. Cell. 1997, 90: 19-30. 10.1016/S0092-8674(00)80310-4.
Green RE, Krause J, Ptak SE, Briggs AW, Ronan MT, Simons JF, Du L, Egholm M, Rothberg JM, Paunovic M, Pääbo S: Analysis of one million base pairs of Neanderthal DNA. Nature. 2006, 444: 330-336. 10.1038/nature05336.
Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH-Y, Hansen NF, Durand EY, Malaspinas A-S, Jensen JD, Marques-Bonet T, Alkan C, Prüfer K, Meyer M, Burbano HA, Good JM, Schultz R, Aximu-Petri A, Butthof A, Höber B, Höffner B, Siegemund M, Weihmann A, Nusbaum C, Lander ES, Russ C, et al: A draft sequence of the Neandertal genome. Science. 2010, 328: 710-722. 10.1126/science.1188021.
Krause J, Orlando L, Serre D, Viola B, Prüfer K, Richards MP, Hublin J-J, Hänni C, Derevianko AP, Pääbo S: Neanderthals in central Asia and Siberia. Nature. 2007, 449: 902-904. 10.1038/nature06193.
Reich D, Green RE, Kircher M, Krause J, Patterson N, Durand EY, Viola B, Briggs AW, Stenzel U, Johnson PLF, Maricic T, Good JM, Marques-Bonet T, Alkan C, Fu Q, Mallick S, Li H, Meyer M, Eichler EE, Stoneking M, Richards M, Talamo S, Shunkov MV, Derevianko AP, Hublin J-J, Kelso J, Slatkin M, Pääbo S: Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature. 2010, 468: 1053-1060. 10.1038/nature09710.
Burbano HA, Hodges E, Green RE, Briggs AW, Krause J, Meyer M, Good JM, Maricic T, Johnson PLF, Xuan Z, Rooks M, Bhattacharjee A, Brizuela L, Albert FW, Rasilla M de la, Fortea J, Rosas A, Lachmann M, Hannon GJ, Pääbo S: Targeted investigation of the Neandertal genome by array-based sequence capture. Science. 2010, 328: 723-725. 10.1126/science.1188046.
Krause J, Fu Q, Good JM, Viola B, Shunkov MV, Derevianko AP, Pääbo S: The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature. 2010, 464: 894-897. 10.1038/nature08976.
Meyer M, Kircher M, Gansauge M-T, Li H, Racimo F, Mallick S, Schraiber JG, Jay F, Prüfer K, de Filippo C, Sudmant PH, Alkan C, Fu Q, Do R, Rohland N, Tandon A, Siebauer M, Green RE, Bryc K, Briggs AW, Stenzel U, Dabney J, Shendure J, Kitzman J, Hammer MF, Shunkov MV, Derevianko AP, Patterson N, Andrés AM, Eichler EE, et al: A high-coverage genome sequence from an archaic Denisovan individual. Science. 2012, 338: 222-226. 10.1126/science.1224344.
Bos KI, Schuenemann VJ, Golding GB, Burbano HA, Waglechner N, Coombes BK, McPhee JB, DeWitte SN, Meyer M, Schmedes S, Wood J, Earn DJD, Herring DA, Bauer P, Poinar HN, Krause J: A draft genome of Yersinia pestis from victims of the Black Death. Nature. 2011, 478: 506-510. 10.1038/nature10549.
Griffith F: The significance of pneumococcal types. J Hyg (Lond). 1928, 27: 113-159. 10.1017/S0022172400031879.
Zinder ND, Lederberg J: Genetic exchange in Salmonella. J Bacteriol. 1952, 64: 679-699.
McClintock B: The origin and behavior of mutable loci in maize. Proc Natl Acad Sci USA. 1950, 36: 344-355. 10.1073/pnas.36.6.344.
Acknowledgements
JU wishes to thank Josh Witten for useful suggestions and for text editing. The editors of Genome Biology would like to thank all contributing Editorial Board members for their time and for the thoughtful nature of their responses.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Doolittle, W., Fraser, P., Gerstein, M.B. et al. Sixty years of genome biology. Genome Biol 14, 113 (2013). https://doi.org/10.1186/gb-2013-14-4-113
Published:
DOI: https://doi.org/10.1186/gb-2013-14-4-113