
Abstract
Organisms display astonishing levels of cell and molecular diversity, including genome size, shape, and architecture. In this chapter, we review how the genome can be viewed as both a structural and an informational unit of biological diversity and explicitly define our intended meaning of genetic information. A brief overview of the characteristic features of bacterial, archaeal, and eukaryotic cell types and viruses sets the stage for a review of the differences in organization, size, and packaging strategies of their genomes. We include a detailed review of genetic elements found outside the primary chromosomal structures, as these provide insights into how genomes are sometimes viewed as incomplete informational entities. Lastly, we reassess the definition of the genome in light of recent advancements in our understanding of the diversity of genomic structures and the mechanisms by which genetic information is expressed within the cell. Collectively, these topics comprise a good introduction to genome biology for the newcomer to the field and provide a valuable reference for those developing new statistical or computation methods in genomics. This review also prepares the reader for anticipated transformations in thinking as the field of genome biology progresses.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
- Organism diversity
- Viruses
- Prokaryotes
- Eukaryotes
- Organelles
- DNA
- RNA
- Protein
- Regulatory DNA
- Epigenetics
- Plasmids
- Transcription
- Translation
- DNA replication
- Chromatin
- Gene structure
1 Introduction
Following the introduction of the concept of the genome in 1920 [1], the field of genome science has grown to encompass a vast range of interconnected topics (e.g., nucleic acid chemistry, molecular structure, replication and expression biochemistry, mutational processes, evolutionary dynamics, and interactions with cellular processes). Although the notion of the genome as a fundamental biological unit has been with us for nearly a century, it is only within the last decade that genomics has emerged as a transformative discipline within biology and the health sciences [2]. Its rapid development was in large part due to advances in massively parallel next-generation sequencing [3], which yielded unprecedented levels of genomic data. Those data revealed extensive natural variation in the way that genomes are structured and processed. This led modern biologists to reevaluate the fundamental definition of the genome.
The typical definition of the genome is often dualistic, referencing both structural features and its function to store and transmit biological information [4]. For example, the US National Institutes of Health (NIH) uses the following definition: “A genome is an organism’s complete set of DNA, including all of its genes. Each genome contains all of the information needed to build and maintain that organism. In humans, a copy of the entire genome—more than three billion DNA base pairs—is contained in all cells that have a nucleus.” This conception, as with many others, is structural with regard to physical features (viz., genes and DNA base pairs) and informational with regard to its role in carrying out cellular functions (viz., to build and maintain the organism). Through increased knowledge of genome diversity, the field has come to realize that both conceptions of the genome are sometimes insufficient [4]. We now understand that the physical structures of the genome can be transient and that the expression of information contained within a genome is often conditioned on non-genomic factors. The science of genome biology is entering a new era based on a deeper understanding of the relationship between genotype and phenotype [5].
The purpose of this review is to provide a condensed overview of genome biology and to anticipate transformations in thinking that will occur as the field progresses. The remainder of this article is structured into four parts, with the next section providing a brief overview of the diversity of organismal cell types. The two subsequent sections introduce the structural and informational aspects of genomes, respectively. In the final section, we reassess the definition of the genome through selected biological examples and conclude with an updated perspective on the nature of the genome as an informational entity.
2 Organism Diversity and Cell Types
Cells are the smallest living unit of an organism. All cells have three attributes in common: cell membrane, cytoplasm, and genome. Structurally, cells can be divided into two basic types: prokaryotic and eukaryotic cells. Eukaryotic cells tend to be more complex. They possess a nucleus and other membrane-bound organelles, which are specialized components in the cell that perform unique functions (e.g., nucleus, mitochondria, plastids). Conversely, prokaryotic cells lack membrane-bound organelles. Although similar in cell structure, prokaryotes include two fundamentally distinct domains: the eubacteria (true bacteria, often referred to simply as bacteria) and the archaea.
Cellular life is detected in almost every environment on Earth. As life has colonized and adapted to the vast number of niches, cells have evolved an incredible amount of diversity in regard to size [6], form [7], lifestyle [8, 9], and complexity [10]. Understanding the basis of such diversity remains one of the central aims of biology. Readers interested in the latest understanding of Earth’s biodiversity, the unique characteristics of its organisms, and how both extant and extinct forms are related to each other are encouraged to explore the following resources: the University of California Museum of Paleontology “History of life through time” exhibit [11], the Tree of Life Web Project [12], the Encyclopedia of Life [13].
2.1 Viruses
Viruses are infectious agents of living cells that are unable to reproduce in the absence of a host. Viruses are not considered cellular entities since they lack two of the essential attributes that define a cell; they possess neither a cell membrane nor cytoplasm. The discovery of virophages, viruses that parasitize other viruses, resurrected the debate on their classification as living organisms [14]. Some consider viruses to be living entities since they can be hosts to other viruses, with a virophage infection leading to the eventual death of the host virus, implying an initial “living” state [15]. The opposing view asserts that a virus’ inability to reproduce outside of a cellular host makes them nonliving entities [16, 17]. Irrespective of their delineation as living or nonliving, viruses are relevant to this review as they possess genomes and are the most abundant biological replicators in the biosphere [18].
Outside of their host, viruses exist as viral particles (virions) consisting of a protein capsule that protects and encloses their genome. Once a virion has entered a host cell, it “hijacks” the host’s cellular structures and processes to carry out the metabolically active phase of the viral life cycle. At this stage, the virus exhibits physiological properties reminiscent of living cells; they metabolize, grow, and reproduce. There is a wide range of viral lifestyles, with corresponding diversity in viral forms, sizes, hosts, and genomes [16]. The largest known virus, the mimivirus, was originally identified as an infectious agent of an amoeba [19] and can itself become a host for virophages [14]. To put this in context, the virion of a mimivirus can be larger than some prokaryotic cells [16]. At the other end of the scale are viruses such as the circoviruses, some of which have small genomes made up of less than 2000 nucleotides [20]. A more detailed account of viral diversity can be found at the ViralZone website [21].
2.2 Bacteria
The bacterial cell is prokaryotic, and it is relatively simple as compared to eukaryotic cells. It has no membrane-bound organelles, and the chromosome (usually one) is not separated from the other components of the cell. While predominantly unicellular, they often live in biofilms, a community of cells bound together by a secreted polymer matrix [22], displaying a range of cooperative behaviors [23]. They can also exhibit regulated differentiation into different cell types, where two cells with the same genome have different morphology and function [22, 24].
Only a very small fraction of bacterial diversity (less than 1%) can be cultured and grown in the laboratory [25]. The problem of uncultivable bacteria is a consequence of our limited knowledge of their physiological diversity and the interactions necessary for their growth [26]. To this end, efforts are being made to study bacteria in nature [27,28,29] but with limited progress given the immense metabolic diversity of bacteria. Even within the incomplete sampling of cultivable bacteria, there is considerable diversity in cell shape [30], mode of reproduction [9], and cell cycle regulation [31].
The bacterial cell cycle involves the coordination of genome replication and segregation of replicated copies into daughter cells, followed by cell division. In this way, the transmission of genetic material is “vertical” from one cell generation to the next. Under certain conditions, some bacteria, such as E. coli, can initiate a new round of genome replication prior to completion of cell division [32, 33], thereby resulting in an increase in the number of gene copies near the origin of replication as compared to loci replicated later [31]. Other bacteria, such as Caulobacter, maintain a tightly regulated cell cycle to ensure a single replication event per division [34]. Under optimal conditions, some species can complete their cell cycle every 20 min, implying that a single cell could produce more than a billion descendants in a mere 10 h. In addition to vertical transfer, genetic information can be transferred “horizontally” between unrelated cells via the processes of transformation, conjugation, or transduction [35]. An event that transfers gene(s) between different species (or cells) by any of these three processes is referred to as a horizontal gene transfer (HGT) event.
2.3 Archaea
Archaea are single-celled organisms that appear strikingly similar to bacteria under light and electron microscopes. Like bacteria they often have a single circular chromosome and lack a nucleus, and for a long period of time the archaea were wrongly categorized as bacteria. The first indication that the archaea might be a separate domain of life was obtained from phylogenetic analyses of the 16S rRNA gene [36]. Advancements in genome sequencing and analysis yielded further evidence of the evolutionary distinction between the bacterial and archaeal domains [37]. Despite their superficial cellular similarity to bacteria, the archaea have many molecular-level similarities to eukaryotes, leading researchers to hypothesize that the ancestor of the eukaryotes arose within the archaea [38].
Previously, archaea were assumed to be a minor group of organisms inhabiting extreme environments beyond the tolerance of bacteria (salt brines, hydrothermal vents, acidic and anoxic conditions, etc.). Through culture-independent methods, archaea were discovered to be much more widespread and metabolically diverse. Archaea are now known to inhabit the human gut, and through mutualistic community relationships, they play a key role in human health and metabolism [39,40,41]. There is increasing evidence for archaea playing a significant role in global nutrient cycling [42]. They contribute major mechanisms for anaerobic methane oxidation [42], ammonia oxidation [43], and other parts of the nitrogen cycle including nitrogen fixation [44]. The archaea also appear to be ecologically competitive with bacteria, as they make significant contributions to the microbial communities of non-extreme soil, aquatic, and marine environments [43, 45]. Although they can be highly abundant in such environments, archaeal diversity is greatest in the more extreme habitats [45].
Archaea possess an array of bacteria-like, eukaryote-like, and archaea-specific features. The archaeal cell wall is chemically and structurally diverse, yet they systematically lack a cell wall peptidoglycan, murein, that is ubiquitous among the bacteria [46, 47]. Their membrane lipids are chemically different from those found in either bacteria or eukaryotes [48], and they possess many novel enzymes that are required for the biosynthesis of their unique membranes [49, 50]. Consequently, most archeoviruses are unique to archaea [51]. Even structural appendages that initially appeared to be homologous to bacterial appendages are often structurally distinct and have different genetic basis than the bacterial counterparts [52,53,54]. At the biochemical level, the archaea use many sources of energy and are metabolically diverse, probably more so than either bacteria or eukaryotes [55].
2.4 Eukaryotes
All complex multicellular organisms are eukaryotes (animals, plants, fungi, red algae, and brown algae), as are many unicellular organisms [56, 57]. Eukaryotic cells are found in a wide diversity of sizes and shapes [58, 59]. They are generally larger and have a more complex internal organization than the bacteria and archaea. A key characteristic of the eukaryotic intracellular organization is the use of lipid membranes to separate their contents into different compartments [60, 61]. The bulk of the eukaryotic genetic material is surrounded by a nuclear envelope and is thus maintained in a separate organelle, the nucleus. This provides a fundamental perspective on how eukaryotic cells differ from bacterial and archaeal cells and has important consequences on the expression of eukaryotic genetic information.
In addition to the nucleus, other organelles (mitochondria and plastids) contain small genomes that encode additional genes. Both mitochondria and plastids originated from ancient endosymbiosis events between ancestral eukaryotic cells and bacterial organisms. Following these events, the invading bacteria underwent a process of genome reduction in which they transitioned from autonomous organisms to cell-dependent organelles [62].
Despite our familiarity with plants, animals, and fungi, the vast majority of eukaryotic diversity lies outside of those groups and is largely microbial [63]. These “other” eukaryotes are collectively called protists. They do not form a monophyletic group, i.e., protists do not from a phylogenetic group that is comprised of a shared common ancestor and all of its descendants [57, 64]. The term protist is used largely for convenience to classify all eukaryotes that are not plants, animals, or fungi. Protists embody extensive ecological and structural diversity and include several important groups of unicellular eukaryotes involved in human diseases [65]. For example, the unicellular apicomplexan eukaryote Plasmodium is the causative agent of malaria, which affects around 10% of the world population [65]. More positively, protist species are important primary producers and are an essential link in the ocean’s biogeochemical cycles [66].
3 Genome Structure and Organization
The notion of the gene as the physical carrier of hereditary information existed years before its physical and chemical structures were known. In 1902, Sutton provided the first clear support for the chromosomal theory of inheritance, allocating genes to segments on chromosomes [67]. The modern view of the gene is more often focused on a particular chemical sequence of nucleic acids rather than a chromosomal locus, but the two are not independent. The genetic instructions encoded within an organism’s nucleic acid molecules comprise the organism’s genotype. The physical manifestation of such genetic information, which will depend on environmental interactions, comprises the organism’s phenotype.
There are two types of nucleic acids: deoxyribonucleic acid (DNA) and ribonucleic acid (RNA). Both are polymers consisting of chains of nucleotides. Each nucleotide includes three components: a 5-carbon sugar, a phosphate group, and a nitrogenous base. A nitrogenous base together with the sugar (without the phosphate group) is called a nucleoside. The sugar component in RNA, ribose, is a normal sugar with one hydroxyl group (OH) attached to each carbon atom. Deoxyribose, the sugar present in DNA, differs only in the absence of one oxygen atom at the 2′ carbon atom (H instead of OH). This chemical difference is crucial for enabling enzymes to distinguish between RNA and DNA polymers. The 5′ sugar carbon carries a phosphate group and is referred to as the 5′ end of the polynucleotide molecule (DNA or RNA). The 3′ end has a free hydroxyl (OH) group that is available to form chemical bonds with other atoms. As a result, synthesis of DNA and RNA in the cell proceed through the addition of a nucleotide to a 3′ terminal hydroxyl group. The polynucleotides, therefore, exhibit directionality, and synthesis occurs in a 5′ to 3′ direction.
All living cells employ the double helical structure of DNA as a chemical means to store information. Each of the two longitudinal strands is an alternating sequence of phosphate and a 5-carbon sugar. At each sugar, the two strands are bridged by two nitrogenous bases, one purine molecule (of type adenine [A] or guanine [G]) and the other a pyrimidine molecule (of type cytosine [C], thymine [T], or uracil [U]). The chemical bridges between purine and pyrimidine molecules (called base pairs) are held together by hydrogen bonds. Each purine can be complemented by only one pyrimidine: A forms two hydrogen bonds with T (or U in RNA) and C forms three hydrogen bonds with G. These are referred to as the canonical or Watson-Crick pairings. Given this pairing pattern, the sequences of the double-stranded DNA are said to be complementary, and the sequence of one strand can be deduced from the sequence of its complementary strand. The order of the nitrogenous bases in DNA (or RNA) is what confers the meaning of the information encoded in the genome.
A vital feature of genetic information is its ability to be replicated and passed on to daughter cells. The core mechanisms that copy DNA are conserved in all three domains of cellular life: bacteria, archaea, and eukaryotes [68]. Accurate DNA replication is essential to produce viable offspring—too many alterations in the DNA impede the production of functional proteins, thereby increasing the chances of nonviable progeny. Therefore, most DNA replicates with high fidelity. However, mistakes do occur. In humans, on average one error occurs in 30 million bases copied per cell division [69]. The cells produced from these altered genes are called mutants.
Although all living things carry DNA, the processes through which genetic information is physically transferred from DNA to RNA (called transcription) and then used to create a polypeptide molecule with a unique sequence of amino acids (called translation) differ between domains of life. The lack of membrane-bound nuclei in prokaryotes permits the simultaneous occurrence of transcription and translation [70]. In eukaryotes, those processes are separated by the nuclear membrane; DNA is first transcribed to RNA in the nucleus, and the RNA product is subsequently translated to an amino acid sequence in the cytoplasm, ultimately leading to the construction of a protein.
Organisms from all domains of life, and many of the viruses that parasitize them, have a very large genome compared with the size of the cell or compartment to which it is confined. For instance, the human nuclear DNA consists of approximately three billion base pairs; when stretched out, it amounts to about 2 m of total DNA per cell. The average human cell size is merely 10 μm. The impressive ability to store DNA within the cell is possible through a process of genome packaging. In eukaryotes and some archaea, the DNA wraps around histone proteins to form nucleosomes. In humans, this results in a two-million-fold decrease in size, allowing the DNA to compact into the nucleus [68]. Prokaryotic DNA compaction is achieved using a combination of supercoiling, macromolecular crowding, and association with DNA-binding proteins [71]. The degree of the supercoiling used in prokaryotes varies considerably between different species.
Prokaryotic cells tend to have efficient genomes, with most of their genetic material composed of protein-coding regions. Archaeal genomes are, on average, more compact than bacterial genomes [72]. An increase in prokaryotic genome size is therefore often accompanied by an increase in the number of genes encoded. This trend is not evident in eukaryotes, for which there is little association between genome size and the number of protein-coding genes [73]. Consider the E. coli genome, more than 90% of its DNA encodes proteins. This is in stark contrast with the modest 2% protein-coding regions present in human DNA [74]. Most eukaryotic genomes are riddled with non-protein-coding regions (see Subheading 4.2 for an evolutionary mechanism). This results in them having larger genome sizes on average than prokaryotic cells [74].
3.1 Viral Genomes
Viruses use any combination of either RNA or DNA, either single- or double-stranded molecules, in either circular or linear forms, to encode their genetic instructions [75, 76]. The viral genetic material is typically referred to as segments rather than chromosomes. Viral genomes composed of multiple segments are referred to as segmented. When different strains of the same segmented viral species infect a cell, genomes from the different strains can mix to produce hybrids—a process known as reassortment. Hybrid flus such as the H1N1 swine influenza A virus originated in this way [77].
Viral strains package their genomes in various ways. Most DNA and RNA viruses with small genomes (<20 kb) employ energy-independent packaging systems where capsid assembly and genome condensation are coupled. One example is the RNA genome of the HIV retrovirus that, in the mature virion, forms a RNA-protein complex with one of the cleavage products of the Gag polyprotein [78]. Other viruses, such as the lambda bacteriophage, require ATP to pump their genome directly into a preassembled capsid [79]. The latter type of machinery is ubiquitous in bacterial viruses. Alternatively, large viruses package their genome using histone-like proteins that are critical for eukaryotic genome packaging [80]. For a review on genome packaging in viruses, see ref. 81.
3.2 Bacterial Genomes
Despite not being confined within a membrane-bound compartment, the prokaryotic genome will be unevenly distributed throughout the cell. It often clusters in an irregularly shaped viscous region known as the nucleoid that makes up about a quarter of the intracellular volume [82]. The organization and distribution of the nucleoid are dynamic and dependent on the growth rate and presence of antibiotics [83].
It was previously thought that all bacterial cells possessed a single circular chromosome. In 1989, the first linear bacterial chromosome was discovered in the spirochaete Borrelia burgdorferi, the causative agent of Lyme disease [84, 85]. Additionally, recent advancements have revealed that many cells retain multiple circular or linear chromosomes [86]. These often consist of a primary chromosome, which is larger and harbors a higher density of essential genes compared to the secondary chromosome(s) [87].
The replication of bacterial DNA initiates at a well-defined sequence, called the origin of replication. The proteins involved in replication bind to the origin site and DNA synthesis proceeds in both directions. Circular chromosomes require a single origin, and replication is terminated by either a stop signal or when the two replication forks meet [88]. Linear bacterial chromosomes typically have a central origin, and replication proceeds bidirectionally much as in circular chromosomes. However, replication enzymes are unable to synthesize new DNA at the ends of a linear chromosome, and this results in the gradual shortening of DNA after each replication event [89]. Linear chromosomes, therefore, require terminal structures known as telomeres to protect against DNA degradation. Telomeres are characterized by the presence of multiple tandem repeats of short noncoding nucleotide sequences.
Linear prokaryotic chromosomes have evolved two different types of telomeres [90]. The first, best understood in the streptomycetes, uses a terminal protein complex covalently attached to the 5′ end of the DNA molecules. During replication, DNA polymerase binds the first synthesized nucleotide directly to the terminal protein. This replication strategy allows for the complete duplication of the linear molecule with no loss of genetic information [91]. The second type, best studied in the spirochetes, involves the formation of closed hairpin structures at the termini [92]. Replication of the linear DNA proceeds as expected. Once duplication of each DNA strand is completed the newly synthesized DNA are temporarily still attached—forming a structure superficially resembling a circular chromosome. A specific enzyme is then recruited to separate the two linear strands and re-form the telomeres [93]. For an overview of telomeric structures, see ref. 94.
3.3 Archaeal Genomes
Archaeal genomes share features with both bacteria and eukaryotes. Archaea typically possess circular chromosomes reminiscent of bacteria genomes; some have a single chromosome and a single origin of replication, while other species have multiple chromosomes and multiple origins on each [95, 96]. Given that archaea have the prokaryote cell type that lacks membrane-bound organelles (and hence nuclei), they are similar to bacteria in permitting the simultaneous occurrence of transcription and translation. Nonetheless, there are fundamental differences from the bacteria in the processing of genomic information. The initiation of amino acid synthesis in archaea more closely resembles that used in the eukaryotic transcription process. Additionally, the core archaeal transcription machineries are more closely related to eukaryotes [97, 98]. Archaeal and eukaryotic DNA replication and repair systems have also been shown to have many features in common [99].
Relatively little is known about the structure of archaeal genomes [100], but some are packaged into chromatin via histone proteins. Chromatin is a compact and organized chromosome structure that consists of DNA in close association with proteins. Interestingly, this form of chromatin is present in all eukaryotes and missing from bacteria [101]. Among the archaea that use histones (i.e., Thermoproteales and Euryarchaea), the geometry of their histone-mediated chromatin is the same as in eukaryotes [102]. However, archaeal histones are often shorter than the eukaryotic histones [101]. Groups of archaea that lack histones (e.g., Crenarchaea) encode other DNA-binding proteins associated with the architecture of bacterial chromatin [100]. Another family of DNA-binding proteins called Alba (acetylation lowers binding affinity) is ubiquitous among archaea. They are abundant small proteins that facilitate genome compaction, play a key role in determining the architecture of archaeal chromatin, and regulate gene expression on a genomic scale [101]. Alba proteins have been detected in both histone-lacking and histone-containing archaea [103].
3.4 Eukaryotic Genomes
Eukaryotes sequester their linear chromosomes within a membrane-bound nucleus. Linear eukaryotic chromosomes have three essential structural elements: a centromere, a pair of telomeres, and origins of replication. The centromere is the attachment point for spindle microtubules—the filaments responsible for physically moving chromosomes during cell division. Telomeres are the protective ends of a linear chromosome. The origins of replication are the sites where DNA synthesis begins. Eukaryotes typically have multiple linear chromosomes, each with many origins of replication. The larger genome size and slower replication machinery in eukaryotes necessitate the need of multiple origins to speed up the replication process.
In eukaryotic cells, nuclear DNA compaction involves the association of DNA with the protein products of a family of genes, the histones, whose sequence variants provide for a variety of different functions. The eukaryotic chromosome is organized at the lowest level by wrapping the DNA around histones, forming nucleosomes. This structure constitutes the basic unit of the chromatin fiber, which is further organized into higher-order structures mediated by other proteins [104, 105]. Sequence variation in histones, in combination with posttranslational modification of the protein, affects the structural properties of chromosomal nucleosomes and gene expression.
Eukaryotic DNA consists of at least three types of sequences: unique-sequence DNA, moderately repetitive DNA, and highly repetitive DNA. Unique-sequence DNA are regions that are present only once or at most a few times in the genome. Most protein-coding regions fall within this category. Alternatively, more than half of the total DNA in all eukaryotic genomes is made up of repeated sequence motifs that are either moderately or highly repetitive [106]. Moderately repetitive DNA are sequences from 160 to 180 base pairs (bp) in length that are repeated thousands of times [106]. Some of these sequences perform important functions for the cell, such as coding for types of RNA [107]. Highly repetitive DNA are short sequences, less than 60 bp that are present in hundreds of thousands of copies repeated throughout the genome. Repeats that are 2–10 bp are known as microsatellites, whereas motifs that are 10–60 bp are termed minisatellites [108].
Most of the repetitive sequences arise through transposition (see Subheading 4.2). The repeated sequences can be found either in tandem arrays, i.e., appearing adjacent to each other, or interspersed throughout the genome. The evolution and maintenance of nonfunctional repeated sequences have spurred the interest of genome scientists, with some classifying these motifs as selfish-genes that reproduce to propagate themselves and provide no positive contribution to the organism’s phenotype or fitness [106]. Repeats also represent technical challenges for bioinformaticians developing software for sequence alignment and genome assembly. From a computational perspective, repeats create ambiguities that are challenging to resolve. For a review on computational challenges and solutions, see ref. 108.
3.5 Auxiliary DNA Structures
Both prokaryotes and eukaryotes have secondary chromosomal structures. For eukaryotes, this refers to any form of DNA found outside of a nucleus—although the discovery of microDNA extends this classification [109]. Eukaryotic auxiliary DNA often contains essential genes that are necessary for normal cell production. For example, the DNA chromosome located within the mitochondrial organelle encodes genes that are involved in oxidative phosphorylation and the creation of different types of RNA [110]. For prokaryotes, auxiliary DNA refers to any DNA that is not associated with the primary chromosome, and unlike eukaryotes, the genes encoded in such DNA are often dispensable. For example, small circular chromosomes, called plasmids, often contain genes that allow the bacterium to survive various environmental conditions; however they are not usually essential for normal cell function [110].
3.5.1 Mitochondrial DNA
The mitochondrion is a double membrane-bound organelle that is ubiquitous in eukaryotic cells. There is only one known case of a eukaryotic cell able to survive without a mitochondrion [111]. Mitochondria are essential because they are the site of production for most of the cell’s energy, which is produced as ATP by the oxidative phosphorylation metabolic pathway. Additionally, the mitochondrion is the site of iron-sulfur (Fe/S) cluster assembly. Fe/S clusters are protein cofactors that are essential for various extramitochondrial pathways [112]. The mitochondria-lacking eukaryote, a species of Monocercomonoides, is unique in that it lives only within the intestine of the chinchilla and has evolved different strategies for Fe/S cluster formation and obtaining energy absorbed from its environment [111].
Mitochondria are the derivatives of prokaryotic cells that were engulfed by a common ancestor of all eukaryotes. The DNA within these organelles are the remnants of the DNA genome of the ancestral prokaryotic endosymbiont. Thus, the mitochondrial DNA (mtDNA) more closely resembles a prokaryotic genome. For example, in most animals and fungi, mtDNA consists of a single circular chromosome. However, small linear mtDNA chromosomes with defined telomeres have been identified within various protists, animals, and fungi [113, 114]. Additionally, the architecture of mtDNA is not determined by histones but instead by a set of small DNA-binding proteins that induce structures analogous to the bacterial chromatin. Mitochondrial genomes have been categorized into six different types depending on shape, size, structure, and number (see ref. 115).
In humans, the mitochondrial genome encodes 13 of the 80 proteins that are directly involved in oxidative phosphorylation. The remaining proteins are encoded in the nuclear chromosomes [110]. The exact contribution from mitochondrial and nuclear genomes varies across eukaryotes. Nonetheless, in the vast majority of known eukaryotic species, the mtDNA is essential to produce important proteins involved in energy production, demanding that all cells have faithfully inherited the mtDNA.
3.5.2 Plastid DNA
Plastids are similarly derived from an endosymbiosis with a bacterium, with the organelle retaining remnants of that ancestral bacterial genome. Like the mitochondrion, the plastid is a double membrane-bound cytoplasmic organelle. Unlike the mitochondrion, plastids often contain pigment used in photosynthesis. Plastids are found in the cytoplasm of protists and all higher plants. Plastid DNA (ptDNA) is highly reduced relative to the genomes of extant photosynthetic bacteria. In part, the reduction in genome size is due to gene loss with some regions excised and incorporated into the host nuclear DNA [116]. The ptDNA encodes important proteins that are essential for cell viability [117]. Almost all plastids have circular DNA, with the alveolate Chromera velia being the single known case of linear ptDNA. The linear extrachromosomal ptDNA has a telomere arrangement resembling those of linear mtDNA [117, 118].
Genes encoded in ptDNA are involved in the synthesis and storage of various cellular components, including those necessary for photosynthesis. Plastids have diverged to carry out different functions with multiple types identified. For example, chloroplasts are specialized for carrying out photosynthesis; chromoplasts contain pigments that provide petal colors, whereas amyloplasts are used for bulk storage of starch [117].
3.5.3 Nucleomorph DNA
A nucleomorph is a vestigial eukaryotic nucleus found in cryptomonads and chlorarachniophytes, which are both plastid-containing algae. The nucleomorph is located in these organisms between the inner and outer membranes of the plastid and is believed to be derived from the nucleus of an endosymbiotic algal cell engulfed by a larger eukaryotic cell [119]. Thus, the plastid organelle in this case evolved from two endosymbiotic events: a prokaryote was engulfed by a eukaryote which thereby became photoautotrophic and that cell was then engulfed by another eukaryote. The nucleomorph genomes are extremely small compared to the typical nuclear genome, being comprised of mostly single-copy housekeeping genes and having no mobile elements. The nucleomorph genome of the cryptomonads suggests that it was derived from a red algal ancestor, whereas the nucleomorph genome of the chlorarachniophytes suggests a green algal ancestor [119].
3.5.4 Plasmid DNA
Plasmids are present in bacteria, archaea, and eukaryotes [120]. Most plasmids are circular, although linear plasmids have been identified [121]. The genes carried on plasmids tend to be associated with functions that enable or enhance survival and growth under specific conditions. They can be horizontally transferred between prokaryotic cells and represent an important vehicle for sharing genetic information [122]. For example, a plasmid that has evolved an antibiotic resistance gene(s) can be transferred to neighboring bacteria promoting their rapid adaptation to various stresses associated with an antibiotic environment.
The eubacteria E. coli is estimated to have more than 270 plasmids having different distributions among and within cells; some promote mating, while others contain genes that kill other bacteria. The number of plasmids known and sequenced is much higher in bacteria as compared to archaea, with the lowest number having been identified in eukaryotes [122]. In recent years, plasmids have been used extensively in genetic engineering as a means of introducing and modifying target genes [122, 123].
3.5.5 MicroDNA
In 2012, Shibata et al. discovered a new form of extrachromosomal DNA in eukaryotes, called microDNA [122]. In contrast with other auxiliary DNA, microDNA is derived from non-repetitive sequences that are often associated with functional genes. They are circular DNA between 200 and 400 bp and are found in the nuclei of mammalian cells [122]. microDNA is thought to be associated with the repair and maintenance processes of nuclear DNA. It is not yet clear if microDNA plays a functional role in these processes or if they are merely an unavoidable by-product. For the time being, detection of specific microDNA is being proposed as a screening measure to aid the successful eradication of tumors in humans and as a potential method for cancer diagnosis and prognosis [124].
4 Genomic Storage and Processing of Information
It was not possible to understand how hereditary information was encoded and transmitted across generations without first having knowledge of the structure of DNA. Knowledge of DNA structure led to a structure-oriented conception of genomes as linear sequences of ordered nucleotides. Once protein synthesis was linked to gene sequences, the structural view of the genome began to be supplanted by the informational view [125]. Genetic information was initially viewed as a static property belonging to the specific sequence of ordered subunits. However, others have argued that the static view of information is not satisfactory (e.g., [4, 125]). Barbieri [125] contends that “it is only when a sequence provides a guideline to a copymaker that it becomes information for it. It is only an act of copying, in other words, that brings organic information into existence.” Based on Barbieri’s viewpoint, information is not always a property of a specific structure (e.g., DNA or RNA); rather his view is that such molecules are information relevant only when they are used to perform a biological function. A DNA sequence, for example, is said to have information if it is transcribed or interacts with a protein in a biologically relevant way. Similarly, an mRNA transcript also encodes information as it is translated into a protein. Also then, a protein could be viewed as an informational entity in the sense that it is necessary to carry out a biological function. Therefore, under this new conception, as well as the static view, it is clear that biological information can be manifest in different biological molecules; an observation that has complicated the notion of the genome as the fundamental unit of biological information [4].
We now understand that storage of the genetic information required to sustain life does not need to be restricted to biological molecules. This was vividly illustrated in the laboratory when a bacterial genome was chemically sequenced, its information stored within a computer (a completely different medium composed of binary states), then resynthesized in the form of a new DNA chromosome, and that synthetic DNA ultimately used as the sole means to maintain a living cell [126]. Although the information required for life can be stored independently of the chemical structure of the DNA, it cannot be expressed in a biologically useful form without various proteins and RNA molecules. Thus, expression of information encoded within a genome (bringing that information into existence) is contingent on its cellular context. In this section, we examine different ways in which information may be contained within a genome and mechanisms that result in biologically useful expression of that information.
4.1 Gene Expression
Mere knowledge of the DNA sequence of a genome is often insufficient to predict phenotype. The amount and timing of gene expression play a key role. For example, human cells with a nucleus have copies of almost identical DNA sequences. Yet cells perform varying functions, and they organize to create the multiple organs that constitute the human body. Cells achieve this primarily by differentially regulating the rate of transcription and/or translation of genes.
DNA transcription and protein translation comprise elementary levels of information transfer from genotype to phenotype. Maintaining control of these processes is fundamental for all organisms. Genetic elements involved in regulating gene expression are referred to as regulatory elements. They often represent sequences found on the DNA or RNA. In this way, regulatory information can be encoded directly within the nucleic acid sequence. Direct structural proximity if often not necessary, as regulatory elements may be found proximal or distal to the genes they affect. In humans, approximately 8% of nuclear DNA is composed of elements involved directly in regulation such as promoters, enhancers, silencers, and insulators (defined in Subheading 4.1.1; [127, 128]).
If all genetic and regulatory information is encoded in the DNA sequence, why can’t any cell with a complete genome be used to produce a viable organism? The specificity of cells suggests that additional regulatory markers also exist outside of the primary DNA sequence. This type of regulation is epigenetic (above the genes) and is essential for normal development. Epigenetic information is derived from chemical modifications of the chromosome (e.g., DNA methylation or histone modification) that do not change the primary sequence of chromosomal DNA and can be passed from one generation to the next [129, 130]. It is only through the collective actions of all cellular processes that gene products contribute to biochemical pathways and participate in the network of regulatory interactions to produce a complex organism or phenotype.
4.1.1 Transcriptional Regulation
DNA transcription is the chemical process through which information is transferred from DNA to RNA. The transcribed RNA may itself carry out some biological function or may be part of an intermediate information-carrying class of RNA known as messenger RNA (mRNA). mRNA along with other RNA molecules (tRNA and rRNA) are part of the machinery used to synthesize proteins. The flow of genetic information from DNA to RNA to protein is present in all forms of life. However, it is important to note that information transfer is not exclusively unidirectional. The enzyme reverse transcriptase can transfer genetic information from an RNA template into DNA.
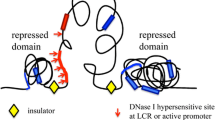
The basic model of transcriptional regulation requires that regulatory proteins called transcriptional factors (TFs) bind specific DNA sequences in regulatory modules (RMs). TFs are protein products that are themselves subjected to regulation of gene expression. RMs are defined according to both the primary DNA sequence to which TFs bind and their role in the process of regulating gene expression. One type of RMs are promoters. They are specific motifs on DNA that are necessary regulatory elements for RNA transcription in prokaryotes and eukaryotes. They bind the basal transcriptional machinery, RNA polymerase and general TFs. Enhancers are RMs that bind activator proteins and enhance the affinity of RNA polymerase to the promoter region. They, therefore, result in an upregulation of transcription of a gene or set of genes. Enhancers often act by stabilizing RNA polymerase binding through structural histone modifications [131]. Silencers are regulatory elements that when bound to repressor proteins function to prevent gene transcription. Silencers and enhancers are often distance-independent, meaning that they can act on gene(s) that are proximal or distal to their location [132]. Enhancers can be thought of as on-switches for gene expression, whereas silencers are the off-switches.
4.1.2 Translational Regulation
The fate of all mRNAs, transcribed from protein-coding genes, is not the same. The mRNA is often subjected to translational regulation depending on cellular and environmental conditions. These regulatory mechanisms affect the rate of protein synthesis. In prokaryotes and eukaryotes, most translational regulation involves structural changes in the mRNA molecule that impact its accessibility [133, 134]. The mRNAs can be sequestered in stress granules or localized in specific regions of a cell’s cytoplasm [135,136,137]. Another mechanism of translational regulation is RNA interference (RNAi). This regulation strategy is common in eukaryotes and involves short noncoding RNAs—microRNA (miRNA) or small interfering RNA (siRNA)—that bind with imperfect complementarity to their target mRNA transcripts. The binding of miRNA (or siRNA) to mRNA destabilizes (or degrades) the target mRNA, thereby inhibiting its translation. The imperfect pairing allows a single RNAi molecule to affect the expression of multiple genes. In the human genome, almost 50% of mRNA transcripts are regulated by one or more miRNAs [138].
In prokaryotes, transcription and translation are more tightly coupled than in eukaryotes, and this allows prokaryotes to regulate their gene expression primarily by controlling the amount of transcription. Nevertheless, prokaryotes can still conduct translational regulation. They can employ fundamentally different types of translational regulatory machinery: the recently discovered CRISPR-Cas system. Although the CRISPR loci were first identified in prokaryotes in 1987 [139], it was only recently described as a bacterial immune defense system [140]. The CRISP-Cas system is most commonly known to target external DNA (viral or plasmid) and degrade it before it can be transcribed or translated. Recent advancement suggests that some CRISPR-Cas systems are more general and have the capacity to target RNA molecules. This was first discovered in Pyrococcus furiosus [141]; similar RNA targeting was later found in Sufolobus solfataricus [142]. Throughout these advancements, CRISPR-Cas system was still strictly viewed as an immune response to target and degrade external nucleic acid molecules. It was only in 2016 that a CRISPR-Cas system was discovered that targets cellular mRNAs and thereby participates in translational regulation [143].
4.1.3 Epigenetics
The term epigenetics was coined in 1942 by Waddington [144]. He defined it as changes in an organism’s phenotype without an underlying alteration of its genome. It is now understood that epigenetic effects cause variation in phenotypes not associated with a change in the primary sequence but by chemical alterations of the DNA. Consider this analogy: throughout this review, whenever a word was being defined it was written in this format. If this chapter was rewritten with all bolds and italics removed, the informational content would be unaltered; however, the emphasis would be different. These “decorative” changes in font are akin to chemical epigenetic markers appended to the DNA. DNA methylation is a type of chemical decoration that is analogous to striking through a phrase. Specifically, it corresponds to the addition of a methyl group to parts of the DNA that results in gene silencing [145]. This additional information is not directly encoded within the primary DNA sequence but is manifested through chemical changes in nucleotides [145]. Thus, DNA methylation is one form of epigenetic control of gene expression. Epigenetic factors may also have an impact on regulation by changing protein-DNA binding. In eukaryotes, epigenetic factors may bind to consecutive histones moving them closer to each other. This results in local DNA compaction and prevents the expression of the gene(s) in this location.
Importantly, an organism’s exposure to certain environmental conditions can impact the epigenetic markers on its genome. Because epigenetic mechanisms ultimately affect the physiological form of the chromosome, such environmental exposures can lead to heritable changes in gene expression with no change to the underlying DNA sequence. It was initially thought that these alterations are not heritable and that following fertilization all epigenetic markers are removed from the zygote genome. Accumulating evidence suggests that such erasure of epigenetic marks occurs for most but not all genes [129, 130].
4.2 Mobile Genetic Elements
Also known as transposons or jumping genes, mobile genetic elements are sequences that can move around within a genome independently of the complex networks which otherwise regulate gene expression [146]. Through their movement, transposons often cause mutations either by inserting into a gene and disturbing its function or by promoting DNA rearrangement. If a transposon is inserted within a protein-coding region, then it will undoubtedly affect the expression of this gene by altering the final protein product. Transposons may also be inserted into regulatory regions resulting in over- or under-expression of certain gene(s). The capability of these DNA sequences to produce new copies of themselves elsewhere in a genome is called transposition. The two types of transposition are:
-
Copy-and-paste (replicative) transposition: a new copy of the transposable element is inserted into a new site, while the old copy remains integrated into the original site [147]. This type of transposition requires transfer of information into an RNA intermediate (retrotransposons) and subsequent retro-transcription into DNA. This mechanism results in an increase in the number transposon copies.
-
Cut-and-paste (non-replicative or conservative) transposition: the transposable element is excised from the old site and is inserted into a new site in the genome. The number of transposons is not increased in this case [147].
Transposable elements are found in all cell types. The kinds of transposable elements vary within and between prokaryotes and eukaryotes. They are often viewed as genetic parasites since they rely on a host cell for information processing systems (replication, transcription, and/or translation). In humans, about 44% of the genome is comprised of sequences that are related to transposable elements [148]. These mobile genetic elements had an important impact on eukaryotic evolution [149, 150]. For example, siRNA regulation is believed to have evolved to regain control of the expression of transposable elements [151]. For a review of the regulatory mechanisms of transposable elements, see ref. 152.
5 The Role of the Genome as an Informational Entity in Biology
Although the information contained within a genome is necessary to maintain a living cell, it is not sufficient on its own. Expression of biologically useful information requires a complex network of cellular components for processing and regulation of the genome. This dependency on external cellular components permits considerable flexibility in how the information is stored. As we have seen, the information essential for eukaryotic life is partitioned between chromosomes located in nuclear and organelle compartments, with some nuclear-encoded proteins being transported to the organelle for assembly with other proteins synthesized within the organelle [110]. Thus, as long as the cellular mechanisms for expression and processing are in place, genomic information can be physically dispersed within the cell. The Cryptophytes have taken this to an extreme, having their genomic information distributed across four cellular compartments: the nucleus, nucleomorph, mitochondria, and plastids [153]. Clearly, the physical location of the genome is not a constraint to information storage and processing. Furthermore, the storage of that information need not remain in a particular physical location. In the case of temperate phages, genomic information is transferred, for a period of time, to the genome of its host where it is maintained by its host’s replication processes [154]. These examples, and others (e.g., [126]), underscore the importance of viewing the genome foremost as an informational entity irrespective of its physical location.
In a well-argued critique of conventional notions of the genome, Goldman and Landweber [4] argue that viewing DNA as the sole source of information leads to additional difficulties. Recall that the NIH definition refers to the genome as containing all of the information needed to build and maintain that organism. We now understand that even the cell and its associated cytoplasm are not always sufficient for realization of all functional capabilities encoded within a genome. In other words, the genome, as conventionally defined, appears to be an incomplete informational entity [4]. Genome research has identified a variety of extracellular informational entities that can influence, and in some cases are even essential to, the creation and maintenance of an organism. Below we review selected examples of this phenomenon prior to reassessing the definition of the genome in light of modern genome science.
Marine cyanobacteria (Prochlorococcus and Synechococcus) are among the most abundant photosynthetic organisms in the world’s oceans. The viruses that infect them (cyanophages) were discovered to possess copies of some of their hosts photosynthesis genes (e.g., PsbA and PsbD: [155, 156]). Through the process of HGT, the cyanophages acquired host genes, which they express after infection to optimize their own gene expression and broaden their host range [157]. As novel as this discovery was, it was completely unexpected that the cyanobacteria and their phages continued to exchange genetic variation through homologous recombination [157]. Through such exchanges, the PsbA and PsbD genes participate in gene pools that extend beyond the photosynthetic species boundaries [157]. Given that cyanobacteria contribute as much as 30% of carbon fixation worldwide, those findings suggest that viral gene pool dynamics have influenced the evolution of oceanic photosynthesis on a global scale. This case demonstrates that to fully understand the origin and distribution of photosynthetic diversity, one must be aware that relevant genetic information can reside outside of the genomes of the photosynthetic organisms.
The bacterial genus Listeria is comprised of ecologically divergent lineages that share gene pools through the process of homologous recombination [158, 159]. Listeria monocytogenes is a pathogen closely related to the nonpathogenic species L. innocua. L. monocytogenes evolved as a pathogen through the process of HGT [160] and then subsequently evolved into ecologically divergent lineages differing in population structure and ability to respond to environmental stress [161]. Among Listeria, recombination is frequent enough to permit natural selection to act independently of the variability present at unlinked loci, thereby promoting or impeding exchangeability of genes among species and ecotypes residing in different niches [159]. This is just one example of the “mosaic genome” model of prokaryotic genome evolution, where the combined effects of recombination, drift, and selection lead to genomes comprised of a mosaic of differentially extendible trans-species gene pools. A wide variety of bacterial species are now thought to have genome dynamics consistent with the mosaic genome model [159, 162,163,164,165]. In some cases, the process of genomic divergence can even become decoupled from the process of ecological divergence [159, 163]. Thus, the physical genomes of some species of prokaryotes are incomplete informational entities.
The single-celled stichotrichous ciliates Oxytricha and Stylonychia have two nuclei that store genomic information in very different forms [166]. One nucleus, called the macronucleus, contains information in the form required for growth and maintenance of a cell. Hence, the macronuclear DNA is often referred to as “active.” The second nucleus, called the micronucleus, contains the same information in a “stored” form, which is used to produce the active form of the DNA in the next generation. However, information storage in the micronucleus is extremely complex. Protein-coding genes expressed by the macronucleus are partitioned into small segments, inverted, and scrambled among ~1 GB of other DNA sequences within the micronucleus. Furthermore, the production of a working macronucleus in the next generation cannot be accomplished without information contained within both small RNA molecules (piRNA) and long RNA templates (lncRNA), which are passed across generations via the cytoplasm of the maternal macronucleus [167, 168]. The piRNA are crucial to the elimination of DNA during the development of an active macronucleus, and the lncRNA mediate (1) unscrambling of the inactive micronuclear DNA, (2) regulation of gene dosage in the macronucleus, and (3) epigenetic transfer of somatic (macronuclear) alterations that are not found within the germ-line (micronuclear) DNA [167]. Thus, without those RNA molecules, the DNA genome of the stichotrichous ciliates is an incomplete informational entity [4]. Furthermore, emerging work on both Oxytricha and Stylonychia suggests that epigenetic modification of their DNA may play a role in the production of active macronuclear DNA [166, 169,170,171]
Complex microbial communities live in close association with the human body and have a strong impact on human health and disease. Host genetic variation is known to influence the composition of those communities [172], and, conversely, microbial variability is thought to influence various host disease states [173]. This association is so intimate that the microbiome has been referred to as an additional “human organ” [174], and substantial amounts of missing heritability associated with many complex human diseases are now being attributed, in part, to a failure to adequately account for microbial genetic variation [175]. Taking inflammatory bowel disease (IBD) as an example, host human genetic variation accounts for less than 50% of its estimated heritability [176]. This result implies that there exists undiscovered context dependence of human genetic variation for IBD. We have since come to understand that there is extensive inter-individual variation in the genetic composition of the gut microbiome and this metagenomic variation can influence healthy and dysregulated human immune responses [177] and is predictive of IBD patient outcomes [178]. Because the development of the IBD phenotype is related to gut microbiome variability, and because genetically similar human hosts can have different microbiomes, heritability estimates for human DNA variation will be impacted [175]. In other words, the expression of similar IBD phenotypes in humans is a function of both human and microbial genetics. Regardless of whether such interactions should be formally included within any future conception of the genome, this example illustrates how the human genome is also an incomplete informational entity with respect to prediction of healthy and disease states.
Goldman and Landweber [4] suggest that the notion of the genome should be reconceptualized in light of our modern, and deeper, understanding of genomic diversity and the mechanisms of information storage and processing. We agree and follow Goldman and Landweber [4] when they call for a “more expansive definition of the genome as an informational entity, often but not always manifest as DNA, encoding a broad set of functional capabilities that, together with other sources of information, produce and maintain the organism.” At first glance, this appears to be consistent with the controversial idea that a collection of functionally integrated organisms, called a holobiont, is a fundamental unit of biological organization and their set of genomes, called a hologenome, is itself a unit subject to evolution by natural selection [179]. However, we cannot go this far. We expect that any hologenome composed of informational entities having even a little independence is analogous to intra-genomic epistasis with just a little recombination. In the latter case, adaptive coevolution is not very effective at moving the system on its fitness landscape via compensatory substitutions [180]. Further, when informational entities are largely independent, either through high recombination (as observed in Listeria) or through independent replication (as within the human gut microbiome), the process of genomic divergence can become decoupled from ecological dynamics. Thus, we cannot agree with the notion of the hologenome as a unit of selection. Rather, we view the genome as a potential mosaic of gene pools subject to different evolutionary dynamics, and we follow Goldman and Landweber [4] by considering it foremost as an informational entity, which may be incomplete and which does not have to manifest exclusively as the DNA within a species boundary.
References
Lederberg J, McCray AT (2001) Ome SweetOmics--a genealogical treasury of words. Scientist 15(7):8
Patra S, Andrew AA (2015) Human, social, and environmental impacts of human genetic engineering. J Biomed Sci 4:2
Behjati S, Tarpey PS (2013) What is next generation sequencing? Arch Dis Child Educ Pract Ed 98(6):236–238
Goldman AD, Landweber LF (2016) What is a genome? PLoS Genet 12(7):e1006181
Tyler-Smith C, Yang H, Landweber LF, Dunham I, Knoppers BM, Donnelly P et al (2015) Where next for genetics and genomics? PLoS Biol 13(7):e1002216
Mueller RL (2015) Genome biology and the evolution of cell-size diversity. Cold Spring Harb Perspect Biol 7(11):a019125
Kysela DT, Randich AM, Caccamo PD, Brun YV (2016) Diversity takes shape: understanding the mechanistic and adaptive basis of bacterial morphology. PLoS Biol 14(10):e1002565
Minelli A, Fusco G (2010) Developmental plasticity and the evolution of animal complex life cycles. Philos Trans R Soc Lond B Biol Sci 365(1540):631–640
Forster SC (2017) Illuminating microbial diversity. Nat Rev Microbiol 15(10):578
Carroll SB (2001) Chance and necessity: the evolution of morphological complexity and diversity. Nature 409(6823):1102
History of life through time UCMP. www.ucmp.berkeley.edu/exhibits/historyoflife.php
The tree of life web project. tolweb.org
The encyclopedia of life. eol.org
Claverie J, Abergel C (2009) Mimivirus and its virophage. Annu Rev Genet 43:49–66
Pearson H (2008) ‘Virophages’ suggests viruses are alive. Nature 454(7205):677
Forterre P (2010) Defining life: the virus viewpoint. Orig Life Evol Biosph 40(2):151–160
Koonin EV, Starokadomskyy P (2016) Are viruses alive? The replicator paradigm sheds decisive light on an old but misguided question. Stud Hist Philos Biol Biomed Sci 59:125–134
Koonin EV (2010) The wonder world of microbial viruses. Expert Rev Anti-Infect Ther 8(10):1097–1099
Raoult D, Audic S, Robert C, Abergel C, Renesto P, Ogata H et al (2004) The 1.2-megabase genome sequence of Mimivirus. Science 306(5700):1344–1350
Finsterbusch T, Mankertz A (2009) Porcine circoviruses—small but powerful. Virus Res 143(2):177–183
Swiss Institute of Bioinformatics. ViralZone. http://www.expasy.org
Nadell CD, Drescher K, Foster KR (2016) Spatial structure, cooperation and competition in biofilms. Nat Rev Microbiol 14(9):589–600
Rosenberg SM (2009) Life, death, differentiation, and the multicellularity of bacteria. PLoS Genet 5(3):e1000418
Flores E, Herrero A (2010) Compartmentalized function through cell differentiation in filamentous cyanobacteria. Nat RevMicrobiol 8(1):39
Lasken RS, McLean JS (2014) Recent advances in genomic DNA sequencing of microbial species from single cells. Nat Rev Genet 15(9):577–584
Stewart EJ (2012) Growing unculturable bacteria. J Bacteriol 194(16):4151–4160
Qin Y, Hou J, Deng M, Liu Q, Wu C, Ji Y, He X (2016) Bacterial abundance and diversity in pond water supplied with different feeds. Sci Rep 6:35232
Jovel J, Patterson J, Wang W, Hotte N, O’Keefe S, Mitchel T et al (2016) Characterization of the gut microbiome using 16S or shotgun metagenomics. Front Microbiol 7:459
Peterson BF, Scharf ME (2016) Metatranscriptome analysis reveals bacterial symbiont contributions to lower termite physiology and potential immune functions. BMC Genomics 17(1):772
Young KD (2006) The selective value of bacterial shape. Microbiol Mol Biol Rev 70(3):660–703
Willis L, Huang KC (2017) Sizing up the bacterial cell cycle. Nat Rev Microbiol 15(10):606–620
Haeusser DP, Levin PA (2008) The great divide: coordinating cell cycle events during bacterial growth and division. Curr Opin Microbiol 11(2):94–99
Thanbichler M (2010) Synchronization of chromosome dynamics and cell division in bacteria. Cold Spring Harb Perspect Biol 2(1):a000331
Brown PJ, Hardy GG, Trimble MJ, Brun YV (2008) Complex regulatory pathways coordinate cell-cycle progression and development in Caulobacter crescentus. Adv Microb Physiol 54:1–101
Frost LS, Leplae R, Summers AO, Toussaint A (2005) Mobile genetic elements: the agents of open source evolution. Nat Rev Microbiol 3(9):722
Woese CR, Fox GE (1977) Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci U S A 74(11):5088–5090
Hug LA, Baker BJ, Anantharaman K, Brown CT, Probst AJ, Castelle CJ et al (2016) A new view of the tree of life. Nat Microbiol 1:16048
Williams TA, Foster PG, Cox CJ, Embley TM (2013) An archaeal origin of eukaryotes supports only two primary domains of life. Nature 504:231–236
Eckburg PB, Lepp PW, Relman DA (2003) Archaea and their potential role in human disease. Infect Immun 71(2):591–596
Nakamura N, Lin HC, McSweeney CS, Mackie RI, Gaskins HR (2010) Mechanisms of microbial hydrogen disposal in the human colon and implications for health and disease. Annu Rev Food Sci Technol 1:363–395
Saengkerdsub S, Ricke SC (2014) Ecology and characteristics of methanogenic archaea in animals and humans. Crit Rev Microbiol 40(2):97–116
Jarrell KF, Walters AD, Bochiwal C, Borgia JM, Dickinson T, Chong JP (2011) Major players on the microbial stage: why archaea are important. Microbiology 157(4):919–936
Prosser JI, Nicol GW (2008) Relative contributions of archaea and bacteria to aerobic ammonia oxidation in the environment. Environ Microbiol 10(11):2931–2941
Leigh JA (2000) Nitrogen fixation in methanogens: the archaeal perspective. Curr Issues Mol Biol 2:125–131
DeLong EF (1998) Everything in moderation: archaea as ‘non-extremophiles’. Curr Opin Genet Dev 8(6):649–654
Kandler O, König H (1998) Cell wall polymers in archaea (archaebacteria). Cell Mol Life Sci 54(4):305–308
Vollmer W, Bertsche U (2008) Murein (peptidoglycan) structure, architecture and biosynthesis in Escherichia coli. Biochim Biophys Acta 1778(9):1714–1734
Kates M (1992) Archaebacterial lipids: structure, biosynthesis and function. Biochem Soc Symp 58:51–72
Sato T, Atomi H (2011) Novel metabolic pathways in archaea. Curr Opin Microbiol 14(3):307–314
Lombard J, López-García P, Moreira D (2012) Phylogenomic investigation of phospholipid synthesis in archaea. Archaea 2012:630910
Forterre P (2013) The common ancestor of archaea and eukarya was not an archaeon. Archaea 2013:372396
Zillig W (1991) Comparative biochemistry of archaea and bacteria. Curr Opin Genet Dev 1(4):544–551
Moissl C, Rachel R, Briegel A, Engelhardt H, Huber R (2005) The unique structure of archaeal ‘hami’, highly complex cell appendages with nano-grappling hooks. Mol Microbiol 56(2):361–370
Szabo Z, Stahl AO, Albers SV, Kissinger JC, Driessen AJ, Pohlschroder M (2007) Identification of diverse archaeal proteins with class III signal peptides cleaved by distinct archaeal prepilin peptidases. J Bacteriol 189(3):772–778
Siebers B, Schönheit P (2005) Unusual pathways and enzymes of central carbohydrate metabolism in archaea. Curr Opin Microbiol 8(6):695–705
Coelho SM, Peters AF, Charrier B, Roze D, Destombe C, Valero M, Cock JM (2007) Complex life cycles of multicellular eukaryotes: new approaches based on the use of model organisms. Gene 406(1):152–170
Adl SM, Simpson AG, Lane CE, Lukeš J, Bass D, Bowser SS et al (2012) The revised classification of eukaryotes. J Eukaryot Microbiol 59(5):429–514
Mathur J (2004) Cell shape development in plants. Trends Plant Sci 9(12):583–590
Mogilner A, Keren K (2009) The shape of motile cells. Curr Biol 19(17):R771
Fagarasanu A, Rachubinski RA (2007) Orchestrating organelle inheritance in Saccharomyces cerevisiae. Curr Opin Microbiol 10(6):528–538
Bornens M (2008) Organelle positioning and cell polarity. Nat RevMol Cell Biol 9(11):874
Dyall SD, Brown MT, Johnson PJ (2004) Ancient invasions: from endosymbionts to organelles. Science (New York, NY) 304(5668):253–257
Corliss JO (2002) Biodiversity and biocomplexity of the protists and an overview of their significant roles in maintenance of our biosphere. Acta Protozool 41(3):199–220
Schlegel M, Hülsmann N (2007) Protists–a textbook example for a paraphyletic taxon. Organisms Divers Evol 7(2):166–172
Parfrey LW, Walters WA, Knight R (2011) Microbial eukaryotes in the human microbiome: ecology, evolution, and future directions. Front Microbiol 2:153
Caron DA, Alexander H, Allen AE, Archibald JM, Armbrust EV, Bachy C et al (2017) Probing the evolution, ecology and physiology of marine protists using transcriptomics. Nat Rev Microbiol 15(1):6–20
Sutton WS (1902) On the morphology of the chromoso group in Brachystola magna. Biol Bull 4(1):24–39
O’Donnell M, Langston L, Stillman B (2013) Principles and concepts of DNA replication in bacteria, archaea, and eukarya. Cold Spring Harb Perspect Biol 5(7):10
Dolgin E (2009) Human mutation rate revealed. Nat News. https://doi.org/10.1038/news.2009.864
Gowrishankar J, Harinarayanan R (2004) Why is transcription coupled to translation in bacteria? Mol Microbiol 54(3):598–603
Griswold A (2008) Genome packaging in prokaryotes: the circular chromosome of E. coli. Nat Educ 1(1):57
Koonin EV, Wolf YI (2008) Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res 36(21):6688–6719
Hou Y, Lin S (2009) Distinct gene number-genome size relationships for eukaryotes and non-eukaryotes: gene content estimation for dinoflagellate genomes. PLoS One 4(9):e6978
Cooper GM (2000) The cell: a molecular approach, 2nd edn. Sinauer Associates, Sunderland, MA
Gelderblom HR (1996) Structure and classification of viruses. In: Baron S (ed) Medical microbiology, 4th edn. The University of Texas Medical Branch at Galveston, Galveston, TX
Kay A, Zoulim F (2007) Hepatitis B virus genetic variability and evolution. Virus Res 127(2):164–176
Trifonov V, Khiabanian H, Rabadan R (2009) Geographic dependence, surveillance, and origins of the 2009 influenza A (H1N1) virus. N Engl J Med 361(2):115–119
Ganser-Pornillos BK, Yeager M, Sundquist WI (2008) The structural biology of HIV assembly. Curr Opin Struct Biol 18(2):203–217
Sun S, Rao VB, Rossmann MG (2010) Genome packaging in viruses. Curr Opin Struct Biol 20(1):114–120
Thomas V, Bertelli C, Collyn F, Casson N, Telenti A, Goesmann A et al (2011) Lausannevirus, a giant amoebal virus encoding histone doublets. Environ Microbiol 13(6):1454–1466
Chelikani V, Ranjan T, Kondabagil K (2014) Revisiting the genome packaging in viruses with lessons from the “Giants”. Virology 466:15–26
Teif VB, Bohinc K (2011) Condensed DNA: condensing the concepts. Prog Biophys Mol Biol 105(3):208–222
Chai Q, Singh B, Peisker K, Metzendorf N, Ge X, Dasgupta S, Sanyal S (2014) Organization of ribosomes and nucleoids in Escherichia coli cells during growth and in quiescence. J Biol Chem 289(16):11342–11352
Baril C, Richaud C, Baranton G, Saint Girons I (1989) Linear chromosome of Borrelia burgdorferi. Res Microbiol 140(7):507–516
Ferdows MS, Barbour AG (1989) Megabase-sized linear DNA in the bacterium Borrelia burgdorferi, the lyme disease agent. Proc Natl Acad Sci U S A 86(15):5969–5973
Rocha EP (2008) The organization of the bacterial genome. Annu Rev Genet 42:211–233
Cooper VS, Vohr SH, Wrocklage SC, Hatcher PJ (2010) Why genes evolve faster on secondary chromosomes in bacteria. PLoS Comput Biol 6(4):e1000732
Worning P, Jensen LJ, Hallin PF, Stærfeldt H, Ussery DW (2006) Origin of replication in circular prokaryotic chromosomes. Environ Microbiol 8(2):353–361
Nandakumar J, Cech TR (2013) Finding the end: recruitment of telomerase to telomeres. Nat Rev Mol Cell Biol 14(2):69–82
Cui T, Moro-oka N, Ohsumi K, Kodama K, Ohshima T, Ogasawara N et al (2007) Escherichia coli with a linear genome. EMBO Rep 8(2):181–187
Hopwood DA (2006) Soil to genomics: the Streptomyces chromosome. Annu Rev Genet 40:1–23
Casjens S (1999) Evolution of the linear DNA replicons of the Borrelia spirochetes. Curr Opin Microbiol 2(5):529–534
Chaconas G, Kobryn K (2010) Structure, function, and evolution of linear replicons in Borrelia. Annu Rev Microbiol 64:185–202
Fulcher N, Derboven E, Valuchova S, Riha K (2014) If the cap fits, wear it: an overview of telomeric structures over evolution. Cell Mol Life Sci 71(5):847–865
Samson RY, Bell SD (2011) Cell cycles and cell division in the archaea. Curr Opin Microbiol 14(3):350–356
Samson RY, Bell SD (2014) Archaeal chromosome biology. J Mol Microbiol Biotechnol 24(5–6):420–427
Bell SD, Jackson SP (2001) Mechanism and regulation of transcription in archaea. Curr Opin Microbiol 4(2):208–213
Reeve JN (2003) Archaeal chromatin and transcription. Mol Microbiol 48(3):587–598
Kelman LM, Kelman Z (2003) Archaea: an archetype for replication initiation studies? Mol Microbiol 48(3):605–615
Bell SD, White MF (2010) Archaeal chromatin organization. Bacterial chromatin. Springer, New York, pp 205–217
White MF, Bell SD (2002) Holding it together: chromatin in the archaea. Trends Genet 18(12):621–626
Mattiroli F, Gu Y, Yadav T, Balsbaugh JL, Harris MR, Findlay ES et al (2017) DNA-mediated association of two histone-bound complexes of yeast Chromatin Assembly Factor-1 (CAF-1) drives tetrasome assembly in the wake of DNA replication. elife 6:e22799
Forterre P, Confalonieri F, Knapp S (1999) Identification of the gene encoding archeal-specific DNA-binding proteins of the Sac10b family. Mol Microbiol 32(3):669–670
Zlatanova J, Caiafa P (2009) CCCTC-binding factor: to loop or to bridge. Cell Mol Life Sci 66(10):1647–1660
Tark-Dame M, van Driel R, Heermann DW (2011) Chromatin folding--from biology to polymer models and back. J Cell Sci 124(Pt 6):839–845
Biscotti MA, Olmo E, Heslop-Harrison JP (2015) Repetitive DNA in eukaryotic genomes. Chromosom Res 23(3):415–420
Rubin GM, Spradling AC (1982) Genetic transformation of drosophila with transposable element vectors. Science (New York, NY) 218(4570):348–353
Treangen TJ, Salzberg SL (2011) Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet 13(1):36–46
Shibata Y, Kumar P, Layer R, Willcox S, Gagan JR, Griffith JD, Dutta A (2012) Extrachromosomal microDNAs and chromosomal microdeletions in normal tissues. Science (New York, NY) 336(6077):82–86
Chen XJ, Butow RA (2005) The organization and inheritance of the mitochondrial genome. Nat RevGenet 6(11):815
Karnkowska A, Vacek V, Zubáčová Z, Treitli SC, Petrželková R, Eme L et al (2016) A eukaryote without a mitochondrial organelle. Curr Biol 26(10):1274–1284
Stehling O, Lill R (2013) The role of mitochondria in cellular Iron-sulfur protein biogenesis: mechanisms, connected processes, and diseases. Cold Spring Harb Perspect Biol 5(8):a011312
Nosek J, Tomáška Ľ (2003) Mitochondrial genome diversity: evolution of the molecular architecture and replication strategy. Curr Genet 44(2):73–84
Smith DR, Keeling PJ (2013) Gene conversion shapes linear mitochondrial genome architecture. Genome Biol Evol 5(5):905–912
Kolesnikov AA, Gerasimov ES (2012) Diversity of mitochondrial genome organization. Biochemistry 77(13):1424
Green BR (2011) Chloroplast genomes of photosynthetic eukaryotes. Plant J 66(1):34–44
Rogalski M, do Nascimento Vieira L, Fraga HP, Guerra MP (2015) Plastid genomics in horticultural species: importance and applications for plant population genetics, evolution, and biotechnology. Front Plant Sci 6:586
Janouškovec J, Sobotka R, Lai D, Flegontov P, Koník P, Komenda J et al (2013) Split photosystem protein, linear-mapping topology, and growth of structural complexity in the plastid genome of Chromera velia. Mol Biol Evol 30(11):2447–2462
Archibald JM (2007) Nucleomorph genomes: structure, function, origin and evolution. BioEssays 29(4):392–402
Funnell BE, Phillips GJ (2004) Plasmid biology. ASM Press, Washington, DC
Ravin NV (2011) N15: the linear phage–plasmid. Plasmid 65(2):102–109
Shintani M, Sanchez ZK, Kimbara K (2015) Genomics of microbial plasmids: classification and identification based on replication and transfer systems and host taxonomy. Front Microbiol 6:242
Burgess DJ (2017) Genetic engineering: CREATE-ing genome-wide designed mutations. Nat Rev Genet 18(2):69
Kumar P, Dillon LW, Shibata Y, Jazaeri AA, Jones DR, Dutta A (2017) Normal and cancerous tissues release extrachromosomal circular DNA (eccDNA) into the circulation. Mol Cancer Res 15(9):1197–1205
Barbieri M (2016) What is information? Philos Trans R Soc A 374(2063):20150060
Gibson DG, Glass JI, Lartigue C, Noskov VN, Chuang RY, Algire MA et al (2010) Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329(5987):52–56
ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489(7414):57–74
Rands CM, Meader S, Ponting CP, Lunter G (2014) 8.2% of the human genome is constrained: variation in rates of turnover across functional element classes in the human lineage. PLoS Genet 10(7):e1004525
Trerotola M, Relli V, Simeone P, Alberti S (2015) Epigenetic inheritance and the missing heritability. Hum Genomics 9(1):17
Tang WW, Dietmann S, Irie N, Leitch HG, Floros VI, Bradshaw CR et al (2015) A unique gene regulatory network resets the human germline epigenome for development. Cell 161(6):1453–1467
Atkinson TJ, Halfon MS (2014) Regulation of gene expression in the genomic context. Comput Struct Biotechnol J 9(13):1–9
Narlikar L, Ovcharenko I (2009) Identifying regulatory elements in eukaryotic genomes. Brief FunctGenomic Proteomic 8(4):215–230
Hershey JW, Sonenberg N, Mathews MB (2012) Principles of translational control: an overview. Cold Spring Harb Perspect Biol 4(12):a011528
Meyer MM (2017) The role of mRNA structure in bacterial translational regulation. Wiley Interdiscip Rev RNA 8(1):e1370
Chao JA, Yoon YJ, Singer RH (2012) Imaging translation in single cells using fluorescent microscopy. Cold Spring Harb Perspect Biol 4(11):a012310
Lasko P (2012) mRNA localization and translational control in Drosophila oogenesis. Cold Spring Harb Perspect Biol 4(10):a012294
Decker CJ, Parker R (2012) P-bodies and stress granules: possible roles in the control of translation and mRNA degradation. Cold Spring Harb Perspect Biol 4(9):a012286
Chekulaeva M, Filipowicz W (2009) Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr Opin Cell Biol 21(3):452–460
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A (1987) Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 169(12):5429–5433
Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV (2006) A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct 1(1):7
Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L et al (2009) RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 139(5):945–956
Zhang J, Rouillon C, Kerou M, Reeks J, Brugger K, Graham S et al (2012) Structure and mechanism of the CMR complex for CRISPR-mediated antiviral immunity. Mol cell 45(3):303–313
Liu Y, Chen Z, He A, Zhan Y, Li J, Liu L et al (2016) Targeting cellular mRNAs translation by CRISPR-Cas9. Sci Rep 6:29652
Waddington CH (1942) The epigenotype. Endeavour 1:18–20
Allis CD, Jenuwein T (2016) The molecular hallmarks of epigenetic control. Nat Rev Genet 17:487–500
Goodier JL, Kazazian HH (2008) Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell 135(1):23–35
Ahmed A (2009) Alternative mechanisms for Tn5 transposition. PLoS Genet 5(8):e1000619
Mills RE, Bennett EA, Iskow RC, Devine SE (2007) Which transposable elements are active in the human genome? Trends Genet 23(4):183–191
Huda A, Jordan IK (2009) Epigenetic regulation of mammalian genomes by transposable elements. Ann N Y Acad Sci 1178(1):276–284
López-Flores I, Garrido-Ramos MA (2012) The repetitive DNA content of eukaryotic genomes. In: Repetitive DNA, vol 7. Karger Publishers, Basel, pp 1–28
Ivics Z, Li MA, Mátés L, Boeke JD, Nagy A, Bradley A, Izsvák Z (2009) Transposon-mediated genome manipulation in vertebrates. Nat Methods 6(6):415–422
Chuong EB, Elde NC, Feschotte C (2017) Regulatory activities of transposable elements: from conflicts to benefits. Nat Rev Genet 18(2):71–86
Curtis BA, Tanifuji G, Burki F, Gruber A, Irimia M, Maruyama S et al (2012) Algal genomes reveal evolutionary mosaicism and the fate of nucleomorphs. Nature 492(7427):59–65
Howard-Varona C, Hargreaves KR, Abedon ST, Sullivan MB (2017) Lysogeny in nature: mechanisms, impact and ecology of temperate phages. ISME J 11:1511–1520
Lindell D, Sullivan MB, Johnson ZI, Tolonen AC, Rohwer F et al (2004) Transfer of photosynthesis genes to and from Prochlorococcus viruses. Proc Natl Acad Sci U S A 101:11013–11018
Zeidner G, Bielawski JP, Shmoish M, Scanlan DJ, Sabehi G, Béjà O (2005) Potential photosynthesis gene recombination between Prochlorococcus and Synechococcus via viral intermediates. Environ Microbiol 7(10):1505–1513
Sullivan MB, Lindell D, Lee JA, Thompson LR, Bielawski JP, Chisholm SW (2006) Prevalence and evolution of core photosystem II genes in marine cyanobacterial viruses and their hosts. PLoS Biol 4(8):e234
Orsi RH, Sun Q, Wiedmann M (2008) Genome-wide analyses reveal lineage specific contributions of positive selection and recombination to the evolution of Listeria monocytogenes. BMC Evol Biol 8(1):233
Dunn KA, Bielawski JP, Ward TJ, Urquhart C, Gu H (2009) Reconciling ecological and genomic divergence among lineages of Listeria under an “extended mosaic genome concept”. Mol Biol Evol 26(11):2605–2615
Buchrieser C, Rusniok C, Kunst F, Cossart P, Glaser P (2003) Comparison of the genome sequences of Listeria monocytogenes and Listeria innocua: clues for evolution and pathogenicity. Pathog Dis 35(3):207–213
Nightingale KK, Windham K, Wiedmann M (2005) Evolution and molecular phylogeny of Listeria monocytogenes isolated from human and animal listeriosis cases and foods. J Bacteriol 187(16):5537–5551
Lawrence JG (2002) Gene transfer in bacteria: speciation without species? Theor Popul Biol 61(4):449–460
Nesbø CL, Dlutek M, Doolittle WF (2006) Recombination in Thermotoga: implications for species concepts and biogeography. Genetics 172(2):759–769
Retchless AC, Lawrence JG (2007) Temporal fragmentation of speciation in bacteria. Science 317(5841):1093–1096
Papke RT, Zhaxybayeva O, Feil EJ, Sommerfeld K, Muise D, Doolittle WF (2007) Searching for species in haloarchaea. Proc Natl Acad Sci U SA 104(35):14092–14097
Wang Y, Wang Y, Sheng Y, Huang J, Chen X, AL-Rasheid KA, Gao S (2017) A comparative study of genome organization and epigenetic mechanisms in model ciliates, with an emphasis on Tetrahymena, Paramecium and Oxytricha. Eur J Protistol 61(Pt B):376–387
Yerlici VT, Landweber LF (2014) Programmed genome rearrangements in the Ciliate Oxytricha. Microbiol Spectr 2:6. https://doi.org/10.1128/microbiolspec.MDNA3-0025-2014
Pilling OA, Rogers AJ, Gulla-Devaney B, Katz LA (2017) Insights into transgenerational epigenetics from studies of ciliates. Eur J Protistol 61(Pt B):366–375
Bulic A, Postberg J, Fischer A, Jönsson F, Reuter G, Lipps HJ (2013) A permissive chromatin structure is adopted prior to site-specific DNA demethylation of developmentally expressed genes involved in macronuclear differentiation. Epigenetics Chromatin 6(1):5
Bracht JR, Fang W, Goldman AD, Dolzhenko E, Stein EM, Landweber LF (2013) Genomes on the edge: programmed genome instability in ciliates. Cell 152(3):406–416
Forcob S, Bulic A, Jönsson F, Lipps HJ, Postberg J (2014) Differential expression of histone H3 genes and selective association of the variant H3. 7 with a specific sequence class in Stylonychia macronuclear development. Epigenetics Chromatin 7(1):4
Blekhman R, Goodrich JK, Huang K, Sun Q, Bukowski R, Bell JT et al (2015) Host genetic variation impacts microbiome composition across human body sites. Genome Biol 16(1):191
Clemente JC, Ursell LK, Parfrey LW, Knight R (2012) The impact of the gut microbiota on human health: an integrative view. Cell 148(6):1258–1270
Baquero F, Nombela C (2012) The microbiome as a human organ. Clin Microbiol Infect 18(s4):2–4
Sandoval-Motta S, Aldana M, Martínez-Romero E, Frank A (2017) The human microbiome and the missing heritability problem. Front Genet 8:80
Gordon H, Moller FT, Andersen V, Harbord M (2015) Heritability in inflammatory bowel disease: from the first twin study to genome-wide association studies. Inflamm Bowel Dis 21(6):1428
Dunn KA, Moore-Connors J, MacIntyre B, Stadnyk A, Thomas NA, Noble A et al (2016) The gut microbiome of pediatric Crohn’s disease patients differs from healthy controls in genes that can influence the balance between a healthy and dysregulated immune response. Inflamm Bowel Dis 22(11):2607–2618
Dunn KA, Moore-Connors J, MacIntyre B, Stadnyk AW, Thomas NA, Noble A et al (2016) Early changes in microbial community structure are associated with sustained remission after nutritional treatment of pediatric Crohn’s disease. Inflamm Bowel Dis 22(12):2853–2862
Bordenstein SR, Theis KR (2015) Host biology in light of the microbiome: ten principles of holobionts and hologenomes. PLoS Biol 13(8):e1002226
Gavrilets S (2004) Fitness landscapes and the origin of species (MPB-41), vol 41. Princeton University Press, Princeton, NJ
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2019 The Author(s)
About this protocol

Cite this protocol
Youssef, N., Budd, A., Bielawski, J.P. (2019). Introduction to Genome Biology and Diversity. In: Anisimova, M. (eds) Evolutionary Genomics. Methods in Molecular Biology, vol 1910. Humana, New York, NY. https://doi.org/10.1007/978-1-4939-9074-0_1
Download citation
DOI: https://doi.org/10.1007/978-1-4939-9074-0_1
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-4939-9073-3
Online ISBN: 978-1-4939-9074-0
eBook Packages: Springer Protocols