
Abstract
Metazoans contain multiple complex microbial ecosystems in which the balance between host and microbe can be tipped from commensalism to pathogenicity. This transition is likely to depend both on the prevailing environmental conditions and on specific gene-gene interactions placed within the context of the entire ecosystem.
Similar content being viewed by others


Metazoans and higher plants are not single-species organisms, but are complex ecosystems composed of a multicellular eukaryotic host, with its unique genetic complement [1], and a multitude of 'microbiomes'. Each microbiome is composed of multiple prokaryotic and eukaryotic symbionts, and the microbiomes and the host collectively make up the 'symbiome' (Table 1) [2]. Symbiotic relationships within these ecosystems exist between each of the microbial strains and the host, and also between and among the members of each microbiome. These interdependencies run the gamut from mutualism (in which both or all species benefit) to commensalism (where one party benefits and does no appreciable harm to the others) to parasitism (where one of the species benefits at the expense of the other(s)). Finally, a pathogenic relationship exists if the parasite produces a morbid condition in the host. These divisions are themselves an oversimplification of what is, in all likelihood, a continuum: where a given strain of microorganism falls within this spectrum depends not only on its genomic complement but also on the makeup of the microbiome as well as the individual host's genetics and other environmental factors.
Pathogenicity is not only dependent on qualitative issues such as the presence of specific species, strains, or genes, but also on their relative abundances. Thus, the differential growth of one microbe may result in others transitioning into or out of pathogenic status. It is therefore likely that many pathogens did not initially evolve as pathogens, but simply take on this role as a result of a lack of ability of the host to maintain homeostasis [3]. Interestingly, not all bacteria associated with pathogenic processes cause disease by their presence; some bacteria are pathogenic by their absence, such as the vaginal lactobacilli whose loss results in an increased pH, which permits overgrowth by invasive species [4–6]. What makes a pathogen, therefore, is the addition, or deletion, of metabolic capabilities in the symbiome that results in a disruption of homeostasis.
Genetic heterogeneity among bacterial populations makes for challenging taxonomy
Bacterial plurality embodies the following concepts: bacteria within a species display enormous phenotypic and genotypic heterogeneity [7]; microbial colonization is nearly universally polyclonal [8–11]; and microbiomes occupying the same niche in different hosts are vastly different with respect to phylogenetic structure [12–14]. Thus, the hologenome (see Table 1 for a definition) is not fixed, but varies with age, health, diet, and other environmental factors. In spite of this plasticity, however, we hope to be able to characterize a set of common features associated with a healthy hologenome as opposed to a disease-state hologenome [15] - the goal of the NIH Microbiome Roadmap Project [16]. We hypothesize that disease-state hologenomes will often display reduced complexity (for example, Clostridium difficile overgrowth in the intestine following antibiotic treatment [17], or a reduced gut microflora associated with patients with inflammatory bowel disease [18]) in a manner analogous to damaged sites in the environment that have been shown to have reduced microbial complexity [19–21].
For many bacterial pathogens, such as the non-typeable Haemophilus influenzae (NTHi) [22, 23], Pseudomonas aeruginosa [24, 25], Staphylococcus aureus (RJ Boissy, unpublished data), Streptococcus agalactiae [26], and Streptococcus pneumoniae [27, 28], whole-genome sequencing has shown that the supragenome is several times larger than the core genome (see Table 1 for definitions). Thus, for these species there are more distributed genes (see Table 1) than core genes. This leads to the realization that bacterial species-level diagnostics are woefully inadequate as prognosticators of disease potential. Therefore, it was not surprising that disease phenotyping for multiple independent isolates of NTHi [29] and pneumococcus (Streptococcus pneumoniae) [30] revealed a spectrum of diseases - from localized chronic infections to universal lethality.
Similarly, species within the Enterobacteriaceae each reveal a broad spectrum of symbiotic relationships with their hosts. The species Escherichia coli contains both mutualistic strains that have a role in host nutrition, and other strains associated with either chronic urinary disease or acute enterohemorrhagic infections [31, 32]. Similarly, pathogenic strains of Enterococcus faecium have emerged from a commensal species, as we discuss below. Whole-genome sequencing of the divergent strains in these species has revealed massive gene loss and gene gain, resulting in intra-species genomes that vary by more than 30% in size [32].
Bacterial species are usually defined by their 16S rRNA gene. Whereas this is useful for determining phylogenetic relationships based on vertically acquired genetic traits, it does not account for horizontally acquired traits, that is, genes acquired by transfer from other species, which are the major driving force in bacterial evolution [23]. Thus, 16S-rRNA-based phylogenies lump together strains that have widely divergent gene distributions, metabolic capabilities, and pathogenic characters [23, 26, 28–33]. A species definition based on possession of a core genome has been proposed [7], but even this is too inclusive to be useful in clinical diagnostics. With the increasing availability of whole-genome sequencing and comparative genomic hybridization (CGH), it should be possible to obtain and analyze very large amounts of bacterial genomic data, which could be cross-indexed with strain-specific disease virulence information to develop effective clinical prognostic indicators.
Genes and gene combinations determine pathogenicity
As discussed above, within-species comparative genomics combined with disease phenotyping can identify classes of virulence genes that are associated with different pathogenic profiles [22–32]. These findings strongly implicate specific distributed genes and gene combinations as the determinants of which bacterial strains are likely to act as pathogens. Both genotypic and phenotypic heterogeneity have been demonstrated for the pneumococcus, with some strains associated with chronic indolent infections whereas others are associated with invasive or systemic disease [30]. Similarly, the NTHi display a broad spectrum of phenotypes [29] as well as having a highly plastic genome [22, 23], making it likely that correlation studies would find virulence-specific genetic and metabolic pathways.
This view is a departure from classical medical microbiology in which a species-level diagnosis is used to make a prognosis. Thus, diagnostics development would profit from large-scale bacterial genotype-phenotype correlation studies designed to provide information on the distributed genes, which are the genes most frequently associated with disease states. Such disease-associated genes may be largely confined to a single species, or may be passed among related species, or may be more widely transmitted across broader taxonomic lineages. Examples of species-specific distributed genes include the various heme-acquiring genes found among the NTHi, and the multiple IgA-cleaving proteases isolated among the pneumococci. Within the order Enterobacteriaceae, the shiga-like toxin genes have been isolated from multiple species, and at higher taxonomic levels, gene cassettes for antibiotic resistance and for natural competence (that is, the ability to take up DNA from the environment) have been passed between Gram-negative and Gram-positive bacteria.
The ability to carry out whole-genome sequencing of relatively large numbers of bacterial strains using 454-based sequencing technology [34] provides a means of rapidly and inexpensively characterizing the species' core genomes and supragenomes. Once a relatively complete species supra-genome is available [23, 28], microarrays can be constructed containing probes for each distributed gene. These CGH arrays can then be used to interrogate the genomes of large numbers of clinical isolates with different disease phenotypes, providing the information to perform quantitative trait locus-based gene-association studies for the identification of disease-specific virulence genes. Such a statistical approach to bacterial genetics is new, as until now there have been insufficient sequence data for such an approach. The application of this technology would also provide a comprehensive means of characterizing the functional roles of the plurality of unannotated genes that exist in even the best-studied bacterial species.
How do pathogens evolve and where do they come from?
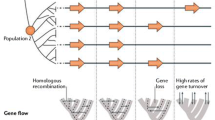
The distributed genome hypothesis [35, 36] states that bacterial pathogens arise and acquire virulence traits primarily via horizontal gene transfer (Figure 1). More recently, it has become clear that many bacteria are multicellular organisms during part of their life cycle [37], and this has led to the recognition that bacteria possess a number of virulence traits that are expressed only at the population level and are not operational at the single-cell level [38]. These hypotheses are based on the observation that nearly all classes of pathogenic bacteria maintain highly energy-demanding mechanisms for accessing foreign DNA [39], in spite of the fact that most of these species maintain small genomes. The importance of this observation is that in a background of processes that favor gene deletion [40], the maintenance of multiple horizontal gene transfer mechanisms indicates that these traits are highly selected for. The distributed genome hypothesis also posits that chronic pathogens utilize the distribution of non-core genes among strains of a species as a survival strategy, whereby the continuous recombination of genetic characters between strains serves as a supra-virulence factor that improves population survival through the generation of new strains with novel combinations of genes. Thus, this population-level gene reassortment acts as a counterpoint to the adaptive immune response of vertebrates, providing a means for pathogens to constantly present the host with novel antigens obtained from any of the constituent species of the symbiome.

The distributed genome hypothesis. (a) Schematic showing the distributed (non-core) genes of a species supragenome in a population pool with individual strains below each containing the same set of core genes (green helix). (b) Schematic showing each of the strains of a species with the core genome and a unique distribution of non-core genes.
Many pathogenic bacteria have complex life cycles that include stages in the environment and passage through multiple hosts. These organisms, therefore, come in contact with many different selective pressures at various stages of their life cycle, and some of the adaptations that provide protection from predation or competition in one stage can induce pathogenicity in another stage. One way in which pathogens evolve is that environmental organisms acquire genes through horizontal transfer that give them an advantage within their non-pathogenic ecosystem. A classic example is the evolution of pathogenic forms of Vibrio cholerae, non-pathogenic progenitor strains of which are principally found in aquatic ecosystems. Pathogenic strains originate from non-pathogenic strains through a multistep process that includes the acquisition of the type IV toxin-co-regulated pilus (TCP). This acquisition is followed by infection with the filamentous phage CTXϕ, which uses the pilus as a point of entry and provides the genes encoding cholera toxin [41]. Studies of cholera epidemics suggest that this general series of genomic rearrangements occurs independently in each epidemic in response to competition among extant environmental strains. These studies led Faruque et al. [41] to hypothesize that "continual emergence of new toxigenic strains and their selective enrichment during cholera outbreaks constitute an essential component of the natural ecosystem for the evolution of epidemic V. cholerae strains to ensure its continued existence."
Legionella pneumophila, a bacterium that lives intra-cellularly, also probably evolved its pathogenic characters outside the human host. In humans, L. pneumophila grows and replicates in human alveolar macrophages to cause pneumonia, particularly in immunocompromised hosts. The ability to live within phagocytic cells is the critical virulence factor for this organism and is encoded by the icm/dot secretion system [42], which originally evolved to permit the bacterium's survival within free-living grazing protozoa. Similarly, E. coli O157, although notorious as a highly virulent enterohemorrhagic pathogen of humans, is primarily a commensal microorganism of cattle that also lives in the environment. Although E. coli O157 can be transmitted from person to person, this is not its principal means of propagation; thus, it is likely that its virulence in humans is a byproduct of other evolutionary forces. Many E. coli strains, including O157, that contain a lambda-like prophage carrying the shiga-like toxin genes (stx) have been shown to have a survival advantage in the presence of the ubiquitous bactivorous protozoan Tetrahymena pyriformis [43]. These investigations showed that most of the survival advantage of the stx-containing strains can be attributed to better survival within the protozoan's food vacuoles. Thus, for both L. pneumophila and O157 it would appear that the primary virulence factors associated with human disease actually evolved to play a critical role in the organisms' survival in other stages of their life cycles. Interestingly, however, the shiga toxin of O157 causes diarrhea in humans, which could lead to increased spread of this strain through fecal contamination. Thus, it is tempting to speculate that acquisition of shiga toxins may be under multiple unrelated evolutionary pressures.
Competition among microorganisms can also generate strains that are pathogenic in their host as a side effect of the intermicrobial arms race. Microorganisms rarely live in isolation, and the myriad interactions amongst co-colonizing species and strains impose a constant selective pressure that ensures the continual evolution of new strains. Thus the same bacterial horizontal gene transfer mechanisms that provide a counterpoint to the host's adaptive immune response also serve to generate more competitive strains for interspecies competition, with some of these antibacterial mechanisms also resulting in increased virulence towards the host. There is abundant evidence that the numerous bacterial species colonizing the human respiratory mucosa are in competition with each other. Both NTHi and the pneumococcus form biofilms on the middle-ear mucosa that are associated with chronic otitis media but, even when both species are present in the same sample, they do not form mixed biofilms [44]. NTHi can also induce an anti-pneumo-coccal host response during mixed infections that is characterized by increased recruitment of neutrophils into the paranasal spaces [45]. This favors H. influenzae - in spite of the fact that in mixed laboratory culture the pneumo-coccus predominates. Conversely, H. influenzae is competed against by S. pneumoniae. Both H. influenzae and Neisseria meningitidis use sialylation of lipooligosaccharides as a mechanism to evade host immune surveillance through mimicry, whereas S. pneumoniae expresses NanA, which desialylates the cell surface of both these bacteria [46]. NanA also alters multiple surface carbohydrates and removes sialic acid residues from human epithelial cells [47]. Disruption of NanA decreases the ability of the pneumococcus to establish a persistent infection, as it can no longer expose the sialylated host-cell receptors needed for attachment [48]. Thus, NanA plays a role in pathogenesis as well as in inter-species competition.
A single molecule is, however, not always advantageous in interactions both with the host and between competing microorganisms. The pore-forming toxin of S. pneumoniae, pneumolysin, increases access of the peptidoglycan of H. influenzae cell walls to cytoplasmic immune molecules that initiate an anti-pneumococcal response, thus providing an advantage to H. influenzae [49]. Thus, the balance between fitness in different environmental settings is critical when considering how pathogens evolve. Mutations that offer a fitness advantage in one environment may confer a disadvantage in another. This is perhaps best understood in respect of microbial drug resistance, where mutations that confer an advantage in the presence of drugs are often deleterious (resulting in slower growth rates) in its absence.
In the monitoring of emerging pathogens it will become increasingly important to recognize the genes and regulatory systems that facilitate transition into a new niche or that balance gene expression within a strain such that it can survive in different environments. In a recent study, Giraud and colleagues [50] created gnotobiotic mice by colonizing germ-free mice with E. coli. In each of eight independent experiments, after habituation, the bacteria were shown to have mutations in the EnvZ-OmpR two-component response regulator, a signal transduction system that controls an entire regulon. This strongly implicates this locus as providing a fitness advantage in this particular environment [50]. This is likely to be the case for many master regulators, and given such an important role in adaptation one might expect these genes to be mostly part of the core genome. In the pneumococcus, however, only a subset of the predicted two-component signal-response systems are core-encoded. Thus, it remains to be determined whether the distributed two-component systems affect pneumococcal fitness under any particular environmental condition, and how the presence, absence, and mutation of these master regulators provides an advantage for one strain over another.
Many pathogens evolve in situ from species that are commensals in the eukaryotic host. This is not surprising, as these organisms are already adapted for survival within the extant symbiome and acquisition of virulence genes can produce a pathogen de novo. Examples of adaptation to a new niche selecting for virulence are commonly observed within the genus Salmonella. Salmonella enterica subspecies I is well adapted to warm-blooded vertebrates. There are more than 1,000 serotypes of this subspecies with different degrees of host adaptation. The level of host specificity among the serotypes correlates with their capacity to cause disease. Mononuclear phagocytes are barriers to the host range of S. enterica, and mechanisms enabling survival of the bacteria within these cells allow adaptation to individual host species [51]. The serotype Typhimurium is successful in mice, and survives well in murine, but not human, macrophages; the reverse is true for the serotype Typhi, which causes disease in humans. In contrast, other subspecies of S. enterica are mainly associated with cold-blooded vertebrates. It is thought that these subspecies survive in the alimentary tract of reptiles, where they are well adapted as commensal organisms [51].
Another example of pathogenic strains evolving from non-pathogenic ones via horizontal gene transfer is the case of Enterococcus faecium. This bacterium has recently evolved from a commensal into a frequently isolated nosocomial (hospital-acquired) pathogen in intensive care units [52]. Comparative genomics has shown that the pathogenic strains have arisen from multiple backgrounds, but all show evidence of having acquired insertion elements (a type of transposable element) that are not present in the commensal strains. Thus, the creation of a new environmental niche, the intensive care unit, has facilitated the evolution of a new subpopulation of this species. The degree of genetic variation among strains in the 'hospital clade' of E. faecium (as assessed by pulsed-field gel electrophoresis and multi-locus sequence typing) was compared with the degree of variation among all other strains. This revealed that the diversity indices (ratio of average genetic similarities) were higher for the hospital clade [52], strongly suggesting increased genomic plasticity within this population that is likely to facilitate its further adaptation.
Host mutations are associated with the development of bacterial pathogenicity
An example of specific host-bacterium gene combinations resulting in pathogenesis (and the evolution of a pathogen from a commensal) involves the human genetic disease cystic fibrosis. This disease is caused by mutations in the human CFTR gene that lead to the loss of a chloride channel, resulting in highly viscous pulmonary mucus that prevents the normal activity of the 'mucociliary escalator', which is designed to sweep bacteria out of the airways. The disease first becomes apparent with colonization and chronic infection by NTHi, which leads inexorably to secondary infection by the opportunistic environmental bacterium P. aeruginosa, which establishes a chronic infection involving a biofilm. The pseudomonal infection is ultimately lethal (although modern medical practice can extend life for decades). What is most interesting is that as the P. aeruginosa infection transitions from acute to chronic, there is significant evolution of the bacterial genome [53–56] that makes P. aeruginosa much more pathogenic in the lungs of cystic fibrosis patients. Proof of this hypothesis came with the observation that preadolescents with cystic fibrosis who attended the same clinics and summer camps as older adolescents with the disease were experiencing very rapid clinical progression. Molecular typing of the P. aeruginosa isolates revealed that the young children were being infected with the highly evolved chronic pathogens, adapted to the cystic fibrotic lung, from the older people [56]. In the final analysis, sequential colonization by multiple bacterial species, none of which is highly pathogenic in the healthy host, evolves into what becomes a lethal infection in the presence of a defective host gene. Thus, the cystic fibrosis lung illustrates the concept that the entire composition of the hologenome is important in defining pathogenicity and virulence.
Novel pathogens are constantly emerging from environmental and commensal bacterial flora as a result of competitive selective pressures and ubiquitous horizontal gene transfer. Many, perhaps most, virulence traits did not arise originally to damage the host, but rather as a means to compete with other microbes or to prevent predation, or as a means to obtain nutrients from the host. Humans come into contact with a large range of ecological niches through agriculture, aquaculture, and other harvesting, commercial and recreational activities. Given the enormous numbers of microbial species in each of these niches, and the vast size of the accessible supragenomes available to each of these species, novel pathogens are likely to be a permanent feature of human existence.
References
McVean G, Spencer CC, Chaix R: Perspectives on human genetic variation from the HapMap Project. PLoS Genet. 2005, 1: e54-10.1371/journal.pgen.0010054.
Rosenberg E, Koren O, Reshef L, Efrony R, Zilber-Rosenberg I: The role of microorganisms in coral health, disease and evolution. Nat Rev Microbiol. 2007, 5: 355-362. 10.1038/nrmicro1635.
Goodacre R: Metabolomics of a superorganism. J Nutr. 2007, 137 (1 Suppl): 259S-266S.
Boskey ER, Telsch KM, Whaley KJ, Moench TR, Cone RA: Acid production by vaginal flora in vitro is consistent with the rate and extent of vaginal acidification. Infect Immun. 1999, 67: 5170-175.
Sobel JD: Is there a protective role for vaginal flora?. Curr Infect Dis Rep. 1999, 1: 379-383. 10.1007/s11908-999-0045-z.
Boskey ER, Cone RA, Whaley KJ, Moench TR: Origins of vaginal acidity: high D/L lactate ratio is consistent with bacteria being the primary source. Hum Reprod. 2001, 16: 1809-1813. 10.1093/humrep/16.9.1809.
Ehrlich GD, Hu FZ, Shen K, Stoodley P, Post JC: Bacterial plurality as a general mechanism driving persistence in chronic infections. Clin Orthop Relat Res. 2005, 437: 20-24. 10.1097/00003086-200508000-00005.
Murphy TF, Sethi S, Klingman KL, Brueggemann AB, Doern GV: Simultaneous respiratory tract colonization by multiple strains of nontypeable Haemophilus influenzae in chronic obstructive pulmonary disease: implications for antibiotic therapy. J Infect Dis. 1999, 180: 404-409. 10.1086/314870.
Mukundan D, Ecevit Z, Patel M, Marrs CF, Gilsdorf JR: Pharyngeal colonization dynamics of Haemophilus influenzae and Haemophilus haemolyticus in healthy adult carriers. J Clin Microbiol. 2007, 45: 3207-3217. 10.1128/JCM.00492-07.
Sá-Leão R, Simões AS, Nunes S, Sousa NG, Frazão N, de Lencastre H: Identification, prevalence and population structure of non-typable Streptococcus pneumoniae in carriage samples isolated from preschoolers attending day-care centres. Microbiology. 2006, 152: 367-376. 10.1099/mic.0.28596-0.
Sá-Leão R, Nunes S, Brito-Avô A, Alves CR, Carriço JA, Saldanha J, Almeida JS, Santos-Sanches I, de Lencastre H: High rates of transmission of and colonization by Streptococcus pneumoniae and Haemophilus influenzae within a day care center revealed in a longitudinal study. J Clin Microbiol. 2008, 46: 225-234. 10.1128/JCM.01551-07.
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI: Evolution of mammals and their gut microbes. Science. 2008, 320: 1647-1651. 10.1126/science.1155725.
Lozupone CA, Knight R: Species divergence and the measurement of microbial diversity. FEMS Microbiol Rev. 2008, 32: 557-578. 10.1111/j.1574-6976.2008.00111.x.
McKenna P, Hoffmann C, Minkah N, Aye PP, Lackner A, Liu Z, Lozupone CA, Hamady M, Knight R, Bushman FD: The macaque gut microbiome in health, lentiviral infection, and chronicenterocolitis. PLoS Pathog. 2008, 4: e20-10.1371/journal.ppat.0040020.
Relman DA: New technologies, human-microbe interactions, and the search for previously unrecognized pathogens. J Infect Dis. 2002, 186 (Suppl 2): S254-S258. 10.1086/344935.
Human Microbiome Project. [http://nihroadmap.nih.gov/hmp]
Job ML, Jacobs NF: Drug-induced Clostridium difficile-associated disease. Drug Safety. 1997, 17: 37-46.
Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, Roca J, Dore J: Reduced diversity of faecal microbiota in Crohn's disease revealed by a metagenomic approach. Gut. 2006, 55: 205-211. 10.1136/gut.2005.073817.
Müller AK, Westergaard K, Christensen S, Sørensen SJ: The diversity and function of soil microbial communities exposed to different disturbances. Microb Ecol. 2002, 44: 49-58. 10.1007/s00248-001-0042-8.
Gans J, Wolinsky M, Dunbar J: Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science. 2005, 309: 1387-1390. 10.1126/science.1112665.
Curtis TP, Sloan WT: Microbiology. Exploring microbial diversity - a vast below. Science. 2005, 309: 1331-1333. 10.1126/science.1118176.
Shen K, Antalis P, Gladitz J, Sayeed S, Ahmed A, Yu S, Hayes J, Johnson S, Dice B, Dopico R, Keefe R, Janto B, Chong W, Goodwin J, Wadowsky RW, Erdos G, Post JC, Ehrlich GD, Hu FZ: Identification, distribution, and expression of novel (nonRd) genes in ten clinical isolates of nontypeable Haemophilus influenzae. Infect Immun. 2005, 73: 3479-3491. 10.1128/IAI.73.6.3479-3491.2005.
Hogg JS, Hu FZ, Janto B, Boissy R, Gladitz J, Swierczek N, Hayes J, Keefe R, Yu S, Post JC, Hu FZ, Ehrlich GD: Characterization and modeling of the Haemophilus influenzae core and supra-genome based on the complete genomic sequences of Rd and 12 clinical non-typeable16 strains. Genome Biol. 2007, 8: R103-10.1186/gb-2007-8-6-r103.
Shen K, Sayeed S, Antalis P, Gladitz J, Ahmed A, Dice B, Janto B, Dopico R, Keefe R, Hayes J, Johnson S, Yu S, Ehrlich N, Jocz J, Kropp L, Tadique E, Wong R, Wadowsky RM, Slifkind M, Preston RA, Erdos G, Post JC, Ehrlich GD, Hu FZ: Extensive genomic plasticity in Pseudomonas aeruginosa revealed by identification and distribution studies of novel (nonPAO1) genes among clinical isolates. Infect Immun. 2006, 74: 5272-5283. 10.1128/IAI.00546-06.
Mathee K, Narasimhan G, Valdes C, Qiu X, Matewish JM, Koehrsen M, Rokas A, Yandava CN, Engels R, Zeng E, Olavarietta R, Doud M, Smith RS, Montgomery P, White JR, Godfrey PA, Kodira C, Birren B, Galagan JE, Lory S: Dynamics of Pseudomonas aeruginosa genome evolution. Proc Natl Acad Sci USA. 2008, 105: 3100-3105. 10.1073/pnas.0711982105.
Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, Deboy RT, Davidsen TM, Mora M, Scarselli M, Margarit y Ros I, Peterson JD, Hauser CR, Sundaram JP, Nelson WC, Madupu R, Brinkac LM, Dodson RJ, Rosovitz MJ, Sullivan SA, Daugherty SC, Haft DH, Selengut J, Gwinn ML, Zhou L, Zafar N, et al: Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial "pan-genome". Proc Natl Acad Sci USA. 2005, 102: 13950-13955. 10.1073/pnas.0506758102.
Shen K, Antalis P, Gladitz J, Dice B, Janto B, Keefe R, Hayes J, Ahmed A, Dopico R, Ehrlich N, Jocz J, Kropp J, Yu S, Nistico L, Greenberg DP, Barbadora K, Post JC, Ehrlich GD, Hu FZ: Characterization, distribution and expression of novel genes among eight clinical isolates of Streptococcus pneumoniae. Infect Immun. 2006, 74: 321-330. 10.1128/IAI.74.1.321-330.2006.
Hiller NL, Janto B, Hogg JS, Boissy R, Yu S, Powell E, Keefe R, Ehrlich NE, Shen K, Hayes J, Barbadora K, Klimke W, Dernovoy D, Tatusova T, Parkhill J, Bentley SD, Post JC, Ehrlich GD, Hu FZ: Comparative genomic analyses of seventeen streptococcus pneumoniae strains: insights into the pneumococcal supragenome. J Bacteriol. 2007, 189: 8186-8195. 10.1128/JB.00690-07.
Buchinsky FJ, Forbes M, Hayes J, Hu FZ, Greenberg P, Post JC, Ehrlich GD: Phenotypic plurality among clinical strains of non-typeable Haemophilus influenzae determined by symptom severity in the Chinchilla laniger model of otitis media. BMC Microbiol. 2007, 7: 56-10.1186/1471-2180-7-56.
Forbes ML, Horsey E, Hiller NL, Buchinsky FJ, Hayes JD, Compliment JM, Hillman T, Ezzo S, Shen K, Keefe R, Barbadora K, Post JC, Hu FZ, Ehrlich GD: Strain-specific virulence phenotypes of Streptococcus pneumoniae assessed using the Chinchilla laniger model of otitis media. PLoS ONE. 2008, 3: e1969-10.1371/journal.pone.0001969.
Kaper JB, Nataro JP, Mobley HL: Pathogenic Escherichia coli. Nat Rev Microbiol. 2004, 2: 123-140. 10.1038/nrmicro818.
Perna NT, Plunkett G, Burland V, Mau B, Glasner JD, Rose DJ, Mayhew GF, Evans PS, Gregor J, Kirkpatrick HA, Pósfai G, Hackett J, Klink S, Boutin A, Shao Y, Miller L, Grotbeck EJ, Davis NW, Lim A, Dimalanta ET, Potamousis KD, Apodaca J, Anantharaman TS, Lin J, Yen G, Schwartz DC, Welch RA, Blattner FR: Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature. 2001, 409: 529-533. 10.1038/35054089.
Bushman F: Lateral DNA Transfer. 2002, Cold Spring Harbor: Cold Spring Harbor Laboratory Press
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, et al: Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005, 437: 376-380.
Ehrlich GD: The biofilm and distributed genome paradigms provide a new theoretical structure for understanding chronic bacterial infections. 41st Interscience Conference on Antimicrobial Agents and Chemotherapy: December 16-19 2001. 2001, Chicago, Illinois. American Society for Microbiology, 524-
Shen K, Wang X, Post JC, Ehrlich GD: Molecular and translational research approaches for the study of bacterial pathogenesis in otitis media. Evidence-based Otitis Media. Edited by: Rosenfeld R, Bluestone CD. 2003, Hamilton, BC: Decker, 91-119. 2
Shapiro JA: Thinking about bacterial populations as multicellular organisms. Annu Rev Microbiol. 1998, 52: 81-104. 10.1146/annurev.micro.52.1.81.
Hu FZ, Ehrlich GD: Population-level virulence factors amongst pathogenic bacteria: relation to infection outcome. Future Microbiol. 2008, 3: 31-42. 10.2217/17460913.3.1.31.
Redfield RJ, Findlay WA, Bosse J, Kroll JS, Cameron AD, Nash JH: Evolution of competence and DNA uptake specificity in the Pasteurellaceae. BMC Evol Biol. 2006, 6: 82-10.1186/1471-2148-6-82.
Andersson DI: Shrinking bacterial genomes. Microbe. 2008, 3: 124-130.
Faruque SM, Sack DA, Sack RB, Colwell RR, Takeda Y, Nair GB: Emergence and evolution of Vibrio cholerae O139. Proc Natl Acad Sci USA. 2003, 100: 1304-1309. 10.1073/pnas.0337468100.
Brüggemann H, Cazalet C, Buchrieser C: Adaptation of Legionella pneumophila to the host environment: role of protein secretion, effectors and eukaryotic-like proteins. Curr Opin Microbiol. 2006, 9: 86-94. 10.1016/j.mib.2005.12.009.
Steinberg KM, Levin BR: Grazing protozoa and the evolution of the Escherichia coli O157:H7 Shiga toxin-encoding prophage. Proc Biol Sci. 2007, 274: 1921-1929. 10.1098/rspb.2007.0245.
Hall-Stoodley L, Hu FZ, Stoodley P, Nistico L, Link TR, Burrows A, Post JC, Ehrlich GD, Kerschner JE: Direct detection of bacterial biofilms on the middle-ear mucosa of children with chronic otitis media. JAMA. 2006, 296: 202-211. 10.1001/jama.296.2.202.
Lysenko ES, Ratner AJ, Nelson AL, Weiser JN: The role of innate immune responses in the outcome of interspecies competition for colonization of mucosal surfaces. PLoS Pathog. 2005, 1: e1-10.1371/journal.ppat.0010001.
Shakhnovich EA, King SJ, Weiser JN: Neuraminidase expressed by Streptococcus pneumoniae desialylates the lipopolysaccharide of Neisseria meningitidis and Haemophilus influenzae: a paradigm for interbacterial competition among pathogens of the human respiratory tract. Infect Immun. 2002, 70: 7161-7164. 10.1128/IAI.70.12.7161-7164.2002.
Tong HH, James M, Grants I, Liu X, Shi G, DeMaria TF: Comparison of structural changes of cell surface carbohydrates in the eustachian tube epithelium of chinchillas infected with a Streptococcus pneumoniae neuraminidase-deficient mutant or its isogenic parent strain. Microb Pathog. 2001, 31: 309-317. 10.1006/mpat.2001.0473.
Tong HH, Blue LE, James MA, DeMaria TF: Evaluation of the virulence of a Streptococcus pneumoniae neuraminidase-deficient mutant in nasopharyngeal colonization and development of otitis media in the chinchilla model. Infect Immun. 2000, 68: 921-924. 10.1128/IAI.68.2.921-924.2000.
Ratner AJ, Aquilar JL, Shchepetov M, Lysenko ES, Weiser JN: Nod1 mediates cytoplasmic sensing of combinations of extracellular bacteria. Cell Microbiol. 2007, 9: 1343-1351. 10.1111/j.1462-5822.2006.00878.x.
Giraud A, Arous S, De Paepe M, Gaboriau-Pouthiau V, Bambou JC, Rakotobe S, Lindner AB, Taddei F, Cerf-Bensussan N: Dissecting the genetic components of adaptation of Escherichia coli to the mouse gut. PLoS Genet. 2008, 4: e2-10.1371/journal.pgen.0040002.
Baumler AJ, Tsolis RM, Ficht TA, Adams LG: Evolution of host adaptation in Salmonella enterica. Infect Immun. 1998, 66: 4579-4587.
Leavis HL, Willems RJ, van Wamel WJ, Schuren FH, Caspers MP, Bonten MJ: Insertion sequence-driven diversification creates a globally dispersed emerging multiresistant subspecies of E. faecium. PLoS Pathog. 2007, 3: e7-10.1371/journal.ppat.0030007.
Jelsbak L, Johansen HK, Frost AL, Thøgersen R, Thomsen LE, Ciofu O, Yang L, Haagensen JA, Høiby N, Molin S: Molecular epidemiology and dynamics of Pseudomonas aeruginosa populations in lungs of cystic fibrosis patients. Infect Immun. 2007, 75: 2214-2224. 10.1128/IAI.01282-06.
Ciofu O, Riis B, Pressler T, Poulsen HE, Høiby N: Occurrence of hypermutable Pseudomonas aeruginosa in cystic fibrosis patients is associated with the oxidative stress caused by chronic lung inflammation. Antimicrob Agents Chemother. 2005, 49: 2276-2282. 10.1128/AAC.49.6.2276-2282.2005.
Mathee K, Ciofu O, Sternberg C, Lindum PW, Campbell JI, Jensen P, Johnsen AH, Givskov M, Ohman DE, Molin S, Høiby N, Kharazmi A: Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: a mechanism for virulence activation in the cystic fibrosis lung. Microbiology. 1999, 145: 1349-1357.
Ojeniyi B, Frederiksen B, Hoiby N: Pseudomonas aeruginosa cross-infection among patients with cystic fibrosis during a winter camp. Pediatr Pulmonol. 2000, 29: 177-181. 10.1002/(SICI)1099-0496(200003)29:3<177::AID-PPUL4>3.0.CO;2-U.
Acknowledgements
This work was supported by Allegheny General Hospital and Allegheny Singer Research Institute, as well as by grants from the Health Resources and Services Administration and the NIH-NIDCD: DC02148, DC04173, and DC05659. We thank Mary O'Toole for help with the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Ehrlich, G.D., Hiller, N.L. & Hu, F.Z. What makes pathogens pathogenic. Genome Biol 9, 225 (2008). https://doi.org/10.1186/gb-2008-9-6-225
Published:
DOI: https://doi.org/10.1186/gb-2008-9-6-225