
Abstract
Background
Chromosome four of Drosophila melanogaster, known as the dot chromosome, is largely heterochromatic, as shown by immunofluorescent staining with antibodies to heterochromatin protein 1 (HP1) and histone H3K9me. In contrast, the absence of HP1 and H3K9me from the dot chromosome in D. virilis suggests that this region is euchromatic. D. virilis diverged from D. melanogaster 40 to 60 million years ago.
Results
Here we describe finished sequencing and analysis of 11 fosmids hybridizing to the dot chromosome of D. virilis (372,650 base-pairs) and seven fosmids from major euchromatic chromosome arms (273,110 base-pairs). Most genes from the dot chromosome of D. melanogaster remain on the dot chromosome in D. virilis, but many inversions have occurred. The dot chromosomes of both species are similar to the major chromosome arms in gene density and coding density, but the dot chromosome genes of both species have larger introns. The D. virilis dot chromosome fosmids have a high repeat density (22.8%), similar to homologous regions of D. melanogaster (26.5%). There are, however, major differences in the representation of repetitive elements. Remnants of DNA transposons make up only 6.3% of the D. virilis dot chromosome fosmids, but 18.4% of the homologous regions from D. melanogaster; DINE-1 and 1360 elements are particularly enriched in D. melanogaster. Euchromatic domains on the major chromosomes in both species have very few DNA transposons (less than 0.4 %).
Conclusion
Combining these results with recent findings about RNAi, we suggest that specific repetitive elements, as well as density, play a role in determining higher-order chromatin packaging.
Similar content being viewed by others


Background
FDNA in the eukaryotic interphase nucleus can broadly be distinguished as packaged into two different forms of chromatin, heterochromatin and euchromatin [1]. Classically, heterochromatin has been described as the fraction that remains highly condensed in interphase, has high affinity for DNA-specific dyes, and is commonly seen around the periphery of the nucleus [2]. Heterochromatic regions of the genome have very low rates of meiotic recombination and generally replicate late in S phase. These regions are rich in repetitive sequences, including remnants of transposable elements and retroviruses, as well as simple repeats (satellite DNA). Heterochromatin tends to be gene poor, and those genes found in heterochromatin tend to be larger (longer transcripts) than genes found in euchromatin [3]. Introns of heterochromatic genes have a much higher density of transposable elements than introns of euchromatic genes, accounting for this shift [4]. The less densely packaged euchromatin contains most of the actively transcribed genes. In contrast to this general picture of repeat distribution, Pardue et al. [5] have found by in situ hybridization that the frequency of (dC-dA)·(dG-dT) dinucleotide repeats is higher in euchromatin than in heterochromatin.
Several biochemical marks have been identified that distinguish heterochromatin from euchromatin, including a distinctive pattern of histone modification and the association of particular chromosomal proteins [6]. High concentrations of heterochromatin protein 1 (HP1) are found primarily in pericentric heterochromatin and associated with telomeres in organisms from the yeast Schizosaccharomyces pombe to mammals [7, 8]. Histones in euchromatic domains are typically hyperacetylated, particularly the amino-terminal tails of H3 and H4. In contrast, methylation of histone H3 at lysine 9 (producing H3K9me) is a consistent mark of heterochromatin [9]. HP1 binds to H3K9me through its chromo domain and to SU(VAR)3-9, a methyltransferase that specifically modifies histone H3 at K9, through its chromo shadow domain [9, 10]. These interactions are thought to contribute to heterochromatin maintenance and spreading [1]. The functional significance of this chromatin packaging is demonstrated by the observation that loss-of-function mutations in the gene for HP1, including one that disrupts binding of HP1 to H3K9me, result in a loss of silencing of reporter genes placed in or near heterochromatin (suppression of position effect variegation) [11].
Chromosome four of Drosophila melanogaster, also known as the dot chromosome or the F element, is unique in its chromatin composition. The banded portion (amplified during polytenization) is 1.2 Mb long with 82 genes; this gene density is similar to that of the euchromatic regions of the major (euchromatic) chromosome arms [12, 13]. However, the fourth chromosome also displays many characteristics of heterochromatin, including late replication [14] and a complete lack of meiotic recombination [15]. The banded region of chromosome 4 is known to have an approximately ten-fold higher density of repetitive elements (for example, remnants of retroviruses, transposable elements) in comparison with the long arms of chromosomes 2, 3, and X [16–19], but has little or no (dC-dA)·(dG-dT) dinucleotide repeats [5], again resembling heterochromatin rather than euchromatin. Immunofluorescent staining of polytene chromosomes with antibodies directed against HP1 shows an abundance of HP1 in a banded pattern on chromosome four [20]. A very similar pattern is seen with antibodies directed against H3K9me [9, 21].
A transposable P element containing an hsp70-driven white (w) gene has been a useful reporter of chromatin packaging, giving a uniform red eye phenotype when inserted into the euchromatic arms but a variegating phenotype when inserted into the pericentric heterochromatin or into telomere associated sequences [22]. The variegating phenotype is associated with packaging into a nucleosome array showing more uniform spacing, accompanied by a loss of DNase hypersensitive (DH) sites [23]. Transposition events resulting in insertions on the fourth chromosome produce both variegating and solid red eye phenotypes. The data suggest that while the fourth chromosome of D. melanogaster is largely heterochromatic, it also includes some euchromatic domains [23].
P element transposition-induced deletions and duplications of small genomic regions around the genes Hcf and CG2052 on chromosome four have been shown to cause switching of eye phenotypes from red to variegating and vice versa [24]. Mapping of the breakpoints has shown that the small deletions and duplications lead to changes in the distance of the reporter from a particular DNA transposon, 1360 (also known as hoppel or PROTOP_A). In the region of the fourth chromosome studied, if the inserted P element is within approximately 10 kilobases (kb) of a 1360 element, the white reporter gene has a greater than 90% chance of exhibiting variegating expression, suggesting it is in a heterochromatic domain. If the reporter is more than 10 kb away from a 1360 element, it has a greater than 90% chance of generating a red eye phenotype, suggesting that it is in a euchromatic domain. Therefore, Sun et al. [24] have suggested that proximity to the 1360 element can influence the chromatin packaging state. Recent results from fungi and plants [25], as well as Drosophila [26] have shown that heterochromatin formation is dependent on the RNA interference (RNAi) system. Small double-stranded (ds)RNAs have been recovered from many of the repetitive elements in Drosophila, including 1360 [27], and might target repetitive elements in the genome for silencing by initiation and spreading of heterochromatin packaging.
The small dot chromosome exists in many species of Drosophila [28]. It has long been recognized that phenotypes of similar mutations map to the dot chromosomes of both D. melanogaster and D. virilis [29, 30]. Podemski et al. [31] have shown that probes for several genes from the D. melanogaster fourth chromosome, including ci and Caps, hybridize to the dot chromosome in D. virilis. D. virilis is a member of a Drosophila genus that diverged from D. melanogaster 40 to 60 million years ago [32]. In addition to the sex chromosomes, it has four large autosomes, rather than the two of D. melanogaster; thus, the dot chromosome of D. virilis is chromosome six. The polytenized regions of both dot chromosomes are similar in size. In this study, we will refer to chromosome six of D. virilis and chromosome four of D. melanogaster as dot chromosomes. Our analysis concerns the banded 1.2 Mb region of these chromosomes, estimated to contain approximately 80 genes.
Prior reports indicated that the dot chromosome of D. virilis does not share the heterochromatic characteristics of the dot chromosome of D. melanogaster, despite the fact that it maintains a similar proximity to the heterochromatic chromocenter, as seen in polytene nuclei. In situ hybridizations performed by Lowenhaupt et al. [33] demonstrated that the (dC-dA)·(dG-dT) dinucleotide repeat frequency of the D. virilis dot chromosome is similar to that in its euchromatic arms. In contrast to the observations using D. melanogaster, recombination is observed on the D. virilis dot chromosome [30, 34]. Further, the polytenized portion of the dot chromosome in D. virilis fails to stain with antibodies directed against HP1 [20] (Figure 1b).

Immunofluorescent staining of the polytene chromosomes. Polytene chromosomes from (a) D. melanogaster and (b) D. virilis are shown. Top left, phase contrast; others as labeled. Panels on the right provide a close-up of the chromocenter and the dot chromosome. In the merge picture, yellow represents equal staining, red represents more H3K9me staining, and green represents more HP1 staining. The dot chromosome is indicated with an arrow. In D. melanogaster, antibodies for HP1 and H3K9me stain both the chromocenter and the dot chromosome, although the HP1 staining is slightly stronger than the H3K9me staining on the dot. In D. virilis, both antibodies stain the chromocenter but neither stains the dot chromosome.
Comparative genomics has been invaluable in discovering new functional and regulatory elements in the genomes of a cluster of yeast species, using Saccharomyces cerevisiae as the reference point [35]. We believe this comparative approach will be equally valuable as comparisons of Drosophila species become possible [36, 37]. If the gene compositions of the dot chromosomes of D. melanogaster and D. virilis are similar, what other differences in the DNA sequence could lead to the apparent difference in higher-order chromatin structure? To address this question, we have generated a finished, clone-based sequence for a sample from the D. virilis dot chromosome and from the long chromosome arms; finished sequence leads to more accurate inferences about repetitive sequences [38]. By comparing similar regions of the two dot chromosomes, we show that while the overall repeat density of the dot chromosomes is similar, the density of DNA transposon remnants is significantly higher in D. melanogaster than in D. virilis; the difference is particularly striking for the DINE-1 elements and 1360 elements, discussed above. These results, combined with recent findings about RNAi, lead us to suggest that the difference in chromatin packaging between the dot chromosomes of these two species of Drosophila could be a function of the density and distribution of a subclass of repetitive elements.
Results
Immunofluorescent staining indicates that the D. virilis dot chromosome is largely euchromatic, in contrast to the heterochromatic D. melanogasterdot chromosome
The dot chromosome of D. melanogaster is largely heterochromatic, with some interspersed domains of euchromatin [24]. Immunofluorescent staining of D. melanogaster polytene chromosomes using HP1 antibody shows a banded pattern on the dot chromosome. Many species in the Drosophila genus closely related to D. melanogaster share this staining pattern, including D. simulans, D. yakuba, and D. pseudoobscura (data not shown). In D. melanogaster, staining with an antibody against histone H3 methylated at lysine 9 (anti-H3K9me) coincides with the HP1 staining, at a level slightly less than seen in the pericentric heterochromatin [21] (Figure 1a). In contrast, the dot chromosome of D. virilis does not stain with either anti-HP1 or anti-H3K9me (Figure 1b), supporting the inference that the banded portion of the dot chromosome of D. virilis is generally euchromatic.
Identification of fosmids from the dot chromosome of D. virilis
The chromosomes of D. virilis tend to map to corresponding portions of the chromosomes of D. melanogaster [39]. We compared the recently posted genomic sequence for D. pseudoobscura [37, 40] with the D. melanogaster dot chromosome genes to look for regions of sufficient sequence similarity to act as conserved hybridization probes. The desired probes (see Materials and methods) were radiolabeled and used to screen a D. virilis genomic library (BDVIF01 fosmids, Tucson strain 15010-1001.10, available spotted on a single filter) at low stringency. Positive clones were verified and characterized by in situ hybridizations to the polytene chromosomes from third instar larval salivary glands of D. virilis. Sample results are shown in Figure 2. Eleven fosmids were recovered with homology to the dot chromosome of D. virilis, and seven fosmids were recovered with homology to the major chromosome arms. Based on the in situ hybridization results, the order of the fosmid clones on the dot chromosome is as follows: contigs 30, 103, and 106 appear to cluster near the centromere; contigs 67, 72, and 91 are in the middle of the chromosome; and contigs 50 and 113 hybridize near the telomere. There is also a minor signal with the contig 30 probe near the telomere; this may be the result of a repetitive element present in multiple regions in the chromosome.

In situ hybridizations of fosmids to D. virilis polytene chromosomes. Fosmid DNA was labeled and used for in situ hybridization on denatured polytene chromosomes from D. virilis. Three examples are shown (left to right: contigs 106, 72, 113) demonstrating hybridization to a specific band on the dot chromosome (arrowhead). In some cases, signal is associated with the chromocenter, presumably due to repetitive sequences shared with the band on the dot. In situ hybridizations were performed with at least one fosmid from every contig from the dot chromosome with similar results (data not shown). See Table 1 for the chromosome locations of the other fosmids.
Fosmid sequencing and annotation
The 18 fosmids recovered from the screen were sequenced in collaboration with the Genome Sequencing Center at Washington University School of Medicine. Plasmid subclone libraries were prepared and approximately 600 subclones from each fosmid were end sequenced. The sequences were assembled and finished to high quality by Washington University undergraduate students in the Bio 4342 'Research Explorations in Genomics' course, using phred, phrap, and consed [41–43]. Finished sequences had an estimated error rate of less than 0.01%, and showed in silico restriction digests that matched digests obtained from the starting fosmid with a minimum of two enzymes. Students annotated the finished sequences by looking for genes, repetitive elements, and other features as described in Materials and methods. Four pairs of fosmids have significant sequence overlap; each pair was collapsed into a single contig of non-redundant sequence (contigs 30, 50, 67, and 80).
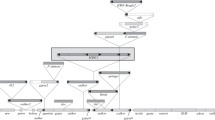
Initial annotation focused on gene finding. D. virilis is evolutionarily close enough to D. melanogaster that the protein coding regions are well conserved. Gene prediction algorithms and local alignment search tools (such as GENSCAN and BLAST; see Materials and methods) were used to annotate genes and determine intron-exon boundaries. In most cases, it was possible to identify the entire coding region of the gene, but the high level of sequence divergence made defining untranslated regions impossible [36]. Comparison of the D. virilis contigs with homologous regions of the D. melanogaster dot chromosome identified specific regions where synteny has been maintained, as well as those regions where inversions have occurred. Figure 3 shows a comparison of two D. virilis contigs with the homologous regions from the D. melanogaster chromosomes. Detailed annotation results and comparisons between the other individual D. virilis fosmids and their homologous regions in D. melanogaster are available as Additional data file 1 (dot chromosome sequences) and Additional data file 2 (non-dot chromosome sequences). Note that the strain of D. virilis used here is a different strain from that recently sequenced (by Agencourt Bioscience Corporation, Beverly, MA, USA). The two strains differ by about 1% base substitutions, with numerous insertions or deletions (indels), but show similar organization at the gene level (CDS, unpublished observation). The clone-based sequencing used here results in more accurate inferences in regions that are highly repetitive; the sequences most likely to be missed in whole genome shotgun techniques are the repeats [38].

Map for two sample contigs from D. virilis (Dv) in comparison with homologous regions of the D. melanogaster (Dm) genome. Shown are two contigs from D. virilis with the corresponding regions from D. melanogaster. Coding sequences (dark blue boxes) are indicated above each diagram. In the case of D. melanogaster, the thick dark blue bar indicates open reading frames (ORFs), and the thin aqua bar indicates UTRs; only ORFs are identified for D. virilis. Repeat sequences are shown below: red boxes are DNA transposon fragments, while other repetitive elements are represented as yellow boxes. (a) Contig 112 represents a clone from one of the large chromosomes of D. virilis. While the orientations of Egfr and CG10440 are the same with respect to each other, there is a large tandem repeat between the two genes in D. virilis, but not in D. melanogaster. (b) Contig 67 represents a clone from the dot chromosome of D. virilis. The structure of the genomic region is similar to the corresponding region in D. melanogaster, but there is more intergenic space in D. virilis, whereas in D. melanogaster, there are more transposable elements in the introns. All of the fosmids described here with homologous regions in D. melanogaster have been annotated in a similar manner; the maps are available in the Additional data files. Scale: one division equals 5 kb.
Table 1 shows all contigs sequenced, giving their total sizes, listing annotated genes, and providing clone names (BACPAC Center). In situ hybridization results identified the fosmids as either on the dot chromosome or on a major D. virilis chromosome. In parentheses following each gene is the chromosome position of the gene in the genome of D. melanogaster. Figure 4 maps the contigs from the dot chromosome of D. virilis to the dot chromosome of D. melanogaster based on the presence of orthologous genes. Three of the contigs (67, 106, and 113) are completely syntenic with respect to the D. melanogaster dot chromosome. One contig, 103, is completely syntenic with respect to its genes from the dot chromosome, but also contains CG5367, a gene from the second chromosome of D. melanogaster. Four contigs (30, 72, 50, and 91) contain genes that are exclusively from the dot chromosome of D. melanogaster but show evidence of a high number of inversions with respect to the D. melanogaster chromosome. For example, contig 30 contains both pan and Caps, genes that come from opposite sides of the banded portion of the D. melanogaster dot chromosome. (This rearrangement was also observed in earlier studies [31].) Of the 28 genes identified in the D. virilis dot chromosome clones, only one lies elsewhere in the D. melanogaster genome. In the D. virilis contigs from major chromosomes, four (contigs 13, 112, 121, 122) are completely syntenic compared to homologous gene regions from D. melanogaster, and two (contigs 11 and 80) show inversions within the chromosomes. Only one major chromosome contig (80) contains a gene that is found on the dot chromosome in D. melanogaster. Contig 80 maps to a major arm of D. virilis; it contains D. melanogaster dot chromosome gene CG1732 flanked by several genes from D. melanogaster chromosome 3. In total, the fosmids sequenced represent 372,650 bp of sequence from the dot chromosome of D. virilis and 273,110 bp of sequence from the major chromosomes. D. virilis contigs 72 and 91 from the dot chromosome and 11 and 80 from the major arms showed so much rearrangement that it was impossible to define precise homologous area(s) from D. melanogaster. These contigs were not used in comparisons for intron size, percent DNA transcribed, or in any of the repeat density calculations. Maps representing locations and sizes of genes and repeats in each contig are available in Additional data files 1 and 2.

Map of the D. virilis (Dv) dot chromosome contigs in relation to the dot chromosome of D. melanogaster (Dm). Shown at the bottom is a map of the genes on the D. melanogaster dot chromosome. Colored bars with labels represent genes for which we have identified a (complete or partial) homologue in the D. virilis fosmids sequenced. Colored boxes above the scale bar are schematic (not to scale) representations of the D. virilis contigs. Immediately above the scale bar is a representation of those sequenced contigs that contain syntenic regions from D. virilis, where genes are in the same order and orientation as in D. melanogaster. In the uppermost portion of the figure are the contigs mapping to the D. virilis dot chromosome that are rearranged with respect to the D. melanogaster dot chromosome. Boxes are color-coded to represent the genes present in the contig, with dashed lines connecting to show the extent of rearrangement. Notably, contig 30 contains both pan and Caps, which lie on opposite sides of the banded portion of the D. melanogaster dot chromosome.
Average intron size and percent DNA transcribed
While centromeric regions are rich in satellite DNA and relatively gene poor [3], gene density (defined as the number of genes per Mb) in the banded portion of the dot chromosome is similar to the major chromosomes of D. melanogaster [19] (66.5 genes/Mb for the dot and 74.6 genes/Mb for the major chromosomes for the regions analyzed here). This is also true for the regions of the D. virilis genome we have sequenced (62.2 genes/Mb for the dot and 67.3 genes/Mb for major chromosomes). Observation of those few heterochromatic genes that have been cloned and sequenced (for example, light [44]) suggests that these genes may have larger introns on average, and this has been reported for D. melanogaster dot chromosome genes [19]. Average intron size, defined as total intron length divided by total number of introns, is 448 bp (± 126 bp) for our sample from the major D. virilis chromosomes and 405 bp (± 110 bp) for the corresponding regions of D. melanogaster. D. virilis dot chromosome genes in our sample have an average intron length of 890 bp (± 179 bp); in homologous regions of the D. melanogaster genome, it is 859 bp (± 115 bp). Figure 5 shows a graph that compares the intron size cumulative distribution functions of the dot chromosomes with the major chromosomes. Due to the non-normal distribution of intron sizes, the non-parametric Kolmogorov-Smirnov (KS) test is used to evaluate the statistical significance in the pairwise comparisons. The KS test indicates that the difference in the distribution of intron sizes between the two dot chromosomes is not statistically significant (D = 0.1237, p = 0.2816). However, the distribution of intron sizes for the dot chromosomes is significantly different from those for the major chromosomes for both species (D = 0.223, p = 0.0496 and D = 0.245, p = 0.0291 for D. virilis and D. melanogaster, respectively).

Distribution of intron sizes in D. virilis compared to D. melanogaster. Introns from all D. virilis and D. melanogaster genes in the contigs studied were separated into groups based on size. The number on the x axis represents the minimal intron size; an intron is counted in that bin if it has that many bases or fewer. The y axis tallies the percent of total introns that fall into that bin. The two dot chromosomes have significantly similar intron size distributions, which differ significantly from those of the major chromosome arms.
Percent DNA transcribed, defined as primary transcript length over total sequence length, is more similar between the homologous chromosomes than between the dot chromosomes and the major chromosomes. (In this instance, 5' and 3' untranslated regions (UTRs) were not scored in calculations of percent DNA transcribed, as these regions could not be identified in the putative D. virilis genes.) The sequenced regions of the D. virilis and comparable regions of the D. melanogaster dot chromosomes have transcript densities of 58.7% and 51.0%, respectively, while transcript densities of the major chromosomes are 22.2% for D. virilis and 25.9% for D. melanogaster. The difference in percent DNA transcribed between the dot and non-dot contigs reflects the larger average size of introns in the dot chromosome genes.
(dC-dA)·(dG-dT) dinucleotide repeat frequency
One marker of euchromatin is the presence of abundant (dC-dA)·(dG-dT) dinucleotide repeats, also known as CA/GT repeats. In situ hybridization shows that these repeats are widely distributed in euchromatin, but that the dot chromosome of D. melanogaster has a much lower density of these repeats [5]. The dot chromosome of D. virilis has a CA/GT repeat frequency similar to its major autosomes, as shown by in situ hybridization [33]. Dinucleotide repeat analysis of the sequences from the D. virilis fosmids in comparison with the homologous regions of the D. melanogaster genome supports the in situ hybridization results. The fosmids from the dot chromosome of D. virilis have CA/GT repeats with an average length of 36 bp and a total density of 0.15%. Regions of the D. melanogaster dot chromosome homologous to these fosmids have only one CA/GT repeat, which is 21 bp long, giving a total CA/GT density of 0.0069%. In the D. virilis clones mapping to major chromosomes, 0.96% of the DNA is made up of CA/GT, with the average repeat being 32 bp long. In homologous regions of the D. melanogaster genome, 0.32% of the DNA is CA/GT, with the average length of dinucleotide regions being 24 bp. Thus, while the D. virilis dot chromosome has a lower level of CA/GT than the major chromosome arms (about six-fold less than D. virilis and about two-fold less than D. melanogaster), it has a approximately 20-fold higher level of this repeat than is found in the dot chromosome of D. melanogaster.
Repeat analysis
Initial analysis of known repetitive elements in the D. virilis contigs was performed using RepeatMasker [45]. RepBase 8.12 [46, 47] contains previously characterized repeats from the D. virilis species group. As a simple initial approach we searched for de novo repeats by comparing the fosmid sequences to each other, looking for regions of high similarity by BLASTN [48]. Most apparently novel repeated sequences identified by this technique were immediately adjacent to known repeats identified by RepeatMasker and were, therefore, assumed to be unmasked extensions of those repeats. A few novel repeats were identified that were not similar to any other known repetitive element, expressed sequence tag (EST), or protein sequence. Using this simple technique, novel repeats constituted less than 1% of the total repetitive DNA; however, given the small size of our dataset (0.65 Mb) it is possible that repetitive elements could be missed.
Figure 6a shows the repeat density of different classes of repetitive elements in the D. virilis contigs and the comparable regions of the D. melanogaster genome using RepeatMasker/RepBase (Drosophila default parameters) plus this simple de novo BLASTN technique. While there is some variation in repeat density between the contigs of a given region (dot chromosome or major chromosome), the totals appear to represent an average value of the contigs studied. Using this analysis, the overall repeat density of the D. virilis dot chromosome contigs is 14.6%; the average of the individual repeat densities is 15.4% ± 7.9%. The overall repeat density of the homologous D. melanogaster regions is 25.3%; the average of the individual repeat densities is 24.7% ± 5.4%. Fosmids from the dot chromosome of D. melanogaster show a consistently higher density of DNA transposons and DINE-1 elements than do the fosmids from the dot chromosome of D. virilis. Comparison of the sample from the dot chromosome of D. melanogaster analyzed here to the entire banded portion of the dot chromosome (using RepeatMasker and RepBase 8.12) shows very similar results (Figure 6a). In contrast, the euchromatic arms of the large chromosomes of D. melanogaster and D. virilis have similar repeat densities, with approximately 6% of the sequence classified as repetitive. (Quesneville et al. [49] estimate the total repeat density of D. melanogaster to be 5.3%.) Other repeat types differed between the two species as well. In our sample from these chromosome arms, D. virilis has more simple repeats and D. melanogaster has more retroelements. Overall, these results suggest that both the higher repeat density and the overrepresentation of DNA transposons contribute to heterochromatin formation on the D. melanogaster dot chromosome. However, because D. virilis is not as well studied as D. melanogaster, it is possible that this approach misses some uncharacterized repeats. To address this issue, we undertook several different strategies.

Repeat analysis of D. virilis contigs compared to the D. melanogaster genome. The repeat density, defined as the percentage of total sequence (in base-pairs) that has been annotated as repetitive has been calculated using the D. virilis fosmid sequence obtained in this study and homologous regions from D. melanogaster (see Materials and methods). D. melanogaster and D. virilis have a very similar low repeat density on the major chromosome arms, and a similar but much higher repeat density on the dot chromosomes. (a) Percent repeat for each type identified by RepeatMasker using RebBase 8.12 with additional repeats identified in a BLASTN all-by-all comparison of the fosmid sequences presented here. (b) Percent repeat for each type identified by RepeatMasker using the Superlibrary (see text for description). The dot chromosome of D. melanogaster has about three times more DNA transposon sequence than does the D. virilis dot chromosome. 'Unknown' repeats are those from both RebBase 8.12 and the D. virilis PILER-DF library that have not been classified as to type.
Recent investigations have developed multiple search tools for de novo identification of novel repetitive sequences in genome assemblies [50, 51]. Using such tools, we created a 'Superlibrary' in which we added sequences from species-specific libraries from both D. melanogaster and D. virilis to the RebBase 8.12 Drosophila transposable element (TE) library to generate a library with as little bias as possible. The additional repeats came from three sources. Two novel repetitive elements that were identified in D. melanogaster using the PILER-TR program were added [50]. We also added a complete set of 66 elements from D. virilis identified by PILER-DF analysis (C Smith and G Karpen, personal communication) of the posted D. virilis whole genome assembly [52]. Finally, a recently identified sequence of DINE-1 from D. yakuba was added [53].
All of the D. virilis and D. melanogaster sequences used in this study were then analyzed for repetitive DNA using RepeatMasker with this Superlibrary. This approach identified a total repeat density of the D. virilis contigs from the dot chromosome of 22.8%, while homologous regions of the D. melanogaster dot chromosome have 26.5% repetitive DNA (Figure 6b). Using the same Superlibrary, the segments from the major chromosomes of D. virilis have a total repeat density of 8.4%, compared to D. melanogaster major chromosomes, which have a density of 6.8%. This analysis shows that the overall density of repeats on the D. virilis and D. melanogaster dot chromosome fosmids is similar, and significantly higher than the density of repeats on the major chromosomes from either species. Other analysis techniques used to assess the difference between the D. virilis and D. melanogaster sequences, including a TBLASTX comparison using a RebBase 8.12 library from which invertebrate sequences had been removed [49, 54], and a Repeat Scout library assembly [51], also showed little difference in the total amount of repetitive sequence found in the D. virilis and D. melanogaster dot sequences (not shown). Thus, all of the follow-up techniques applied indicate that the sequences from the dot chromosomes of both D. virilis and D. melanogaster are enriched for repetitive sequences compared to the sequences derived from the major chromosomes of both species. The analysis of each contig as well as the total representation of each type of repeat is presented in Table 2 and in Figure 6b. The contrast between the results shown in Figure 6a and those shown in Figure 6b illustrates the problem posed by biased repeat libraries, an issue that must be carefully considered in studies of this type. The observation that three different analyses (discussed above) support the results shown in Figure 6b lends confidence to the conclusions derived here.
While the overall density of repetitious elements is similar, there is a major difference in the density of DNA transposons (Table 2). Of the D. melanogaster dot chromosome DNA from our sample, 18.6% consists of remnants of DNA transposons, including sequences from 1360 elements, P elements (artifacts and related fragments), Tc1 elements and DINE-1. Only 6.4% of these regions from the dot chromosome of D. virilis consists of remnants of DNA transposons, about a three-fold reduction. The bulk of the repetitive sequence in the D. virilis dot fosmids tentatively classified as DNA transposons are the dvir.16.2 centroid and the dvir.16.17 centroid, sequences identified in the PILER-DF analysis. Table 3 shows the repeat element and class of the most common repeats in the D. virilis and D. melanogaster dot chromosome contigs studied here, as identified by RepeatMasker/Superlibrary. DNA transposon families are preferentially represented in D. melanogaster, while retroelements (LINEs and LTRs) are more common in D. virilis. Examination of the quantitative results in Table 2 suggests that the dot chromosome of D. virilis has an increase in retroelements (9.1%) in comparison with homologous regions of D. melanogaster (4.2%). However, this difference appears to be due to sample bias, as RepeatMasker/RebBase 8.12 classifies 8.7% of the whole D. melanogaster dot chromosome as retroelements.
DINE-1, also known as DNAREP-1 or INE-1, is a repetitive element that is very common in the genome of D. melanogaster [55]. The density of DINE-1 elements is especially high on the dot chromosome of D. melanogaster, more so than on the major chromosome arms or on the dot chromosome of D. virilis [17, 56]. Using our Superlibrary and repeat identification process, RepeatMasker identifies 0.1% of the D. virilis contigs as sequences with significant similarity to DINE-1 elements, while in the homologous regions of the D. melanogaster dot the density is 10.5%. (The entire D. melanogaster dot has a 9.2% incidence of DINE-1 elements, assessed using RepeatMasker/Superlibrary.) There has been considerable debate as to the origin of DINE-1 elements [56, 57]. Kapitonov and Jurka [57] have recently suggested that DINE-1 is a retrotransposon based on homology to a D. virilis Penelope GenBank accession, but sequences with homology to DINE-1 in this accession fall outside of the canonical Penelope sequence [58] (C Bergman, personal communication). Analysis of DINE-1 elements in D. yakuba suggests a relatively recent burst of transposition in that species. A consensus sequence based on these recent DINE-1 elements contains no long terminal repeats nor a poly-A tail (suggestive of a retroelement), but does have a terminal 12 bp perfect repeat, a characteristic of transposons [53]. Thus, while we have provided separate statistics for this class, we consider DINE-1 elements to be DNA transposon remnants. Separate statistics are also provided in Table 2 for the 1360 DNA transposon fragments, as this class is of particular interest as a potential target for heterochromatin formation, as discussed above. Again, this family is significantly enriched in D. melanogaster dot chromosome fosmids, making up 4.1% of the sample DNA, in comparison to 0.8% in D. virilis dot chromosome fosmids and 0% in the samples from the major chromosome arms.
Transposable elements are much more prevalent in the introns of heterochromatic genes than in the introns of euchromatic genes [4]; this may contribute to the evolution and structure of genes in heterochromatin. Maintaining a focus on total repeat density (and not repeat type), we analyzed the introns of all of the contigs with a repeat database generated by combining the RepeatScout output from both the D. melanogaster and the D. virilis whole genome assemblies. Using RepeatMasker with this library (omitting low complexity and simple repeats), one finds that introns of the D. virilis dot chromosome genes studied here contain 27.0% repetitive elements, while in homologous regions of the D. melanogaster dot, 33.1 % of the introns are made up of repetitive elements. Analysis of the contigs from the major chromosomes of D. virilis and the homologous regions from D. melanogaster did not find any recognizable transposable elements in the introns. Thus the two dot chromosomes are in this respect more similar to each other than they are to the major chromosomes from either species.
Comparing Figures 6a and 6b, it is apparent that the two repeat-finding strategies represented gave very different results. D. melanogaster and D. virilis are fairly close together phylogenetically, but use of the previously defined RepBase library, which has good representation of D. melanogaster repeats, was insufficient to find all of the D. virilis repeats, particularly on the dot chromosome. This result stresses the importance of using techniques such as PILER to find species-specific repeats as new species are sequenced, even when repeat sequences are available from a well-studied nearby species. Relying on existing repeat databases can lead to erroneously low estimates of repeat content.
Discussion
The dot chromosomes of D. melanogaster and D. virilisdiffer in the density of DNA transposons
While one of the conspicuous characteristics of pericentric heterochromatin is a low gene density, previous sequence analysis has shown that the heterochromatic D. melanogaster dot chromosome resembles euchromatic domains in this regard, having a gene density (number of genes per Mb) similar to the long arms of the major autosomes [12, 19]. Interestingly, the D. melanogaster dot chromosome does have an approximately two-fold higher percentage of DNA transcribed (percentage of DNA between the start sites and stop sites for transcription) than the major chromosomes, due primarily to longer introns in the dot chromosome genes. Introns of dot chromosome genes of both species examined here were longer than introns from the major chromosomes (Figure 5), apparently reflecting the higher repeat content of the dot chromosomes (see above). Thus, the heterochromatic D. melanogaster and the euchromatic D. virilis dot chromosomes are very similar to each other in gene density, percent DNA transcribed, and gene/intron size, suggesting that these parameters are not critical in determining chromatin packaging decisions.
Total repeat density (percentage of the DNA in repetitive sequences) for the D. virilis dot chromosome fosmids has a value of 22.8%, while the homologous regions of the dot chromosome of D. melanogaster have a total density of 26.5%. Kaminker et al. [18] analyzed the distribution of transposable elements (not including simple or tandem repeats) in the D. melanogaster genome. This analysis indicated that the number of repetitive elements per Mb is five to ten times higher on the dot chromosome than in the rest of the sequenced genome, which includes very little heterochromatin. The repeat analysis of our region of study agrees with the whole chromosome results from Kaminker et al. [18] in that the level of repetitive elements (predominantly partial or dead TEs) shows a large difference (about three- to four-fold) between the dot chromosomes and the major chromosomes. Our analysis reported here shows that there is only a small difference in the total repeat density on the heterochromatic D. melanogaster dot chromosome and on the euchromatic D. virilis dot chromosome. This finding suggests that the higher density of repetitive elements probably does not play a deciding role in driving the heterochromatic packaging of the dot chromosome in D. melanogaster. This does not preclude the possibility that high repeat densities are a necessary precondition for heterochromatin formation, but argues that a high repeat density is not sufficient in and of itself to drive formation of heterochromatin.
This analysis rather focuses attention on the high level of DNA transposons found in the D. melanogaster dot chromosome, but lacking in the D. virilis dot chromosome. Prominent elements of this type in D. melanogaster include 1360 (aka hoppel or PROTOP_A) and DINE-1. It has previously been suggested that DINE-1 might contribute to heterochromatin packaging on the dot chromosome of D. melanogaster [17, 56]. In our computational analysis, we found that sequences homologous to DINE-1 were also present on the dot chromosome of D. virilis, but at a much lower concentration (0.1%) compared to D. melanogaster (10.5%), in agreement with the in situ hybridization analysis previously reported [56]. Our computer homology searches and analysis by others [57] indicates that portions of the DINE-1 element found in RebBase 8.12 show high similarity to a genomic fragment containing Penelope elements from D. virilis, but this similarity falls outside of the region defined by Evgen'ev et al. to be required for Penelope activity [53, 58] (C Bergman, personal communication). Thus, in our analysis, DINE-1 has been treated as a DNA transposon type, but has been reported separately in Figure 6 for clarity. DINE-1 is absent from the major chromosome arms in this sample. In contrast, Penelope elements are very common in the dot chromosome (approximately 9%), and are present in the major chromosomes of D. virilis (0.4%). Retrotransposons such as Penelope are important in determining chromosome rearrangements [59], but have not been associated with heterochromatin formation.
It has been suggested that the buildup of repetitive elements on the dot chromosome may be due in part to the lack of recombination [19, 60]. However, we find an overabundance of repetitive sequences on the dot chromosomes of both D. melanogaster and D. virilis. Recombination does occur on the dot chromosome of D. virilis, albeit at a lower rate [30]. This observation suggests that there may be a selective advantage in maintaining a higher than average density of repetitive sequences (and larger than average genes) in this small chromosome, regardless of the chromatin packaging status.
DNA transposons may be targets for heterochromatin formation
Work by Sun et al. [24] suggests a particular DNA transposon that may act as an initiator of heterochromatin formation on the dot chromosome of D. melanogaster. If a white reporter P element insertion site is within 10 kb of a 1360 element on the dot chromosome, there is a high probability of a variegating phenotype [24]. Hence remnants of this DNA transposon may serve as a cis-acting determinant of heterochromatin formation on the dot chromosome of D. melanogaster, presumably acting as targets of an RNAi-directed process [26] analogous to that reported in S. pombe [1].
1360 elements are fragments of a DNA transposon that has been recognized in many studies to have a high concentration on the dot chromosome and in the pericentric heterochromatin [17, 19]. Coelho et al. [60] studied 1360 in many different strains of D. melanogaster and found that the association with heterochromatin is very consistent, again suggesting that 1360 elements play an important role in the structure and function of heterochromatin. Given a lack of introns, it has been suggested that 1360 elements are derivatives of a retrotranscription event [61], but because this element has terminal inverted repeats at its ends and encodes a transposase with similarity to the P enzyme [57, 61], it is likely to function as a DNA transposon. 1360 may be a very recent invader of the D. melanogaster genome [57]; some differences in insertion sites are observed in different stocks of D. melanogaster [24, 60]. The origin of RNAi is thought to be as a silencing mechanism for retroviral genome invaders or transposons with multiple exact copies. Thus, it is possible that recent invaders are most likely to be targets of RNAi-induced heterochromatin formation.
Some regions of the D. melanogaster dot chromosome have significantly lower density of DNA transposons other than DINE-1 than the chromosome as a whole, particularly contigs 30.1, 106, and 67 (Table 2). The density of DINE-1 elements in these contigs is similar to that in the rest of the dot chromosome, but the level of other DNA transposons is less than 2%. Interestingly, these regions appear to be euchromatic domains of the dot chromosome of D. melanogaster as shown by a white reporter. The region around contig 30.1 is within 25 kb of an insertion site for a P element with full red eye expression, and the region around contig 67 is within 20 kb of six P elements with full red eye expression [24]. This suggests that the local density of repetitive elements other than DINE-1 may be important in driving changes in chromatin structure, or that another factor might be countering the influence of DINE-1 in these regions.
Why might DNA transposons be a preferred target for heterochromatin formation? DNA transposons contain inverted repeats at each end that facilitate their mobilization within the genome; these could intrinsically lead to dsRNA if both ends are transcribed. This has been reported in C. elegans [62], but we are not aware of any similar reports for D. melanogaster. Using a P element reporter, Dorer and Henikoff [63] showed that DNA transposon mobilization events can lead to tandem and inverse duplications of the P element. These tandem arrays can lead to heterochromatin formation and silencing of a reporter gene within the P element construct. It is also possible that an endogenous transposable element could be present as inverted copies in an intron; transcription could then produce hairpins that could be targeted by the RNAi machinery for degradation and result in heterochromatin-mediated transcriptional gene silencing. (Fragments of LINE elements in inverted orientations in the introns of mammalian genes have been found to be a source of miRNAs [64].) Thus, the mode of mobilization might generate configurations of DNA transposon sequence that make these elements a preferred target for RNAi-directed gene silencing and heterochromatin formation.
Our computational analysis has shown that inverted fragments of TEs can readily be found in introns in both species. Screening for inverted repeats (IRs; using RepeatMasker/RebBase 8.12) located within a single intron and within 100 bp of each other revealed 87 copies of inverted TEs (81 DINE-1, 2 1360 elements, 4 S2_DM elements) within the D. melanogaster genome and three (all Penelope) within our D. virilis fosmids. Some of these candidates are predicted to form stable hairpin structures by mfold [65]. Among these hairpin candidates are the 1360 IR found in an intron of Caps on the D. melanogaster dot chromosome, and the Penelope IR found in an intron of toy on the D. virilis dot chromosome. Transcription through these loci could create hairpin structures that might be subsequently processed by the Drosha and Dicer machinery to produce short dsRNA, leading to initiation of heterochromatin formation. Hence the potential exists for both DNA transposons and retroelements to act as targets for RNAi-directed gene silencing and heterochromatin formation; why the former appears to be favored is not clear.
Empirically, the absence of heterochromatin formation on the D. virilis dot chromosome appears not to be related to a lower density of repetitive elements in this genomic domain, but may be a consequence of the low density of DNA tranposons. Gene density, percent DNA transcribed, and size of introns do not seem to be critical discriminatory factors. However, a higher frequency of CA/GT repeats on the D. virilis dot chromosome is associated with euchromatic chromatin packaging. No mechanism has been suggested to explain the significance of this correlation. Other differences in primary sequence and density of particular transposable elements between the dot chromosomes of these two species, as yet unidentified, could also play a role. Genomic data is forthcoming on many Drosophila species [52]. A comparison of several heterochromatic dot chromosomes with several euchromatic dot chromosomes will no doubt further elucidate the basis of heterochromatin formation in Drosophila.
Materials and methods
Chromosome staining
Immunofluorescent staining of polytene chromosomes from D. melanogaster (Oregon R) and D. virilis (Tucson strain 15010-1001.10) third instar larvae was carried out as described [66]. HP1 antibody was monoclonal mouse C1A9 antibody (cell supernatant) used at a dilution of 1:1. Rabbit antibody for histone H3 methylated at lysine 9 was from Upstate Biotechnology (Lake Placid, NY, USA) used at a dilution of 1:25. Secondary antibodies were labeled with Alexa fluor 488 (Molecular Probes, Eugene, OR, USA) for goat anti-mouse and Alexa fluor 594 (Molecular Probes) for goat anti-rabbit, both used at a dilution of 1:400.
Identification of fosmids from the dot chromosome of D. virilis
Coding sequences from all genes from the dot chromosome of D. melanogaster were used in BLASTN [48] searches against the D. pseudoobscura NCBI trace archive [40]. Similar regions were entered into Block Maker [67] to find regions of highest conservation; PCR primers were designed around these regions using CODEHOP [68]. Criteria for inclusion were a length of at least 200 bp, with 80% homology between D. melanogaster and D. pseudoobscura, with screening to the D. melanogaster genome to ensure that the region did not contain any repetitive elements. PCR was performed using D. melanogaster genomic DNA as the template; the PCR products were subsequently labeled and used to probe the BDVIF01 library (of Tucson strain 15010-1001.10) originally described in Bergman et al. [36], now available spotted on a single filter from BACPAC [69]. Hybridizations and washes were performed at low stringency. Positive clones were verified using Southern blots, slot blots, and restriction mapping. Some of the recovered clones mapped to chromosomes other than the D. virilis dot; these appear to have been identified by cross hybridization resulting from low stringency hybridization and washing conditions.
In situhybridization of fosmids to polytene chromosomes
In situ hybridization probes were digoxygenin-dUTP labeled fosmid inserts. Hybridizations were performed on polytene chromosomes of third instar larvae of D. virilis (Tucson strain 15010-1001.10) as described by Casacuberta and Pardue [70].
Sequencing of D. virilisfosmid clones
D. virilis fosmid DNA was prepared by streaking the glycerol stocks onto selective media agar plates, picking three isolated colonies and preparing a mini-prep of DNA from each. Mini-prep DNA was digested using HindIII and analyzed by agarose gel electrophoresis to compare restriction patterns to those obtained initially from the clones. Colonies verified by the restriction pattern were then inoculated into 200 ml of liquid media and grown in culture. Large-scale fosmid DNA isolation was performed from these cultures. The DNA was then sheared with the Hydroshear™ (manufactured by Genomic Solutions, Ann Arbor, MI, USA) to a nominal size range of 3 to 4 kb, end-repaired, and separated on an agarose sizing gel. The 3 to 4 kb band was excised from the gel and purified by phenol:chloroform extraction. The sheared insert DNA was subcloned into pZero2.1 (Invitrogen, Frederick, MD, USA), electroporated into DH10B cells, and the cells plated onto solid media. For each fosmid project, 768 subclones were picked into glycerol-containing media and archive stocks were grown for 24 hours at 37°C. Of these, 384 subclones were processed through the Genome Sequencing Center production sequencing pipeline, including magnetic bead-based DNA purification, dual end sequencing with Big Dye version 3.1 terminator chemistry (Applied Biosystems Inc., Foster City, CA, USA), and analysis on ABI 3730xl sequencers. Sequence assembly was performed using phred, phrap, and consed to design finishing strategies [42, 43]. All fosmids were finished to the same quality standard as used for the human genome [71]. Sequences were confirmed by comparison of in silico digests of the finished sequence to restriction digest patterns of the purified fosmid DNA for at least two separate restriction enzymes. The nucleotide sequences and predicted protein sequences reported here have been submitted to GenBank (accession numbers DQ378280-DQ378293).
Curation strategy
Gene sequences were initially identified by similarity in BLASTALL [48], comparing finished D. virilis fosmids to D. melanogaster protein, EST, cDNA and genomic sequences in GenBank. D. virilis fosmids were also compared to D. pseudoobscura genomic sequence [40] using BLASTN and TBLASTN [72]. Intron-exon boundaries were determined by visual inspection of coding matches, aided by Genscan [73, 74], in conjunction with other annotated features. BLAT [75] was also used in this process, comparing intron-exon boundaries of predicted coding sequence across D. melanogaster, D. yakuba and D. pseudoobscura. Known protein-coding sequences, exon boundaries and intron sizes were obtained from Ensembl [76] and Flybase [13, 77].
D. virilis retroelements, DNA transposons, low-complexity and simple repeat sequences were masked by RepeatMasker [45] using default parameters for Drosophila sequences. The RepeatMasker version used was from 03/06/04 with crossmatch version 0.990329 (RebBase update 8.12). Repeats unique to the D. virilis fosmids were predicted by BLASTN of each fosmid against a database of all the D. virilis sequences obtained, excluding regions that had significant D. melanogaster EST and protein BLASTALL matches.
The Superlibrary was created by combining repeats identified by the Drosophila TE library in RepBase 8.12, novel repeats identified using PILER-DF on the D. virilis dvirAra08 assembly [78] (C Smith and G Karpen, personal communication), novel D. melanogaster repeats identified using PILER-TR [50] and the D. yakuba DINE-1 element [53]. RepeatMasker was used with the Superlibrary to identify the portion of each fosmid or equivalent region that contained repetitive sequences.
RepeatScout (1.0.1) [51] was run with default parameters against the dmelWGS2 and dvirAra08 assemblies to generate the RepeatScout libraries for D. melanogaster and D. virilis. Tandem repeats and simple repeats in each RepeatScout library were removed using trf [79, 80] and nseg [81, 82]. The results were then combined to create a custom Drosophila RepeatScout library.
Sequence comparison
Sequence comparisons were made of D. virilis fosmids with corresponding regions from the D. melanogaster (Release 3.2) genome, as determined by a set of reproducible guidelines involving syntenic features and equivalent flanking sequence. If a region of non-coding sequence from D. virilis was interrupted in the corresponding D. melanogaster sequence by an annotated gene, then only sequence up to that gene was included in the comparison. Where non-coding sequence flanked a corresponding gene in D. melanogaster, with no other identifying features that would indicate a loss of synteny, the extracted sequence extended an equal number of bases from the gene as seen in D. virilis. Several of the D. virilis fosmids (contigs 11, 72, 80, and 91) had no appreciable synteny with the D. melanogaster genome and were, therefore, not included in repeat comparisons between the two species. The gene density and intron size calculations take into account all fosmids.
Kolmogorov-Smirnov test
The KS test is a non-parametric, distribution-free statistical test that can determine if two datasets differ significantly. It produces a D statistic that represents the maximum difference of the two distributions in an empirical cumulative distribution plot, allowing one to accept or reject a hypothesis that the datasets are from the same distribution. Statistical analysis was done using program R from the R Foundation for Statistical Computing [83].
CA/GT repeat analysis
BLASTN [48] was used to search the contigs for CA/GT repeats. Blast databases containing either all of the D. virilis sequence obtained here or all of the corresponding regions from D. melanogaster were constructed using formatdb with default parameters. The databases were searched with a sequence of 100 (CA) repeats using BLASTALL default parameters except that low complexity filtering was turned off and the (E)xpect value was set to 0.1. The location of all hits was analyzed to remove any duplicate hits prior to assignment to either the dot or major chromosomes.
Additional data files
The following additional data are available with the online version of this paper. Additional data file 1 provides maps of each fosmid from D. virilis and the homologous regions from D. melanogaster (if available) showing the genes and identified repetitive elements for dot chromosome sequences. Additional data file 2 provides maps of each fosmid from D. virilis and the homologous regions from D. melanogaster (if available) showing the genes and identified repetitive elements for non-dot chromosome sequences. Additional data file 3 provides a fasta file pre-formatted for use with RepeatMasker containing the PILER-DF identified repeats from the D. virilis assembly dvirAra08, the D. yakuba DINE-1 element, and the PILER-TR identified novel repeats from D. melanogaster, which were added to RepBase 8.12 Drosophila TE library to generate the Superlibrary used to analyze repeats.
References
Grewal SI, Elgin SC: Heterochromatin: new possibilities for the inheritance of structure. Curr Opin Genet Dev. 2002, 12: 178-187. 10.1016/S0959-437X(02)00284-8.
Heitz E: Das Heterochromatin der Moose. Jahrbucher fur Wissenschaftliche Botanik. 1928, 69: 762-818.
Hoskins RA, Smith CD, Carlson JW, Carvalho AB, Halpern A, Kaminker JS, Kennedy C, Mungall CJ, Sullivan BA, Sutton GG, et al: Heterochromatic sequences in a Drosophila whole-genome shotgun assembly. Genome Biol. 2002, 3: R1-0085. 10.1186/gb-2002-3-12-research0085.
Dimitri P, Junakovic N, Arca B: Colonization of heterochromatic genes by transposable elements in Drosophila. Mol Biol Evol. 2003, 20: 503-512. 10.1093/molbev/msg048.
Pardue ML, Lowenhaupt K, Rich A, Nordheim A: (dC-dA)n.(dG-dT)n sequences have evolutionarily conserved chromosomal locations in Drosophila with implications for roles in chromosome structure and function. EMBO J. 1987, 6: 1781-1789.
Richards EJ, Elgin SC: Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell. 2002, 108: 489-500. 10.1016/S0092-8674(02)00644-X.
James TC, Elgin SC: Identification of a nonhistone chromosomal protein associated with heterochromatin in Drosophila melanogaster and its gene. Mol Cell Biol. 1986, 6: 3862-3872.
Eissenberg JC, Elgin SC: The HP1 protein family: getting a grip on chromatin. Curr Opin Genet Dev. 2000, 10: 204-210. 10.1016/S0959-437X(00)00058-7.
Schotta G, Ebert A, Krauss V, Fischer A, Hoffmann J, Rea S, Jenuwein T, Dorn R, Reuter G: Central role of Drosophila SU(VAR)3-9 in histone H3-K9 methylation and heterochromatic gene silencing. EMBO J. 2002, 21: 1121-1131. 10.1093/emboj/21.5.1121.
Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T: Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001, 410: 120-124. 10.1038/35065138.
Eissenberg JC, James TC, Foster-Hartnett DM, Hartnett T, Ngan V, Elgin SC: Mutation in a heterochromatin-specific chromosomal protein is associated with suppression of position-effect variegation in Drosophila melanogaster. Proc Natl Acad Sci USA. 1990, 87: 9923-9927.
Misra S, Crosby MA, Mungall CJ, Matthews BB, Campbell KS, Hradecky P, Huang Y, Kaminker JS, Millburn GH, Prochnik SE, et al: Annotation of the Drosophila melanogaster euchromatic genome: a systematic review. Genome Biol. 2002, R3: 1-0083. 10.1186/gb-2002-3-12-research0083.
Drysdale RA, Crosby MA: FlyBase: genes and gene models. Nucleic Acids Res. 2005, 33 (Database): D390-395. 10.1093/nar/gki046.
Barigozzi C, Dolfini S, Fraccaro M, Raimondi GR, Tiepolo L: In vitro study of the DNA replication patterns of somatic chromosomes of Drosophila melanogaster. Exp Cell Res. 1966, 43: 231-234. 10.1016/0014-4827(66)90399-5.
Bridges CB: The mutants and linkage data of chromosome four of Drosophila melanogaster. Biol Zh. 1935, 4: 401-420.
Miklos GL, Yamamoto MT, Davies J, Pirrotta V: Microcloning reveals a high frequency of repetitive sequences characteristic of chromosome 4 and the beta-heterochromatin of Drosophila melanogaster. Proc Natl Acad Sci USA. 1988, 85: 2051-2055.
Locke J, Podemski L, Roy K, Pilgrim D, Hodgetts R: Analysis of two cosmid clones from chromosome 4 of Drosophila melanogaster reveals two new genes amid an unusual arrangement of repeated sequences. Genome Res. 1999, 9: 137-149.
Kaminker JS, Bergman CM, Kronmiller B, Carlson J, Svirskas R, Patel S, Frise E, Wheeler DA, Lewis SE, Rubin GM, et al: The transposable elements of the Drosophila melanogaster euchromatin: a genomics perspective. Genome Biol. 2002, 3: R1-0084. 10.1186/gb-2002-3-12-research0084.
Bartolome C, Maside X, Charlesworth B: On the abundance and distribution of transposable elements in the genome of Drosophila melanogaster. Mol Biol Evol. 2002, 19: 926-937.
James TC, Eissenberg JC, Craig C, Dietrich V, Hobson A, Elgin SC: Distribution patterns of HP1, a heterochromatin-associated nonhistone chromosomal protein of Drosophila. Eur J Cell Biol. 1989, 50: 170-180.
Haynes KA, Leibovitch BA, Rangwala SH, Craig C, Elgin SC: Analyzing heterochromatin formation using chromosome 4 of Drosophila melanogaster. Cold Spring Harb Symp Quant Biol. 2004, 69: 267-272. 10.1101/sqb.2004.69.267.
Wallrath LL, Elgin SC: Position effect variegation in Drosophila is associated with an altered chromatin structure. Genes Dev. 1995, 9: 1263-1277.
Sun FL, Cuaycong MH, Craig CA, Wallrath LL, Locke J, Elgin SC: The fourth chromosome of Drosophila melanogaster : interspersed euchromatic and heterochromatic domains. Proc Natl Acad Sci USA. 2000, 97: 5340-5345. 10.1073/pnas.090530797.
Sun FL, Haynes K, Simpson CL, Lee SD, Collins L, Wuller J, Eissenberg JC, Elgin SC: cis -Acting determinants of heterochromatin formation on Drosophila melanogaster chromosome four. Mol Cell Biol. 2004, 24: 8210-8220. 10.1128/MCB.24.18.8210-8220.2004.
Matzke MA, Birchler JA: RNAi-mediated pathways in the nucleus. Nat Rev Genet. 2005, 6: 24-35. 10.1038/nrg1500.
Pal-Bhadra M, Leibovitch BA, Gandhi SG, Rao M, Bhadra U, Birchler JA, Elgin SC: Heterochromatic silencing and HP1 localization in Drosophila are dependent on the RNAi machinery. Science. 2004, 303: 669-672. 10.1126/science.1092653.
Aravin AA, Lagos-Quintana M, Yalcin A, Zavolan M, Marks D, Snyder B, Gaasterland T, Meyer J, Tuschl T: The small RNA profile during Drosophila melanogaster development. Dev Cell. 2003, 5: 337-350. 10.1016/S1534-5807(03)00228-4.
Clayton FE, Guest WC: Overview of chromosomal evolution in the family Drosophilidae. The Genetics and Biology of Drosophila. 1986, 3E: 1-38.
Sturtevant AH, Novitski E: The homologies of the chromosome elements in the genus Drosophila. Genetics. 1941, 26: 517-538.
Gubenko IS, Evgen'ev MB: Cytological and linkage maps of Drosophila virilis chromosomes. Genetica. 1984, 65: 127-139. 10.1007/BF00135277.
Podemski L, Ferrer C, Locke J: Whole arm inversions of chromosome 4 in Drosophila species. Chromosoma. 2001, 110: 305-312.
Powell JR, DeSalle R: Drosophila molecular phylogenies and their uses. Evol Biol. 1995, 28: 87-138.
Lowenhaupt K, Rich A, Pardue ML: Nonrandom distribution of long mono- and dinucleotide repeats in Drosophila chromosomes: correlations with dosage compensation, heterochromatin, and recombination. Mol Cell Biol. 1989, 9: 1173-1182.
Chino M, Kikkawa H: Mutants and crossing over in the dot-like chromosome of Drosophila virilis. Genetics. 1933, 18: 111-116.
Cliften PF, Hillier LW, Fulton L, Graves T, Miner T, Gish WR, Waterston RH, Johnston M: Surveying Saccharomyces genomes to identify functional elements by comparative DNA sequence analysis. Genome Res. 2001, 11: 1175-1186. 10.1101/gr.182901.
Bergman CM, Pfeiffer BD, Rincon-Limas DE, Hoskins RA, Gnirke A, Mungall CJ, Wang AM, Kronmiller B, Pacleb J, Park S, et al: Assessing the impact of comparative genomic sequence data on the functional annotation of the Drosophila genome. Genome Biol. 2002, 3: R1-0086. 10.1186/gb-2002-3-12-research0086.
Richards S, Liu Y, Bettencourt BR, Hradecky P, Letovsky S, Nielsen R, Thornton K, Hubisz MJ, Chen R, Meisel RP, et al: Comparative genome sequencing of Drosophila pseudoobscura : chromosomal, gene, and cis-element evolution. Genome Res. 2005, 15: 1-18. 10.1101/gr.3059305.
Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, Kravitz SA, Mobarry CM, Reinert KH, Remington KA, et al: A whole-genome assembly of Drosophila. Science. 2000, 287: 2196-2204. 10.1126/science.287.5461.2196.
Hartl DL, Nurminsky DI, Jones RW, Lozovskaya ER: Genome structure and evolution in Drosophila : applications of the framework P1 map. Proc Natl Acad Sci USA. 1994, 91: 6824-6829.
Drosophila Genome Project at Baylor College of Medicine. [http://www.hgsc.bcm.tmc.edu/projects/drosophila/]
Ewing B, Hillier L, Wendl MC, Green P: Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8: 175-185.
Ewing B, Green P: Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8: 186-194.
Gordon D, Abajian C, Green P: Consed: a graphical tool for sequence finishing. Genome Res. 1998, 8: 195-202.
Devlin RH, Bingham B, Wakimoto BT: The organization and expression of the light gene, a heterochromatic gene of Drosophila melanogaster. Genetics. 1990, 125: 129-140.
RepeatMasker. [http://repeatmasker.org]
Jurka J: Repeats in genomic DNA: mining and meaning. Curr Opin Struct Biol. 1998, 8: 333-337. 10.1016/S0959-440X(98)80067-5.
Jurka J: Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 2000, 16: 418-420. 10.1016/S0168-9525(00)02093-X.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215: 403-410. 10.1006/jmbi.1990.9999.
Quesneville H, Bergman CM, Andrieu O, Autard D, Nouaud D, Ashburner M, Anxolabehere D: Combined evidence annotation of transposable elements in genome sequences. PLoS Comput Biol. 2005, 1: 166-175.
Edgar RC, Myers EW: PILER: Identification and classification of genomic repeats. Bioinformatics. 2005, 21 (Suppl 1): i152-i158. 10.1093/bioinformatics/bti1003.
Price AL, Jones NC, Pevzner PA: De novo identification of repeat families in large genomes. Bioinformatics. 2005, 21 (Suppl 1): i351-i358. 10.1093/bioinformatics/bti1018.
Assembly/Alignment/Annotation of 12 related Drosophila species. [http://rana.lbl.gov/drosophila]
Yang HP, Hung TL, You TL, Yang TH: Genome-wide comparative analysis of the highly abundant transposable element DINE-1 suggests a recent transpositional burst in Drosophila yakuba. Genetics. 2005, doi:10.1534/genetics.105.051714
Quesneville H, Nouaud D, Anxolabehere D: Detection of new transposable element families in Drosophila melanogaster and Anopheles gambiae genomes. J Mol Evol. 2003, 57 (Suppl 1): S50-59. 10.1007/s00239-003-0007-2.
Singh ND, Arndt PF, Petrov DA: Genomic heterogeneity of background substitutional patterns in Drosophila melanogaster. Genetics. 2005, 169: 709-722. 10.1534/genetics.104.032250.
Locke J, Howard LT, Aippersbach N, Podemski L, Hodgetts RB: The characterization of DINE-1, a short, interspersed repetitive element present on chromosome and in the centric heterochromatin of Drosophila melanogaster. Chromosoma. 1999, 108: 356-366. 10.1007/s004120050387.
Kapitonov VV, Jurka J: Molecular paleontology of transposable elements in the Drosophila melanogaster genome. Proc Natl Acad Sci USA. 2003, 100: 6569-6574. 10.1073/pnas.0732024100.
Pyatkov KI, Shostak NG, Zelentsova ES, Lyozin GT, Melekhin MI, Finnegan DJ, Kidwell MG, Evgen'ev MB: Penelope retroelements from Drosophila virilis are active after transformation of Drosophila melanogaster. Proc Natl Acad Sci USA. 2002, 99: 16150-16155. 10.1073/pnas.252641799.
Evgen'ev M, Zelentsova H, Mnjoian L, Poluectova H, Kidwell MG: Invasion of Drosophila virilis by the Penelope transposable element. Chromosoma. 2000, 109: 350-357.
Coelho PA, Queiroz-Machado J, Hartl D, Sunkel CE: Pattern of chromosomal localization of the Hoppel transposable element family in the Drosophila melanogaster subgroup. Chromosome Res. 1998, 6: 385-395. 10.1023/A:1009277322626.
Reiss D, Quesneville H, Nouaud D, Andrieu O, Anxolabehere D: Hoppel, a P-like element without introns: a P-element ancestral structure or a retrotranscription derivative?. Mol Biol Evol. 2003, 20: 869-879. 10.1093/molbev/msg090.
Sijen T, Plasterk RH: Transposon silencing in the Caenorhabditis elegans germ line by natural RNAi. Nature. 2003, 426: 310-314. 10.1038/nature02107.
Dorer DR, Henikoff S: Expansions of transgene repeats cause heterochromatin formation and gene silencing in Drosophila. Cell. 1994, 77: 993-1002. 10.1016/0092-8674(94)90439-1.
Smalheiser NR, Torvik VI: Mammalian microRNAs derived from genomic repeats. Trends Genet. 2005, 21: 322-326. 10.1016/j.tig.2005.04.008.
Matthews DH, Sabina J, Zuker M, Turner DH: Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol. 1999, 288: 911-940. 10.1006/jmbi.1999.2700.
Stephens GE, Craig CA, Li Y, Wallrath LL, Elgin SCR: Immunofluorescent staining of polytene chromosomes: exploiting genetic tools. Methods Enzymol. 2004, 376: 372-393.
Henikoff S, Henikoff JG, Alford WJ, Pietrokovski S: Automated construction and graphical presentation of protein blocks from unaligned sequences. Gene. 1995, 163: GC17-26. 10.1016/0378-1119(95)00486-P.
Rose TM, Schultz ER, Henikoff JG, Pietrokovski S, McCallum CM, Henikoff S: Consensus-degenerate hybrid oligonucleotide primers for amplification of distantly related sequences. Nucleic Acids Res. 1998, 26: 1628-1635. 10.1093/nar/26.7.1628.
BACPAC Center at CHORI. [http://bacpac.chori.org]
Casacuberta E, Pardue ML: Coevolution of the telomeric retrotransposons across Drosophila species. Genetics. 2002, 161: 1113-1124.
International Human Genome Sequencing Consortium: Finishing the euchromatic sequence of the human genome. Nature. 2004, 431: 931-945. 10.1038/nature03001.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-3402. 10.1093/nar/25.17.3389.
GENSCAN. [http://genes.mit.edu/GENSCAN.html]
Burge C, Karlin S: Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997, 268: 78-94. 10.1006/jmbi.1997.0951.
Kent WJ: BLAT - the BLAST-like alignment tool. Genome Res. 2002, 12: 656-664. 10.1101/gr.229202. Article published online before March 2002.
Ensembl. [http://www.ensembl.org]
Flybase. [http://www.flybase.org]
Drosophila Heterochromatin Genome Project PILER-DF Libraries. [ftp://ftp.dhgp.org/pub/DHGP/PILER-DF/]
Tandem Repeat Finder (trf). [http://tandem.bu.edu/trf/trf.html]
Benson G: Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999, 27: 573-580. 10.1093/nar/27.2.573.
Segment sequence(s) by local complexity (seg). [ftp://ftp.ncbi.nih.gov/pub/seg/nseg/]
Wooton J, Federhen S: Statistics of local complexity in amino acid sequences and sequence databases. Comput Chem. 1993, 17: 149-163. 10.1016/0097-8485(93)85006-X.
R project. [http://www.r-project.org]
Acknowledgements
We would like to thank the Washington University Genome Sequencing Center for generating raw sequences and providing training and support for members of Bio 4342. Ginger Fewell and Catrina Fronick coordinated library construction and sequencing, Darren O'Brien directed wet lab work, while Cynthia Madsen-Strong, J Phillip Latreille, Charlene Pearman, and Joelle Viezer provided training and support in the use of consed. Michael Brent and Mani Arumugam aided in annotation and analysis of fosmid sequences. We would like to thank Alan Templeton for his help in the statistical analysis of intron size and Casey Bergman (University of Manchester, Manchester, UK) for review and advice on repetitive sequence analysis. We would also like to thank Chris Smith and Gary Karpen (Lawrence Berkeley National Lab, Berkeley, CA, USA) for access to and use of their D. virilis whole genome PILER-DF library and HP Yang (National Yang-Ming University, Taipei, Taiwan, ROC) for making available her analysis of D. yakuba DINE-1 sequence prior to publication. Members of the Elgin lab contributed throughout the process with criticism and suggestions. This work was supported primarily by Grant #52003904 from the Howard Hughes Medical Institute to Washington University (for SCRE) with additional funding from NIH grant GM050315 to MLP and NIH grant GM068388 to SCRE.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
13059_2005_1283_MOESM1_ESM.pdf
Additional data file 1: Maps of each fosmid from Drosophila virilis and the homologous regions from Drosophila melanogaster (if available) showing the genes and identified repetitive elements for dot chromosome sequences (PDF 2 MB)
13059_2005_1283_MOESM2_ESM.pdf
Additional data file 2: Maps of each fosmid from D. virilis and the homologous regions from D. melanogaster (if available) showing the genes and identified repetitive elements for non-dot chromosome sequences (PDF 1017 KB)
13059_2005_1283_MOESM3_ESM.txt
Additional data file 3: A fasta file pre-formatted for use with RepeatMasker containing the PILER-DF identified repeats from the D. virilis assembly dvirAra08, the D. yakuba DINE-1 element, and the PILER-TR identified novel repeats from D. melanogaster, which were added to RepBase 8.12 Drosophila TE library to generate the Superlibrary used to analyze repeats (TXT 148 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Slawson, E.E., Shaffer, C.D., Malone, C.D. et al. Comparison of dot chromosome sequences from D. melanogaster and D. virilisreveals an enrichment of DNA transposon sequences in heterochromatic domains. Genome Biol 7, R15 (2006). https://doi.org/10.1186/gb-2006-7-2-r15
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/gb-2006-7-2-r15