
Abstract
Background
Candida glabrata is a pathogenic yeast of increasing medical concern. It has been regarded as asexual since it was first described in 1917, yet phylogenetic analyses have revealed that it is more closely related to sexual yeasts than other Candida species. We show here that the C. glabrata genome contains many genes apparently involved in sexual reproduction.
Results
By genome survey sequencing, we find that genes involved in mating and meiosis are as numerous in C. glabrata as in the sexual species Kluyveromyces delphensis, which is its closest known relative. C. glabrata has a putative mating-type (MAT) locus and a pheromone gene (MFALPHA2), as well as orthologs of at least 31 other Saccharomyces cerevisiae genes that have no known roles apart from mating or meiosis, including FUS3, IME1 and SMK1.
Conclusions
We infer that C. glabrata is likely to have an undiscovered sexual stage in its life cycle, similar to that recently proposed for C. albicans. The two Candida species represent two distantly related yeast lineages that have independently become both pathogenic and 'asexual'. Parallel evolution in the two lineages as they adopted mammalian hosts resulted in separate but analogous switches from overtly sexual to cryptically sexual life cycles, possibly in response to defense by the host immune system.
Similar content being viewed by others

Background
The genus Candida consists of ascomycete yeast species that lack an apparent sexual (teleomorph) stage in their life cycle and seem to reproduce only mitotically. However, data from the C. albicans genome project has recently led researchers to question the asexuality of that species. C. albicans was found to have a mating type-like (MTL) locus similar to the Saccharomyces cerevisiae MAT locus [1]. Natural isolates of C. albicans are diploid MTL a/MTLα heterozygotes, similar to diploid S. cerevisiae but unable to sporulate. By genetic engineering to create MTL a and MTLα hemizygotes, C. albicans was induced to mate in the laboratory and in infected mice, forming tetraploids [2,3]. In addition, Miller and Johnson [4] showed that C. albicans MTL hemizygotes undergo phenotypic switching between the common 'white' form and an 'opaque' form that is a million-fold more active in mating. Further analysis of the almost-complete genome sequence of C. albicans revealed that it contains homologs of most of the S. cerevisiae genes involved in the key sexual processes of meiosis and sporulation as well as mating [5]. These findings have led to the hypothesis that the life cycle of C. albicans includes a cryptic sexual phase, which perhaps is utilized only occasionally or under particular environmental conditions [6] - infrequently enough not to have been detected during more than a century of research into C. albicans, but frequently enough to cause evolutionary conservation of the genes involved in the sexual process. Interestingly, sexual forms of some other Candida species were identified long ago by mycologists but their significance has often gone unrecognized by molecular biologists because the anamorphs and teleomorphs are assigned different names - for example, the sexual form of Candida krusei is called Issatchenkia orientalis [7,8].
Medically, C. albicans is still the major fungal agent of human disease, but C. glabrata is a species of growing concern. The incidence of C. glabrata infections, particularly in the bloodstream, has risen alarmingly over the past decade [9,10]. It has also gained much attention since the discovery of its inherently low susceptibility to the drug fluconazole [11]. The genome of C. glabrata appears to be haploid whereas C. albicans is diploid [12,13]. But similarly to C. albicans, C. glabrata also undergoes phenotypic switching [14,15], raising the interesting question of whether it too may have an undiscovered teleomorph form.
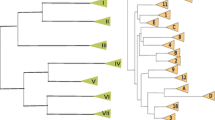
In phylogenetic trees drawn from rDNA sequences, most Candida species including C. albicans fall into one monophyletic group, whereas C. glabrata is much more closely related to S. cerevisiae than to the C. albicans group (Figure 1 and [16,17,18]). Therefore, the 'asexual' life cycles of C. glabrata and C. albicans arose independently from sexual ancestors [16,19]. Phylogenetic analysis also showed unexpectedly that C. glabrata's closest relative is Kluyveromyces delphensis, a sexual species [17,18]. C. glabrata is a commensal resident of the human intestinal tract and an opportunistic pathogen [20,21], whereas K. delphensis was first isolated from dried figs [22] and is often found associated with Drosophila willistoni [23]. The type strain of K. delphensis is homothallic and therefore probably diploid. It has been studied very little at the molecular level.

Phylogenetic relationships among ascomycete yeasts, based on the aligned coding regions of the 5S, 18S, 5.8S and 26S rRNAs. Thick lines show 'asexual' lineages. The tree was constructed by the neighbor-joining method and bootstrap values (1,000 replicates) are shown. A. gossypii, Ashbya gossypii; D. hansenii, Debaryomyces hansenii; P. angusta, Pichia angusta; P. sorbitophila, Pichia sorbitophila; Z. rouxii, Zygosaccharomyces rouxii.
The aim of the present study was to use genome survey sequencing (GSS) to characterize the genomic differences between the closely related asexual C. glabrata and the sexual K. delphensis. However, we find that there are no significant differences between the gene repertoires of these species. C. glabrata has many genes involved in mating, meiosis and sporulation, including a pheromone gene and a putative mating-type locus. This leads us to propose that, like C. albicans, it must have an undiscovered sexual phase in its life cycle.
Results
Genome survey sequencing of C. glabrata and K. delphensis
We constructed plasmid libraries with random genomic inserts of 7-15 kilobases (kb) from C. glabrata and K. delphensis and sequenced both ends of about 3,000 plasmids (> 3 megabases (Mb) of primary sequence data, or approximately 0.2× genome coverage) from each species. Phylogenetic analysis of the combined complete 5S, 18S, 5.8S and 26S rDNA sequences from yeasts (Figure 1) confirms that C. glabrata and K. delphensis are each other's closest known relatives [17,18]. The phylogenetic tree also confirms that C. glabrata is more closely related to S. cerevisiae than to C. albicans, indicating that the two Candida species originated independently from sexual ancestors.
Assembled sequence contigs from C. glabrata and K. delphensis were searched against the complete set of S. cerevisiae proteins using FASTY reciprocal best hits to identify putative orthologs (see Materials and methods). The results of the genome surveys of the two species should be roughly comparable to one another, because the same methods and similar numbers of clones were used in both cases. The genomes can be assumed to be similar in size [24,25], and the two species are equally distantly related to S. cerevisiae. Indeed, they yielded similar numbers of sequence contigs (4,481 from C. glabrata, 4,202 from K. delphensis) and similar numbers of putative genes with unambiguous S. cerevisiae orthologs (1,941 and 2,057, respectively).
Gene functions in C. glabrata and K. delphensis were inferred from the known functions of their S. cerevisiae orthologs, using the 'cellular role' categories of the Yeast Proteome Database (Table 1). Orthologs were found for approximately 40% of the S. cerevisiae genes involved in most cellular roles, reflecting the level of GSS coverage. The numbers of genes found in C. glabrata and K. delphensis for each cellular role are highly similar (Table 1), and the two genomes are not significantly different in any of the 42 categories (p > 0.05 by χ2 test). Importantly, C. glabrata does not have significantly fewer genes than K. delphensis in the categories of mating response and meiosis, which relate to sexual functions.
Mating pathway genes
We identified C. glabrata orthologs of many genes in the S. cerevisiae mating response pathway including the Ste11, Ste7 and Fus3 kinases and the Ste12 transcription factor (Figure 2). Because some components of the mating pathway also participate in other pathways (such as filamentous growth) that might legitimately be expected to be present in an asexual organism, we focus here on genes that have no other known functions apart from mating. The GSS data identified C. glabrata orthologs of 13 S. cerevisiae genes that may be involved exclusively in mating (Table 2). These include an α-factor pheromone gene (MFALPHA2 [26]), STE13, whose sole function appears to be maturation of prepro-α-factor [27], and STE6, whose only known role is in a-factor export [28]. The complete C. glabrata MFALPHA2 gene was sequenced and codes for a signal peptide and three repeats of a candidate mature pheromone sequence WHWV(R/K)(L/I)RKGQGLF (single-letter amino-acid notation) flanked by processing sites for Kex2 [29], Kex1 and Ste13 proteases. The ortholog in K. delphensis was also sequenced and has four copies of the sequence WHWLSVRPGQPIY. The two precursor proteins share 49% sequence identity.

Model of the S. cerevisiae pheromone response pathway (adapted from [30]). Genes whose orthologs were identified in C. glabrata are indicated in gray.
C. glabrata appears capable of responding to pheromones as well as synthesizing them, because it has genes for the polarity-establishment proteins Far1 and Cdc24 [30], for Sgv1 (a kinase acting in the pheromone adaptation pathway [31]), and Akr1 (a protein with an inhibitory effect on the pheromone signal transduction pathway [32]). At the end of the signal transduction cascade it has orthologs of Fus3 (the final MAP kinase in the mating response pathway, which activates Ste12 and Far1 [33]), as well as the nuclear fusion protein Kar5 [34].
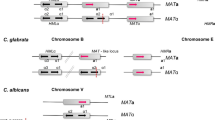
We identified putative mating-type (MAT) loci in both C. glabrata and K. delphensis, containing orthologs of the S. cerevisiae genes for the α1 transcription activator and the α2 repressor (MATALPHA1 and MATALPHA2, respectively), oriented divergently (Figure 3). As expected from the species phylogeny, the level of amino-acid sequence identity between S. cerevisiae and C. glabrata (38% in α1, and 40% in α2) is greater than that between S. cerevisiae and K. lactis or C. albicans in the same proteins [1,35]. Between C. glabrata and K. delphensis, there is 59% amino-acid sequence identity in α1 and 76% in α2.

Gene organization (not to scale) around the MAT locus of S. cerevisiae and the putative MAT loci of K. delphensis and C. glabrata. Dashed horizontal lines indicate the extents of the clones sequenced.
In K. delphensis the α2 and α1 genes are flanked on one side by a series of five genes whose orthologs are beside the MAT locus on S. cerevisiae chromosome III (Figure 3), in the same arrangement except that K. delphensis lacks PHO87. These genes include BUD5, which is almost twice as large in K. delphensis as in S. cerevisiae (1,241 amino acids versus 642). The predicted KdBud5 protein includes an extra SH3 domain near its amino terminus, giving it an overall structure more similar to Cdc25 [36]. On the other side of the α2 and α1 genes in K. delphensis there is a series of five genes whose orthologs are on S. cerevisiae chromosome XII, beginning with YLR186W (EMG1 [37]). The same breakpoint between chromosome III and chromosome XII orthologs is also seen in C. glabrata (Figure 3). It therefore seems likely that a chromosomal rearrangement occurred on the right-hand side of the MAT locus either in an ancestor of K. delphensis and C. glabrata, or in an ancestor of S. cerevisiae. Interestingly, the coding regions of the α1 gene and EMG1 overlap by 28 nucleotides at their 3' ends in both K. delphensis and C. glabrata.
Meiotic genes
The GSS data from C. glabrata also identified orthologs of many S. cerevisiae genes involved in meiosis, a central step in the sexual cycle that leads ultimately to the production of gametes (sporulation). We found C. glabrata orthologs of 19 S. cerevisiae genes whose only known functions are in meiosis or sporulation (Table 3), including the master regulatory switch gene IME1 [38]. S. cerevisiae IME1 expression is induced by the a1/α2 heterodimer representing the genetic signal from a diploid cell, in combination with nutritional signals. We found C. glabrata orthologs of MCK1 and RIM9, which are inducers of IME1 expression, and UME6 which negatively regulates meiosis-specific genes during vegetative growth but is converted into an activator of early meiosis genes when Ime1 is present [39,40]. C. glabrata also has orthologs of IME2, which can promote sporulation in the absence of IME1 [41], and IDS2 and RIM4, whose products promote Ime2-dependent activation of many downstream targets [42,43].
C. glabrata has orthologs of RIM4 and MUM2, both of which are needed for premeiotic DNA replication [44,45], and HOP2, which acts to prevent synapsis between nonhomologous chromosomes [46]. We also found MSH4, which is implicated in synaptonemal complex formation and meiotic recombination [47]. The presence of these genes suggests that critical events required for the unique process of reductional division during meiosis I, such as recombination and chromosome synapsis, occur in C. glabrata.
Similarly, we found orthologs of genes involved in the middle and late stages of meiosis. The middle-stage genes include SPO1, a phospholipase B homolog that promotes spindle-body duplication exclusively during meiosis [48], and SPO22, CSM1 and CSM3 which are less well characterized but show meiosis-specific expression with deletion mutants exhibiting varying degrees of chromosome missegregation [49]. C. glabrata also has a homolog of SMK1, which in S. cerevisiae encodes a MAP kinase involved in a sporulation-specific signal transduction cascade, necessary for proper spore morphogenesis and full expression of late meiotic genes [50,51]. Another surprising finding is an ortholog of DIT1, which is required for dityrosine biosynthesis [52]. In yeasts, the dimerized amino acid dityrosine has only been found on the outer surface layer of the ascospore wall but not in vegetative cell walls [53]. The maintenance of these genes in C. glabrata is highly indicative of an ability to sporulate.
Discussion
The results from survey sequencing of the C. glabrata and K. delphensis genomes show that they have very similar repertoires of genes in all categories of cellular roles (Table 1), including mating and meiosis. More detailed analysis showed that C. glabrata has orthologs of at least 31 genes that in S. cerevisiae have no known functions apart from mating or meiosis (Tables 2 and 3), and that it has intact genes for α-factor and a putative mating-type locus. Together, these results suggest that C. glabrata has an undiscovered sexual cycle. Although it is possible that future studies in S. cerevisiae will discover new roles for some of these genes other than in mating or meiosis, it seems more reasonable to propose that C. glabrata has a sexual cycle than to propose that it is asexual and that all 31 genes have been preserved in its genome because they have undiscovered roles in nonsexual processes. The compact nature of yeast genomes makes it unlikely that all the sexual genes we identified by GSS are pseudogenes, and the MFALPHA2, MATALPHA1 and MATALPHA2 genes certainly are not pseudogenes.
Even though we did not find orthologs of some other genes that are central to mating (for example, STE2/STE3 and MFA1 [54]) or meiosis (for example, NDT80), it should be noted that the genome was only surveyed to 0.2× sequence coverage, so that only 1,941 genes (roughly one-third of the expected number of genes in the genome) were detected in this study. It is interesting that C. glabrata has orthologs of MFALPHA2 and IME1, which were not found in C. albicans [5]. However, this is possibly just due to extensive sequence divergence, rather than gene loss, in C. albicans. The complete sequence of C. glabrata Ime1 has only 27% amino-acid sequence identity to S. cerevisiae Ime1. Very recently, a candidate C. albicans pheromone gene was described [55,56].
Despite the evolutionary distance between them (Figure 1) and gross differences such as the fact that C. glabrata is haploid whereas C. albicans is diploid, there are remarkable parallels between the evolution of C. glabrata and C. albicans. The two species have evolved independently from sexually reproducing yeast ancestors that are unlikely to have been pathogenic, because the majority of lineages in this phylogenetic group are not pathogenic (Figure 1). Thus, in becoming human pathogens, both C. glabrata and C. albicans have adopted a lifestyle where the sexual phase is hidden. Miller and Johnson [4] proposed that, in C. albicans, this is because the white (asexual) form survives better in the mammalian host. By analogy, it is tempting to speculate that one of the forms produced by phenotypic switching in C. glabrata [14,15] might be mating-competent. It is interesting to note that in other species of Candida for which sexual forms (teleomorphs) have been identified, such as Candida krusei, the form isolated in clinical settings is invariably the asexual one [7,8]. We hypothesize that having a sexual cycle may be essential for the long-term evolutionary viability of all yeast species because of the evolutionary advantages conferred by recombination [57,58], but that mating confers a disadvantage on those individuals that mate because they are somehow more vulnerable to the host's immune response. The result of these opposing forces seems to have been the evolution of cryptically sexual pathogens in which the frequency of mating in the population has been reduced to a low but optimal level.
Materials and methods
The type strains of C. glabrata (CBS 138) and K. delphensis (CBS 2170) were purchased from the Centraalbureau voor Schimmelcultures (Utrecht, Netherlands). High-molecular-weight DNA was prepared using standard procedures and partially digested with Sau3AI. Fragments in the size range 7-15 kb were isolated and used to make random genomic libraries in the low copy number Escherichia coli vector pMCL210 (AGOWA, Germany). Sequences were obtained from both ends of the insert for 2,939 C. glabrata (CG) and 2,974 K. delphensis (KD) clones, with a further 449 CG and 290 KD clones sequenced successfully from one end only. The average lengths of sequence reads used for analysis were 548 base-pairs (bp) (CG) and 515 bp (KD). Representation of mitochondrial DNA in the libraries appeared to be very low, even though we did not take any specific measures to exclude it. After data analysis, the inserts of selected plasmid clones were sequenced completely on both strands by primer walking. Sequences have been deposited in GenBank with accession numbers BZ293019-BZ299345 (C. glabrata GSS), BZ299346-BZ305583 (K. delphensis GSS), AY181247-AY181250 (complete sequences of MAT and MFALPHA2 loci), and AJ535506 (C. glabrata IME1).
Trace files from the random genomic clones were base-called using PHRED [59,60] and vector clipping was done by CROSS_MATCH. Clipped sequences shorter than 100 bp were discarded from the dataset. Contigs were assembled using PHRAP with the original trace quality files and are available on request. Contigs were filtered to eliminate mitochondrial DNA as well as known repetitive sequences such as rDNA and Ty elements, which may cause misassembly. This was achieved using the contigs as queries in BLASTN and BLASTX searches [61] against the relevant S. cerevisiae sequences. Any contig with a significant expect value (E-value) of < 1e-5 was excluded from ortholog assignment.
We used a recent annotation of the S. cerevisiae genome [62], containing 5,583 annotated proteins (excluding 'very hypothetical' proteins and pseudogenes), downloaded from [63]. Orthologs of these genes in the C. glabrata and K. delphensis filtered contigs were identified using FASTY version 3.4t05 [64], after a low complexity masking step using the NSEG and PSEG programs [65]. For any gene-sized region in a contig, we considered the S. cerevisiae protein with the strongest FASTY hit to be the ortholog, provided that the E-value for this hit was < 1e-5 and was more than 1e3 times lower than the E-value for the second-best hit to the same region of the contig. For each ortholog identified, its function in S. cerevisiae was examined using the 'cellular role' categories of the Yeast Proteome Database (YPD) of the Incyte BioKnowledge Library [66]. It should be noted that in this functional annotation scheme, a single protein can be classified into more than one cellular role. While we adhered strictly to the YPD classification of genes in our initial analyses (Table 1), we also discovered some genes that can be reclassified on the basis of the literature (for example, SPS2 was classified under 'differentiation' in YPD but was found to be meiosis-specific on further examination) and included these reclassified genes in Tables 2 and 3.
Among the full set of contigs, those that contained rDNA sequences were identified. These were used in conjunction with publicly available rDNA sequences, isolated from the C. glabrata and K. delphensis type strains, to create the complete rDNA repeating unit for both species. The two rDNA sequences were combined with those from 14 other hemiascomycete yeast species used in our previous study [67] and aligned using T_COFFEE [68]. Phylogenetic trees were constructed using the NJ method as implemented in CLUSTALW [69].
Additional data files
Tables of the mating response and meiosis genes identified in C. glabrata and K. delphensis are available as one additional data file.
References
Hull CM, Johnson AD: Identification of a mating type-like locus in the asexual pathogenic yeast Candida albicans. Science. 1999, 285: 1271-1275. 10.1126/science.285.5431.1271.
Magee BB, Magee PT: Induction of mating in Candida albicans by construction of MTLa and MTLα strains. Science. 2000, 289: 310-313. 10.1126/science.289.5477.310.
Hull CM, Raisner RM, Johnson AD: Evidence for mating of the "asexual" yeast Candida albicans in a mammalian host. Science. 2000, 289: 307-310. 10.1126/science.289.5477.307.
Miller M, Johnson A: White-opaque switching in Candida albicans is controlled by mating-type locus homeodomain proteins and allows efficient mating. Cell. 2002, 110: 293-302.
Tzung KW, Williams RM, Scherer S, Federspiel N, Jones T, Hansen N, Bivolarevic V, Huizar L, Komp C, Surzycki R, et al: Genomic evidence for a complete sexual cycle in Candida albicans. Proc Natl Acad Sci USA. 2001, 98: 3249-3253. 10.1073/pnas.061628798.
Gow N: Candida albicans switches mates. Mol Cell. 2002, 10: 217-218.
Calderone RA: Taxonomy and biology of Candida. In Candida and Candidiasis. Edited by: Calderone RA. 2002, Washington, DC: American Society for Microbiology, 15-27.
Kurtzman CP, Fell JW: The Yeasts: A Taxonomic Study. Amsterdam: Elsevier. 1998
Fidel PL, Vazquez JA, Sobel JD: Candida glabrata: review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin Microbiol Rev. 1999, 12: 80-96.
Trick WE, Fridkin SK, Edwards JR, Hajjeh RA, Gaynes RP: Secular trend of hospital-acquired candidemia among intensive care unit patients in the United States during 1989-1999. Clin Infect Dis. 2002, 35: 627-630. 10.1086/342300.
Hitchcock CA, Pye GW, Troke PF, Johnson EM, Warnock DW: Fluconazole resistance in Candida glabrata. Antimicrob Agents Chemother. 1993, 37: 1962-1965.
Whelan WL, Simon S, Beneke ES, Rogers AL: Auxotrophic variants of Torulopsis glabrata. FEMS Microbiol Lett. 1984, 24: 1-4. 10.1016/0378-1097(84)90328-8.
Magee PT, Chibana H: The genomes of Candida albicans and other Candida species. In Candida and Candidiasis. Edited by: Calderone RA. 2002, Washington, DC: American Society for Microbiology, 293-304.
Lachke SA, Srikantha T, Tsai LK, Daniels K, Soll DR: Phenotypic switching in Candida glabrata involves phase-specific regulation of the metallothionein gene MT-II and the newly discovered hemolysin gene HLP. Infect Immun. 2000, 68: 884-895. 10.1128/IAI.68.2.884-895.2000.
Lachke SA, Joly S, Daniels K, Soll DR: Phenotypic switching and filamentation in Candida glabrata. Microbiology. 2002, 148: 2661-2674.
Barns SM, Lane DJ, Sogin ML, Bibeau C, Weisburg WG: Evolutionary relationships among pathogenic Candida species and relatives. J Bacteriol. 1991, 173: 2250-2255.
Cai J, Roberts IN, Collins MD: Phylogenetic relationships among members of the ascomycetous yeast genera Brettanomyces, Debaryomyces, Dekkera, and Kluyveromyces deduced by small-subunit rRNA gene sequences. Int J Syst Bacteriol. 1996, 46: 542-549.
Kurtzman CP, Robnett CJ: Identification and phylogeny of ascomycetous yeasts from analysis of nuclear large subunit (26S) ribosomal DNA partial sequences. Antonie Van Leeuwenhoek. 1998, 73: 331-371. 10.1023/A:1001761008817.
Hendriks L, Goris A, Van de Peer Y, Neefs J-M, Vancanneyt M, Kersters K, Berny J-F, Hennebert GL, De Wachter R: Phylogenetic relationships among ascomycetes and ascomycete-like yeasts as deduced from small ribosomal subunit RNA sequences. Syst Appl Microbiol. 1992, 15: 98-104.
Calderone RA: Candida and Candidiasis. 2002, Washington, DC: American Society for Microbiology
Anderson HW: Yeast-like fungi of the human intestinal tract. J Infect Dis. 1917, 21: 341-385.
van der Walt JP, Tscheuschner IT: Saccharomyces delphensis nov. spec. - a new yeast from South African dried figs. Antonie Van Leeuwenhoek. 1956, 22: 162-166.
Morais PB, Hagler AN, Rosa CA, Mendonca-Hagler LC, Klaczko LB: Yeasts associated with Drosophila in tropical forests of Rio de Janeiro, Brazil. Can J Microbiol. 1992, 38: 1150-1155.
Belloch C, Barrio E, García MD, Querol A: Inter- and intraspecific chromosome pattern variation in the yeast genus Kluyveromyces. Yeast. 1998, 14: 1341-1354. 10.1002/(SICI)1097-0061(199811)14:15<1341::AID-YEA328>3.3.CO;2-L.
Doi M, Homma M, Chindamporn A, Tanaka K: Estimation of chromosome number and size by pulsed-field gel electrophoresis (PFGE) in medically important Candida species. J Gen Microbiol. 1992, 138: 2243-2251.
Singh A, Chen EY, Lugovoy JM, Chang CN, Hitzeman RA, Seeburg PH: Saccharomyces cerevisiae contains two discrete genes coding for the α-factor pheromone. Nucleic Acids Res. 1983, 11: 4049-4063.
Anna-Arriola SS, Herskowitz I: Isolation and DNA sequence of the STE13 gene encoding dipeptidyl aminopeptidase. Yeast. 1994, 10: 801-810.
Browne BL, McClendon V, Bedwell DM: Mutations within the first LSGGQ motif of Ste6p cause defects in a-factor transport and mating in Saccharomyces cerevisiae. J Bacteriol. 1996, 178: 1712-1719.
Bader O, Schaller M, Klein S, Kukula J, Haack K, Mühlschlegel F, Korting HC, Schäfer W, Hube B: The KEX2 gene of Candida glabrata is required for cell surface integrity. Mol Microbiol. 2001, 41: 1431-1444. 10.1046/j.1365-2958.2001.02614.x.
Sheu Y-J, Snyder M: Control of cell polarity and shape. In The Mycota VIII Biology of the Fungal Cell. Edited by: Howard RJ, Gow NAR. 2001, Berlin: Springer-Verlag, 19-53.
Irie K, Nomoto S, Miyajima I, Matsumoto K: SGV1 encodes a CDC28/cdc2-related kinase required for a Gα subunit-mediated adaptive response to pheromone in S. cerevisiae. Cell. 1991, 65: 785-795.
Kao LR, Peterson J, Ji R, Bender L, Bender A: Interactions between the ankyrin repeat-containing protein Akr1p and the pheromone response pathway in Saccharomyces cerevisiae. Mol Cell Biol. 1996, 16: 168-178.
Elion EA, Satterberg B, Kranz JE: FUS3 phosphorylates multiple components of the mating signal transduction cascade: evidence for STE12 and FAR1. Mol Biol Cell. 1993, 4: 495-510.
Brizzio V, Khalfan W, Huddler D, Beh CT, Andersen SS, Latterich M, Rose MD: Genetic interactions between KAR7/SEC71, KAR8/JEM1, KAR5, and KAR2 during nuclear fusion in Saccharomyces cerevisiae. Mol Biol Cell. 1999, 10: 609-626.
Åström SU, Kegel A, Sjöstrand JO, Rine J: Kluyveromyces lactis Sir2p regulates cation sensitivity and maintains a specialized chromatin structure at the cryptic α-locus. Genetics. 2000, 156: 81-91.
Mintzer KA, Field J: The SH3 domain of the S. cerevisiae Cdc25p binds adenylyl cyclase and facilitates Ras regulation of cAMP signalling. Cell Signal. 1999, 11: 127-135. 10.1016/S0898-6568(98)00044-8.
Eschrich D, Buchhaupt M, Kötter P, Entian KD: Nep1p (Emg1p), a novel protein conserved in eukaryotes and archaea, is involved in ribosome biogenesis. Curr Genet. 2002, 40: 326-338. 10.1007/s00294-001-0269-4.
Kassir Y, Granot D, Simchen G: IME1, a positive regulator gene of meiosis in S. cerevisiae. Cell. 1988, 52: 853-862.
Bowdish KS, Mitchell AP: Bipartite structure of an early meiotic upstream activation sequence from Saccharomyces cerevisiae. Mol Cell Biol. 1993, 13: 2172-2181.
Rubin-Bejerano I, Mandel S, Robzyk K, Kassir Y: Induction of meiosis in Saccharomyces cerevisiae depends on conversion of the transcriptional represssor Ume6 to a positive regulator by its regulated association with the transcriptional activator Ime1. Mol Cell Biol. 1996, 16: 2518-2526.
Mitchell AP, Driscoll SE, Smith HE: Positive control of sporulation-specific genes by the IME1 and IME2 products in Saccharomyces cerevisiae. Mol Cell Biol. 1990, 10: 2104-2110.
Sia RAL, Mitchell AP: Stimulation of later functions of the yeast meiotic protein kinase Ime2p by the IDS2 gene product. Mol Cell Biol. 1995, 15: 5279-5287.
Soushko M, Mitchell AP: An RNA-binding protein homologue that promotes sporulation-specific gene expression in Saccharomyces cerevisiae. Yeast. 2000, 16: 631-639. 10.1002/(SICI)1097-0061(200005)16:7<631::AID-YEA559>3.3.CO;2-L.
Deng C, Saunders WS: RIM4 encodes a meiotic activator required for early events of meiosis in Saccharomyces cerevisiae. Mol Genet Genomics. 2001, 266: 497-504. 10.1007/s004380100571.
Engebrecht J, Masse S, Davis L, Rose K, Kessel T: Yeast meiotic mutants proficient for the induction of ectopic recombination. Genetics. 1998, 148: 581-598.
Tsubouchi H, Roeder GS: The Mnd1 protein forms a complex with Hop2 to promote homologous chromosome pairing and meiotic double-strand break repair. Mol Cell Biol. 2002, 22: 3078-3088. 10.1128/MCB.22.9.3078-3088.2002.
Novak JE, Ross-Macdonald PB, Roeder GS: The budding yeast Msh4 protein functions in chromosome synapsis and the regulation of crossover distribution. Genetics. 2001, 158: 1013-1025.
Tevzadze GG, Swift H, Esposito RE: Spo1, a phospholipase B homolog, is required for spindle pole body duplication during meiosis in Saccharomyces cerevisiae. Chromosoma. 2000, 109: 72-85. 10.1007/s004120050414.
Rabitsch KP, Tóth A, Gálová M, Schleiffer A, Schaffner G, Aigner E, Rupp C, Penkner AM, Moreno-Borchart AC, Primig M, et al: A screen for genes required for meiosis and spore formation based on whole-genome expression. Curr Biol. 2001, 11: 1001-1009. 10.1016/S0960-9822(01)00274-3.
Krisak L, Strich R, Winters RS, Hall JP, Mallory MJ, Kreitzer D, Tuan RS, Winter E: SMK1, a developmentally regulated MAP kinase, is required for spore wall assembly in Saccharomyces cerevisiae. Genes Dev. 1994, 8: 2151-2161.
Friesen H, Lunz R, Doyle S, Segall J: Mutation of the SPS1-encoded protein kinase of Saccharomyces cerevisiae leads to defects in transcription and morphology during spore formation. Genes Dev. 1994, 8: 2162-2175.
Briza P, Eckerstorfer M, Breitenbach M: The sporulation-specific enzymes encoded by the DIT1 and DIT2 genes catalyze a two-step reaction leading to a soluble LL-dityrosine-containing precursor of the yeast spore wall. Proc Natl Acad Sci USA. 1994, 91: 4524-4528.
Briza P, Winkler G, Kalchhauser H, Breitenbach M: Dityrosine is a prominent component of the yeast ascospore wall. A proof of its structure. J Biol Chem. 1986, 261: 4288-4294.
Walsh DW, Wolfe KH, Butler G: Genomic differences between Candida glabrata and Saccharomyces cerevisiae around the MRPL28 and GCN3 loci. Yeast. 2002, 19: 991-994. 10.1002/yea.890.
Lan CY, Newport G, Murillo LA, Jones T, Scherer S, Davis RW, Agabian N: Metabolic specialization associated with phenotypic switching in Candida albicans. Proc Natl Acad Sci USA. 2002, 99: 14907-14912. 10.1073/pnas.232566499.
Newport G, Kuo A, Flattery A, Gill C, Blake JJ, Kurtz MB, Abruzzo GK, Agabian N: Inactivation of Kex2p diminishes the virulence of Candida albicans. J Biol Chem. 2003, 278: 1713-1770. 10.1074/jbc.M209713200.
Berbee ML, Taylor JW: Ascomycete relationships: dating the origin of asexual lineages with 18S ribosomal RNA gene sequence data. In The Fungal Holomorph: Mitotic, Meiotic and Pleomorphic Speciation in Fungal Systematics. Edited by: Reynolds DR, Taylor JW. 1993, Wallingford: CAB International, 67-78.
Geiser DM, Timberlake WE, Arnold ML: Loss of meiosis in Aspergillus. Mol Biol Evol. 1996, 13: 809-817.
Ewing B, Hillier L, Wendl MC, Green P: Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8: 175-185.
Ewing B, Green P: Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8: 186-194.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-3402. 10.1093/nar/25.17.3389.
Wood V, Rutherford KM, Ivens A, Rajandream M-A, Barrell B: A re-annotation of the Saccharomyces cerevisiae genome. Comp Funct Genomics. 2001, 2: 143-154. 10.1002/cfg.86.
SCreannotation. [ftp://ftp.sanger.ac.uk/pub/yeast/SCreannotation]
Pearson WR, Wood T, Zhang Z, Miller W: Comparison of DNA sequences with protein sequences. Genomics. 1997, 46: 24-36. 10.1006/geno.1997.4995.
Wootton JC, Federhen S: Analysis of compositionally biased regions in sequence databases. Methods Enzymol. 1996, 266: 554-571.
Hodges PE, Payne WE, Garrels JI: The Yeast Protein Database (YPD): a curated proteome database for Saccharomyces cerevisiae. Nucleic Acids Res. 1998, 26: 68-72. 10.1093/nar/26.1.68.
Wong S, Butler G, Wolfe KH: Gene order evolution and paleopolyploidy in hemiascomycete yeasts. Proc Natl Acad Sci USA. 2002, 99: 9272-9277. 10.1073/pnas.142101099.
Notredame C, Higgins DG, Heringa J: T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000, 302: 205-217. 10.1006/jmbi.2000.4042.
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22: 4673-4680.
Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown PO, Herskowitz I: The transcriptional program of sporulation in budding yeast. Science. 1998, 282: 699-705. 10.1126/science.282.5389.699.
Acknowledgements
We are very grateful to Jure Piskur for help and discussion and to Taisuke Hisatomi for preprints. This study was supported by Science Foundation Ireland.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
13059_2002_520_MOESM1_ESM.doc
Additional data file 1: Tables of the mating response and meiosis genes identified in C. glabrata and K. delphensis. (DOC 40 KB)
Rights and permissions
About this article
Cite this article
Wong, S., Fares, M.A., Zimmermann, W. et al. Evidence from comparative genomics for a complete sexual cycle in the 'asexual' pathogenic yeast Candida glabrata. Genome Biol 4, R10 (2003). https://doi.org/10.1186/gb-2003-4-2-r10
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/gb-2003-4-2-r10