
Abstract
Alternative splicing of pre-mRNAs is central to the generation of diversity from the relatively small number of genes in metazoan genomes. Auxiliary cis elements and trans-acting factors are required for the recognition of constitutive and alternatively spliced exons and their inclusion in pre-mRNA. Here, we discuss the regulatory elements that direct alternative splicing and how genome-wide analyses can aid in their identification.
Similar content being viewed by others


Alternative splicing, the process by which multiple mRNA isoforms are generated from a single pre-mRNA species, is an important means of regulating gene expression. Alternative splicing determines cell fate in numerous contexts, such as sexual differentiation in Drosophila and apoptosis in mammals [1], and aberrant regulation of alternative splicing has been implicated in human disease [1,2]. Additional attention is now being given to alternative splicing in the wake of the sequencing of the human genome. On the basis of the initial drafts of the human genome sequence it was estimated that there are 30,000 to 40,000 genes [3,4] - significantly fewer than expected. Although final gene counts may be higher, there is a disparity between the relatively small number of human genes and the complexity of the human proteome. This suggests that alternative splicing is important in the generation of protein diversity. This article describes what is known about the regulatory elements that direct alternative splicing, how genome-wide analyses are being applied to their identification, and suggests directions for future genomic and large-scale studies.
Prediction of alternative splice forms
Estimates of the extent of alternative splicing in humans made on the basis of alignments of expressed sequence tags (ESTs; sequenced portions of cDNAs) range from 30 to 60% of genes, and even 60% might be an underestimate [3] because there is a lack of ESTs for many tissues and many developmental stages, and ESTs are biased toward the 3' end of an mRNA. The majority of alternative splicing events affect coding regions [5]. Within the coding region, protein domains can be added or removed, the reading frame can be shifted to give rise to an altered protein sequence, or the protein can be truncated by the introduction of a termination codon. Although less common, alternative splicing of 5' and 3' untranslated regions may insert or remove key cis-regulatory elements that affect mRNA localization, stability, and translation. The seven basic types of alternative splicing are illustrated in Figure 1.

Functionally significant examples of different types of alternative splicing. (a) Alternative inclusion of a cassette exon is very common. Neuron-specific inclusion of the N1 exon in the c-src proto-oncogene generates an insertion in the SH3 protein-protein interaction domain that alters its binding to other proteins [34]. (b) Alternative exons may be mutually exclusive, such as exons IIIb and IIIc in the fibroblast growth factor receptor 2 (FGFR-2) gene. Use of IIIb produces a receptor with high affinity for keratinocyte growth factor (KGF), whereas use of IIIc produces a high-affinity FGF receptor. Loss of the IIIb isoform is thought to be important in prostate cancer [35]. (c) The choice of an alternative 5' splice site in the Wilms' Tumor suppressor gene Wt1 results in the insertion of the three amino acids lysine, threonine, and serine (KTS). The +KTS and -KTS forms play distinct roles in kidney and gonad formation, and shift of the balance toward the -KTS form is associated with Frasier syndrome [36]. (d) In the transformer (tra) gene in Drosophila, selection of a female-specific alternative 3' splice site produces a single long open reading frame that gives rise to a regulatory protein that controls female somatic sexual differentiation. In male flies, tra mRNAs lack a long open reading frame, and no protein is made [37]. (e) Alternative terminal exons in the gene encoding calcitonin and calcitonin-gene-related peptide (CGRP) give rise to a hormone involved in calcium homeostasis in the thyroid gland, or a neuropeptide involved in vasodilation in the nervous system [38]. (f) Alternative promoter usage in the myosin light chain (MLC) gene leads to different first exons, which pair with mutually exclusive downstream exons to give rise to distinct protein isoforms, namely MLC1 and MLC3 [39]. This type of alternative splicing pattern results primarily from transcriptional regulation, not from the regulation of splice-site choice per se. (g) Intron retention is one of the rarest forms of alternative splicing in humans. Retention of intron 2 in the human muscle-specific chloride channel 1 (ClC-1) mRNA in myotonic dystrophy (DM) patients introduces a premature stop codon and leads to downregulation of ClC-1 expression, contributing to problems in muscle relaxation (myotonia) [2].
The significance of alternative splicing extends beyond the ability to generate different protein isoforms to the ability to modulate the levels of those isoforms. The proportions of different splice forms produced by alternative splicing may vary in different cell contexts, such as by cell type, developmental stage, or disease state. Numerous examples of cell-type-specific alternative splicing have been found (Figure 1), but the number of alternatively spliced genes identified so far is only a fraction of the number that has been predicted. In the last few years, bioinformatic studies comparing ESTs have greatly increased the number of known alternatively spliced genes (reviewed in [5]), and expanding these comparisons to include human genome sequence data holds the promise of finding many more. Genomic, mRNA, and EST sequences are also being used to better characterize alternative exons and their flanking introns [6]. Furthermore, microarray technologies are now evolving to look at splicing variation as well as overall gene expression in different cell contexts [7,8,9]. In future studies, arrays can be designed to screen for alternative splice forms in different tissues, at different developmental stages, in normal versus disease states, or in different mouse models, such as knockout mice lacking auxiliary splicing regulators. The caveats of microarray analyses are that they often cannot determine whether the splicing of multiple variable regions within an individual transcript is coordinated, and they rely on having sequence data (for example, for exon-exon junctions) prior to probe design. Nonetheless, taken together with bioinformatic approaches, microarrays will help to develop splicing profiles that provide a global picture of how alternative splicing is regulated.
The cis elements and trans-acting factors that regulate alternative splicing
In addition to identifying alternatively spliced mRNAs, genome-wide analyses will help answer many exciting questions about how alternative splicing is regulated. There is much to be learned about the regulatory factors that mediate cell-context-specific alternative splicing and about the cis elements through which they act. Genomic sequence, EST, and alternative splicing databases (Table 1) can be used to identify alternatively spliced RNAs and the regulatory elements that direct splicing decisions.
Auxiliary ciselements
Both constitutive and alternative splicing require the assembly of the basal splicing machinery in spliceosome complexes on consensus sequences present at all boundaries between introns and exons (the 5' and 3' splice sites). The spliceosome has two functions: to recognize and select splice sites, and to catalyze the two sequential transesterification reactions that remove the introns and join the exons together. The efficiency with which the spliceosome acts on an exon is determined by a balance of several features, including the strength of a splice site (that is, its conformity to consensus splice-site sequences), exon size, and the presence of auxiliary cis elements. Exons of ideal size (typically 50 to 300 nucleotides) with strong splice-site sequences are recognized efficiently by the splicing machinery and are constitutively included in the transcript, whereas suboptimal exons require auxiliary elements for recognition. But it is becoming increasingly clear that many constitutive exons also use auxiliary elements to ensure their recognition in all cells expressing the pre-mRNA. For example, a bioinformatic approach was used to identify intronic G-rich elements that facilitate the recognition of small constitutive exons [10]. Alternatively spliced exons are often small and have weak 5' and/or 3' splice sites, but for these exons auxiliary elements serve not only to improve splice site recognition, but also to modulate selection of splice sites used in specific cell contexts.
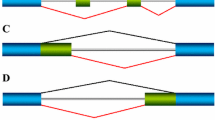
In general, the auxiliary elements that regulate the usage of alternative splice sites share several common features: they are small, variable in sequence, individually weak, and present in multiple copies. They are usually single-stranded, although secondary structure has been implicated in the function of a few elements (see, for example, [11]). Auxiliary elements are often conserved between species and perhaps between similarly regulated genes, but they contain degenerate sequence motifs, making it difficult to identify them. They can be exonic or intronic, and when they are intronic they can lie upstream, downstream, or flanking both sides of the regulated exon. Intronic elements can also be proximal (within 100 nucleotides) or distal (more than one kilobase away from the regulated exon), although they are often located close to the exon. And finally, auxiliary elements can enhance or repress splice-site selection. Depending on their location and their effect on the recognition of alternative splice sites, the elements are referred to as exonic splicing enhancers or silencers or intronic splicing enhancers or silencers (Figure 2). Table 2 lists the intronic splicing enhancers and silencers that have been identified to date; exonic splicing enhancers have recently been cataloged elsewhere [12].

Typical features of alternative exons. Alternative exons are on average less than half the size of constitutive exons and have weak 5' and/or 3' splice sites. Auxiliary elements aid or prevent the recognition of these exons by binding trans-acting factors in different cellular contexts, and how often an exon is included in the mRNA depends on a balance between positive and negative regulation. Enhancer (+) and silencer (-) elements can be found within the alternative exon (yellow box in the center) or the flanking introns (lines). Splicing decisions are controlled by multiple elements, and for a given exon these can be different elements, multiple copies of the same element located at different sites, or a combination of the two (as indicated by the non-yellow colored boxes). Different alternative exons are regulated by different sets of auxiliary elements, but alternative exons that are regulated by the same trans-acting factors have some common elements. Intronic elements can be distal, but are more often located in the introns adjacent to the alternative exon (near the exon-intron boundary), and in some cases can overlap with, or be contained within, the consensus splice site sequences that are recognized by the basal spliceosomal machinery.
Growing evidence suggests that many alternative splice sites are associated with both enhancers and silencers, and that regulation of alternative splicing is often the result of dynamic antagonism between trans-acting factors binding to these elements (reviewed in [13]). Indeed, results from the best-characterized vertebrate experimental systems (see below) argue that for most alternatively spliced transcripts there is no 'default' or unregulated state; instead, the ratio of alternative splice forms observed for a given pre-mRNA results from a balance between positive and negative regulation.
Trans-acting splicing factors
Numerous exonic splicing enhancers have been shown to bind serine/arginine-rich (SR) proteins, a family of essential splicing factors, to promote inclusion of alternative exons with weak splice sites in the pre-mRNA [14]. The effects of SR proteins on alternative splicing are antagonized by the constitutive splicing factor heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1). Overexpression of SR proteins or hnRNP A1 has a general effect on splicing of alternatively spliced pre-mRNAs in vitro or in transient transfection assays, suggesting that alternative splicing of some RNAs can be globally regulated in cell populations by changing the relative ratios of constitutive splicing factors. In at least some cases, hnRNP A1 cooperatively binds exonic splicing silencer sequences within the same exon and blocks binding of the SR proteins to exonic splicing enhancers [15].
Other auxiliary elements mediate their effects by binding to auxiliary splicing factors. For example, muscle-specific splicing elements in the intronic regions flanking the alternative exon 5 of the cardiac troponin T protein regulate inclusion of exon 5 in transcripts in embryonic striated muscle. Positive muscle-specific splicing elements downstream of exon 5 bind members of the CUG-binding protein (CUG-BP) and embryonically lethal abnormal vision-type RNA binding protein 3 (ETR-3)-like factor (CELF) family; binding of CELF splicing factors promotes exon inclusion [16]. Negative elements antagonize this muscle-specific activity by binding the ubiquitously expressed pyrimidine tract-binding protein (PTB) upstream and downstream of exon 5 [17]. The neuron-specific N1 exon of the c-src proto-oncogene is similarly regulated. PTB binds to intronic sequences flanking the N1 exon to inhibit its inclusion in the c-src mRNA in non-neuronal cells [18]. In neurons, a downstream intronic enhancer region binds several factors to derepress the N1 exon, including KH-type splicing-regulatory protein (KSRP), far upstream element binding protein (FBP), and the heterogeneous nuclear ribonucleoproteins hnRNP H and hnRNP F [18]. The neuron-specific splicing of several other targets is similarly repressed by PTB, and is antagonized in at least some of these cases by binding of the neuron-specific activator neurooncological ventral antigen 1 (Nova-1) to enhancer elements [18].
Most cis elements known to regulate alternative splicing were identified using deletion or site-directed mutational analysis of minigenes that were tested in transient transfection assays. This approach is limited by the difficulty in making minigene constructs that preserve the ability to regulate the exon in a cell-culture system (reviewed in [19]). Another caveat of this approach has been that multiple small elements often display functional redundancy, making it hard to identify them by a loss-of-function approach. In contrast, this repetitiveness should be helpful for identification of auxiliary cis elements in large-scale genomic analyses. Sequencing of the human genome has provided large data sets that are invaluable for finding new elements involved in cell-context-specific alternative splicing.
Using genomics to identify alternative splicing elements
There are two basic approaches to using genomic information to investigate alternative splicing elements: first, comparative or computational approaches to identify putative elements followed by validation of the ability of the elements to regulate splicing, and second, experimental approaches to identify sequence motifs followed by searching the genome for natural regulatory sites containing these elements. Both of these approaches have recently been attempted with some early successes.
Computational identification of exonic elements
Large genomic data sets have been used to identify elements by computational analyses. In one study, Fedorov and colleagues [20] found differences in the distribution of pentameric and hexameric nucleotide sequences in the exons of intron-containing and intron-lacking genes, some of which may represent exonic regulatory elements. The differences in nucleotide distributions that they reported were not created by a few strong signals, but rather by the accumulation of multiple weak signals, consistent with our current understanding of exon recognition. The putative exonic elements described by Fedorov et al. [20] did not generally match known exonic splicing enhancers and silencers, but experimentation to test whether these elements do indeed affect splicing was left to future studies.
Fairbrother and colleagues [12] used a computational approach to predict exonic splicing enhancers sequences by statistical analysis, followed by experimental verification of enhancer activity. They looked for hexameric sequences that are enriched in exons relative to introns and that are near weak splice sites relative to strong ones. They identified ten classes of putative exonic splicing enhancers, five of which matched known motifs for such elements and five of which were novel. Representatives of all ten motifs had enhancer activity when tested in minigene constructs in vivo. Fairbrother et al. [12] also showed that nine out of the ten elements tested had greater enhancer activity than corresponding mutant sequences that were statistically predicted to lack enhancer activity due to single base-pair substitutions. The predictive ability of their method was further demonstrated by comparing their predicted motifs to hexamers within the human hypoxanthine phosphoribosyl transferase (HPRT) gene. Numerous natural exonic mutations are known to cause exon skipping in the HPRT gene, mutation of which is associated with Lesch-Nyhan syndrome, a neurogenetic disorder caused by a defect in the purine biosynthesis pathway. Of 30 mutations, more than half disrupt predicted exonic splicing enhancer motifs. The results by Fairbrother et al. [12] suggest that it is possible to predict accurately the splicing phenotype of point mutations in human diseases by computational analysis and demonstrate the striking possibilities for predicting splicing elements from genomic sequence. Interestingly, each of the ten predicted exonic splicing enhancer motifs occurred as often or slightly less often in alternative exons than in constitutive exons, so it is likely that the motifs identified in this study play a role in exon recognition in both alternative and constitutive splicing.
Computational identification of intronic elements
On a small scale, a comparative genomic approach was used to identify intronic elements regulating splicing of a single alternative exon in the transcript for the splicing factor hnRNP A1. Human and mouse hnRNP A1 genomic sequences were aligned and conserved intronic stretches were tested to determine whether they play a role in splicing of the hnRNP A1 pre-mRNA [21]. Brudno and colleagues [22] used a computational approach to find candidate intronic regulatory elements involved in cell-type-specific alternative splicing. In this study, a kilobase of intronic sequence from each side of 25 brain-specific internal alternative exons was contrasted to a larger set of intronic sequences flanking constitutive exons. Brudno et al. [22] found a significant enrichment of the hexanucleotide UGCAUG and related pentamers in the region of the downstream intron proximal to the regulated exon. They also found this sequence at a high frequency downstream of a smaller set of known muscle-specific exons, suggesting that this element may play a broader role in cell-type-specific alternative splicing. Strikingly, UGCAUG was previously identified in intronic splicing enhancers defined by functional assays ([23] and references within), and, furthermore, the splicing factors KSRP and FBP bind to this sequence in the intronic downstream control element of c-src transcripts [18]. Brudno et al. [22] also report that computational analysis of PTB consensus binding sites revealed a statistical over-representation of these sites near brain-specific exons, supporting models in which dynamic antagonism between cell-type-specific activators and the more ubiquitous repressor PTB modulate splicing of alternative exons by binding to positive and negative auxiliary elements, respectively [17,18]. In future studies, this approach could be used to look at other subsets of cell-type-specific alternatively spliced genes, or expanded to encompass more distal intronic regions.
Functional identification of auxiliary elements
The most common experimental approach used to identify RNA motifs associated with regulatory factors is systematic evolution of ligands by exponential enrichment (SELEX), in which preferred functional sites or binding sites are selected and amplified from an initial pool of random RNA sequences. Several studies have identified potential exonic splicing enhancers by functional selection of exonic sequences that enhance splicing in cell-free splicing assays [24,25,26,27] or in cultured cells [28]. Of these, studies from the Krainer laboratory [26,27] are most striking. In this work, Liu and colleagues used a splicing substrate in which the known exonic splicing enhancer was replaced with a pool of random sequences. To select for sequences that mediate splicing via individual SR proteins, they used a splicing complementation assay in which recombinant SR proteins were added to splicing-deficient cytoplasmic extracts. Liu et al. [26,27] identified four sets of exonic splicing enhancer motifs that are specifically activated by SR proteins, namely ASF/SF2, SRp40, SRp55, and SC35. These motifs matched almost a dozen natural exonic splicing enhancers for which binding specificity of individual SR proteins has been determined. The score matrices generated for the four SR proteins were then used as tools to predict exonic splicing enhancers. Disruption of such a predicted enhancer explains aberrant splicing resulting from a single nonsense point mutation in the BRCA1 gene in breast and ovarian cancer patients [29]. The mutation perturbs an exonic splicing enhancer regulated by ASF/SF2, causing exon skipping, which in turn disrupts the carboxy-terminal end of the BRCA1 protein. This mutation had previously been thought to contribute to disease by introducing a premature termination codon, but this analysis [29] demonstrated that the substitution gives rise to a splicing mutation instead.
Similar analysis of a single base-pair substitution within exon 7 of the survival of motor neuron 2 (SMN2) gene showed that exon skipping and subsequent truncation of the SMN2 protein is due to disruption of an exonic splicing enhancer [30]. Truncation of the SMN2 protein prevents compensation for loss of a nearly identical SMN1 gene in patients with spinal muscular atrophy. These studies dramatically demonstrate the importance of exonic splicing enhancers for exon recognition, as well as the significant role disabled elements play in human disease. Thus far, the sequence motifs identified by SELEX for the four SR proteins studied by Liu et al. [26,27] have been compared to established exonic splicing enhancers and point mutations known to affect splicing. In the future, analyses can be extended to include additional SR proteins and the elements identified by SELEX can be compared to databases of genomic sequences, alternatively spliced exons, or other point mutations associated with human diseases.
One study used sequences found by SELEX to identify alternative splicing elements from genomic sequence. Buckanovich and Darnell [31] used SELEX to identify binding sites for the neuron-specific splicing factor Nova-1. The selected sequence of three intact UCAU repeats was shown experimentally to be both necessary and sufficient for high-affinity Nova-1 binding. Although the sequence was selected on the basis of optimal binding and not function, Buckanovich and Darnell [31] were able to identify natural targets of Nova-1-mediated cell-specific splicing, by searching GenBank, a neuron-specific alternatively spliced exon database, and the Nova-1 genomic sequence with the consensus RNA selection sequence. Only two targets (exon 3A of the glycine receptor α2 and exon H of Nova-1) were identified, but at the time of this study, few genomic sequences were available and alternative splicing databases contained few entries. It is interesting to note, however, that Brudno and colleagues [22] found no increase in the frequency of UCAY sequences (where Y is a pyrimidine) in brain-specific alternative introns, suggesting that Nova-1 activity mediated through this element may be important only for a minority of brain-specific exons. Today the approach used by Buckanovich and Darnell [31] could presumably be used to find many natural targets of other cell-context-specific regulators by searching the large genomic and alternative splicing data sets for elements identified by SELEX.
Beyond the genome
Many powerful new tools are emerging that use genomic information and large-scale analyses: investigators can now compare and contrast vast genomic, mRNA, and EST data sets, use computational analyses to predict regulatory targets and elements, and develop array-based expression profiles. A challenge for the new millennium will be to integrate these tools with traditional biochemical and molecular biology approaches to understand how complex processes such as alternative splicing are regulated in specific cellular contexts, such as different cell types or cells at different developmental stages, in response to changing environmental cues, and in human disease. A consolidation of experimental and computational approaches will be required to catalog alternatively spliced genes, to characterize auxiliary cis elements and the trans factors that mediate the use of alternative splice sites, to identify genes with common alternative splicing programs, and to develop profiles of how alternative splicing is regulated in different cellular contexts.
Pioneering studies of this sort have begun in other areas of RNA processing. Darnell and colleagues [32] recently performed SELEX to identify binding motifs for the fragile X mental retardation protein (FMRP), an RNA-binding protein associated with fragile X syndrome that is thought to be involved in regulating mRNA translation, and used the consensus of these motifs to screen 245,000 sets of mammalian genomic sequences to identify natural targets of FMRP. RNAs from six genes bound FMRP and were identified as putative targets regulated by FMRP. In a parallel study, Brown et al. [33] identified a subset of FMRP-associated RNAs by combining microarray analysis of FMRP complexes in the murine brain and polyribosome profiling of cells derived from patients with fragile X syndrome, who harbor a 5' untranslated trinucleotide repeat expansion that leads to transcriptional silencing of the gene encoding FMRP. A total of 14 of the RNAs in this subset were in turn searched for the SELEX-derived FMRP-binding sites, and 7 of 11 putative elements in these RNAs bound with high affinity to FMRP, demonstrating how useful it is to combine these approaches to identify in vivo targets [32]. Similar integrated studies would be valuable for elucidating the elements through which regulatory splicing factors act and the in vivo targets containing these elements. Extending these analyses to model genetic organisms, such as Mus musculus or Drosophila melanogaster, whose genomes have been sequenced, will be especially valuable for defining regulatory networks that coordinate cell-type-specific alternative splicing, allowing us to see the 'big picture' of the transcriptome during development and in models of disease.
References
Lopez A: Alternative splicing of pre-mRNA: Developmental consequences and mechanisms of regulation. Ann Rev Genet. 1998, 32: 279-305. 10.1146/annurev.genet.32.1.279.
Charlet-B N, Savkur R, Singh G, Philips A, Grice E, Cooper T: Loss of the muscle-specific chloride channel in type I myotonic dystrophy lead to misregulated alternative splicing. Mol Cell. 2002, 10: 45-53.
International Human Genome Sequencing Consortium: Initial sequencing and analysis of the human genome. Nature. 2001, 409: 860-921. 10.1038/35057062.
Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al: The sequence of the human genome. Science. 2001, 291: 1304-1351. 10.1126/science.1058040.
Modrek B, Lee C: A genomic view of alternative splicing. Nat Genet. 2002, 30: 13-19. 10.1038/ng0102-13.
Clark F, Thanaraj T: Categorization and characterization of transcript-confirmed constitutively and alternatively splicing introns and exons from human. Hum Mol Gen. 2002, 11: 451-464. 10.1093/hmg/11.4.451.
Shoemaker DD, Schadt EE, Armour CD, He YD, Garrett-Engele P, McDonagh PD, Loerch PM, Leonardson A, Lum PY, Cavet G, et al: Experimental annotation of the human genome using microarray technology. Nature. 2001, 409: 922-927. 10.1038/35057141.
Hu G, Madore S, Moldover B, Jatkoe T, Balaban D, Thomas J, Wang Y: Predicting splice variant from DNA chip expression data. Genome Res. 2001, 11: 1237-1245. 10.1101/gr.165501.
Yeakley J, Fan J-B, Doucet D, Luo L, Wickham E, Ye Z, Chee M, Fu X-D: Profiling alternative splicing on fiber-optic arrays. Nat Biotech. 2002, 20: 353-358. 10.1038/nbt0402-353.
McCullough A, Berget S: G triplets located throughout a class of small vertebrate introns enforce intron borders and regulate splice site selection. Mol Cell Biol. 1997, 17: 4562-4571.
Blanchette M, Chabot B: A highly stable duplex structure sequesters the 5' splice site region of hnRNP A1 alternative exon 7B. RNA. 1997, 3: 405-419.
Fairbrother W, Yeh R-F, Sharp P, Burge C: Predictive identification of exonic splicing enhancers in human genes. Science. 2002, 297: 1007-1013. 10.1126/science.1073774.
Smith C, Valcárcel J: Alternative pre-mRNA splicing: the logic of combinatorial control. Trends Biochem Sci. 2000, 25: 381-388. 10.1016/S0968-0004(00)01604-2.
Cartegni L, Chew S, Krainer A: Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002, 3: 285-298. 10.1038/nrg775.
Zhu J, Mayeda A, Krainer A: Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Mol Cell. 2001, 8: 1351-1361.
Ladd A, Charlet-B N, Cooper T: The CELF family of RNA binding proteins is implicated in cell-specific and developmentally regulated alternative splicing. Mol Cell Biol. 2001, 21: 1285-1296. 10.1128/MCB.21.4.1285-1296.2001.
Charlet-B N, Logan P, Singh G, Cooper T: Dynamic antagonism between ETR-3 and PTB regulates cell type-specific alternative splicing. Mol Cell. 2002, 9: 649-658.
Grabowski PJ, Black DL: Alternative RNA splicing in the nervous system. Prog in Neurobiol. 2001, 65: 289-308. 10.1016/S0301-0082(01)00007-7.
Cooper TA: Defining pre-mRNA cis elements that regulate cell-specific splicing. In Methods in Molecular Biology. Edited by: Haynes S, Totowa NJ. 1999, Humana Press, Inc., 118: 391-403. 10.1385/1-59259-676-2:391.
Fedorov A, Saxonov S, Fedorova L, Daizadeh I: Comparison of intron-containing and intron-lacking human genes elucidates putative exonic splicing enhancers. Nucleic Acids Res. 2001, 29: 1464-1469. 10.1093/nar/29.7.1464.
Chabot B, Blanchette M, Lapierre I, La Branche H: An intron element modulating 5' splice site selection in the hnRNP A1 pre-mRNA interacts with hnRNP A1. Mol Cell Biol. 1997, 17: 1776-1786.
Brudno M, Gelfand M, Spengler S, Zorn M, Dubchak I, Conboy J: Computational analysis of candidate intron regulatory elements for tissue-specific alternative pre-mRNA splicing. Nucleic Acids Res. 2001, 29: 2338-2348. 10.1093/nar/29.11.2338.
Modafferi E, Black D: A complex intronic splicing enhancer from the c-src pre-mRNA activates inclusion of a heterologous exon. Mol Cell Biol. 1997, 17: 6537-6545.
Tian H, Kole R: Selection of novel exon recognition elements from a pool of random sequences. Mol Cell Biol. 1995, 15: 6291-6298.
Schaal T, Maniatis T: Selection and characterization of pre-mRNA splicing enhancers: identification of novel SR protein-specific enhancer sequences. Mol Cell Biol. 1999, 19: 1705-1719.
Liu H-X, Zhang M, Krainer A: Identification of functional exonic splicing enhancer motifs recognized by individual SR proteins. Genes Dev. 1998, 12: 1998-2012.
Liu H-X, Chew S, Cartegni L, Zhang M, Krainer A: Exonic splicing enhancer motif recognized by human SC35 under splicing conditions. Mol Cell Biol. 2000, 20: 1063-1071. 10.1128/MCB.20.3.1063-1071.2000.
Coulter L, Landree M, Cooper T: Identification of a new class of exonic splicing enhancers by in vivo selection. Mol Cell Biol. 1997, 17: 2143-2150.
Liu H-X, Cartegni L, Zhang M, Krainer A: A mechanism for exon skipping caused by nonsense or missense mutations in BRCA1 and other genes. Nat Genet. 2001, 27: 55-58.
Cartegni L, Krainer A: Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002, 30: 377-384. 10.1038/ng854.
Buckanovich R, Darnell R: The neuronal RNA binding protein Nova-1 recognizes specific RNA targets in vitro and in vivo. Mol Cell Biol. 1997, 17: 3194-3201.
Darnell J, Jensen K, Jin P, Brown V, Warren S, Darnell R: Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell. 2001, 107: 489-499.
Brown V, Jin P, Ceman S, Darnell J, O'Donnell W, Tenenbaum S, Jin X, Feng Y, Wilkinson K, Keene J, et al: Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in Fragile X syndrome. Cell. 2001, 107: 477-487.
Brown M, Cooper J: Regulation, substrates and functions of src. Biochim Biophys Acta. 1996, 1287: 121-149. 10.1016/0304-419X(96)00003-0.
Carstens R, Eaton J, Krigman H, Walther P, Carcia-Blanco M: Alternative splicing of fibroblast growth factor receptor 2 (FGF-R2) in human prostate cancer. Oncogene. 1997, 15: 3059-3065. 10.1038/sj.onc.1201498.
Hammes A, Guo J-K, Lutsch G, Leheste J-R, Landrock D, Ziegler U, Gubler M-C, Schedl A: Two splice variants of the Wilms' tumor 1 gene have distinct functions during sex determination and nephron formation. Cell. 2001, 106: 319-329.
Boggs R, Gregor P, Idriss S, Belote J, McKeown M: Regulation of sexual differentiation in D. melanogaster via alternative splicing of RNA from the transformer gene. Cell. 1987, 50: 739-747.
Amara S, Evans R, Rosenfeld M: Calcitonin/calcitonin gene-related peptide transcription unit: tissue-specific expression involves selective use of alternative polyadenylation sites. Mol Cell Biol. 1984, 4: 2151-2160.
Gallego M, Nadal-Ginard B: Myosin light-chain 1/3 gene alternative splicing: cis regulation is based upon a hierarchial compatibility between splice sites. Mol Cell Biol. 1990, 10: 2133-2144.
Stamm S, Zhu J, Nakai K, Stoilov P, Stoss O, Zhang M: An alternative-exon database and its statistical analysis. DNA Cell Biol. 2000, 19: 739-756. 10.1089/104454900750058107.
Drayluk I, Brudno M, Gelfand M, Zorn M, Dubchak I: ASDB: database of alternatively spliced genes. Nucleic Acids Res. 2000, 28: 296-297. 10.1093/nar/28.1.296.
Ji H, Zhou Q, Wen F, Xia H, Lu X, Li Y: AsMamDB: an alternative splice database of mammals. Nucleic Acids Res. 2001, 29: 260-263. 10.1093/nar/29.1.260.
Modrek B, Resch A, Grasso C, Lee C: Genome-wide detection of alternative splicing in expressed sequences of human genes. Nucleic Acids Res. 2001, 29: 2850-2859. 10.1093/nar/29.13.2850.
Honkura T, Ogasawara J, Yamada T, Morishita S: The Gene Resource Locator: gene locus maps for transcriptosome analysis. Nucleic Acids Res. 2002, 30: 221-225. 10.1093/nar/30.1.221.
Kent W, Zahler A: The Intronerator: exploring introns and alternative splicing in Caenorhabditis elegans. Nucleic Acids Res. 2000, 28: 91-93. 10.1093/nar/28.1.91.
Croft L, Schandorff S, Clark F, Burrage K, Arctander P, Mattick J: ISIS, the intron information system, reveals the high frequency of alternative splicing in the human genome. Nat Genet. 2000, 24: 340-341. 10.1038/74153.
Huang Y-H, Chen Y-T, Lai J-J, Yang S-T, Yang U-C: PALS db: putative alternative splicing database. Nucleic Acids Res. 2002, 30: 186-190. 10.1093/nar/30.1.186.
Ponomarenko J, Orlova G, Frolov A, Gelfand M, Ponomarenko M: SELEX_DB: a database on in vitro selected oligomers adapted for recognizing natural sites and for analyzing both SNPs and site-directed mutagenesis data. Nucleic Acids Res. 2002, 30: 195-199. 10.1093/nar/30.1.195.
Burset M, Seledtsov I, Solovyev V: SpliceDB: database of canonical and non-canonical mammalian splice sites. Nucleic Acids Res. 2001, 29: 255-259. 10.1093/nar/29.1.255.
Krause A, Haas S, Coward E, Vingron M: SYSTERS, GeneNest, SliceNest: exploring sequence space from genome to protein. Nucleic Acids Res. 2002, 30: 299-300. 10.1093/nar/30.1.299.
Christoffels A, van Gelder A, Greyling G, Miller R, Hide T, Hide W: STACK: sequence tag alignment and consensus knowledge-base. Nucleic Acids Res. 2001, 29: 234-238. 10.1093/nar/29.1.234.
Spingola M, Grate L, Haussler D, Ares M: Genome-wide bioinformatic and molecular analysis of introns in Saccharomyces cerevisiae. RNA. 1999, 5: 221-234. 10.1017/S1355838299981682.
Deguillien M, Huang S-C, Moriniére M, Dreumont N, Benz E, Baklouti F: Multiple cis elements regulate an alternative splicing event at 4.1R pre-mRNA during erythroid differentiation. Blood. 2001, 98: 3809-3816. 10.1182/blood.V98.13.3809.
Southby J, Gooding C, Smith C: Polypyrimidine tract binding protein functions as a repressor to regulate alternative splicing of α-actinin mutually exclusive exons. Mol Cell Biol. 1999, 19: 2699-2711.
Suzuki H, Jin Y, Otani H, Yasuda K, Inoue K: Regulation of alternative splicing of α-actinin transcript by Bruno-like proteins. Genes Cells. 2002, 7: 133-141. 10.1046/j.1356-9597.2001.00506.x.
Wei N, Lin C, Modafferi E, Gomes W, Black D: A unique intronic splicing enhancer controls the inclusion of the agrin Y exon. RNA. 1997, 3: 1275-1288.
Genetta T, Morisaki H, Morisaki T, Holmes E: A novel bipartite intronic splicing enhancer promotes the inclusion of a mini-exon in the AMP deaminase 1 gene. J Biol Chem. 2001, 276: 25589-25597. 10.1074/jbc.M011637200.
Shibata A, Hattori M, Suda H, Sakaki Y: Identification of cis-acting elements involved in an alternative splicing of the amyloid precursor protein (APP) gene. Gene. 1996, 175: 203-208. 10.1016/0378-1119(96)00150-3.
Hedjran F, Yeakley J, Huh G, Hynes R, Rosenfeld M: Control of alternative pre-mRNA splicing by distributed pentameric repeats. Proc Natl Acad Sci USA. 1997, 94: 12343-12347. 10.1073/pnas.94.23.12343.
Lou H, Yang Y, Cote G, Berget S, Gagel R: An intron enhancer containing a 5 splice site sequence in the human calci-tonin/calcitonin gene-related peptide gene. Mol Cell Biol. 1995, 15: 7135-7142.
Lou H, Gagel R, Berget S: An intron enhancer recognized by splicing factors activates polyadenylation. Genes Dev. 1996, 10: 208-219.
Lou H, Neugebauer K, Gagel R, Berget S: Regulation of alternative polyadenylation by U1 snRNPs and SRp20. Mol Cell Biol. 1998, 18: 4977-4985.
Ryan K, Cooper T: Muscle-specific splicing enhancers regulate inclusion of the cardiac troponin T alternative exon in embryonic skeletal muscle. Mol Cell Biol. 1996, 16: 4014-4023.
Cooper T: Muscle-specific splicing of a heterologous exon mediated by a single muscle-specific splicing enhancer from the cardiac troponin T gene. Mol Cell Biol. 1998, 18: 4519-4525.
Ryan K, Charlet-B N, Cooper T: Binding of PurH to a muscle-specific splicing enhancer functionally correlates with exon inclusion in vivo. J Biol Chem. 2000, 275: 20618-20626. 10.1074/jbc.M909977199.
Côté J, Dupuis S, Jiang Z-H, Wu J: Caspase-2 pre-mRNA alternative splicing: identification of an intronic element containing a decoy 3 acceptor site. Proc Natl Acad Sci USA. 2001, 98: 938-943. 10.1073/pnas.031564098.
Côté J, Dupuis S, Wu J: Polypyrimidine track-binding protein binding downstream of caspase-2 alternative exon 9 represses its inclusion. J Biol Chem. 2001, 276: 8535-8543. 10.1074/jbc.M008924200.
Zhang L, Liu W, Grabowski P: Coordinate repression of a trio of neuron-specific splicing events by the splicing regulator PTB. RNA. 1999, 5: 117-130. 10.1017/S1355838299981530.
Chan R, Black D: The polypyrimidine tract binding protein binds upstream of neural cell-specific c-src exon N1 to repress the splicing of the intron downstream. Mol Cell Biol. 1997, 17: 4667-4676.
Markovtsov V, Nikolic J, Goldman J, Turck C, Chou M-Y, Black D: Cooperative assembly of an hnRNP complex induced by a tissue-specific homolog of polypyrimidine tract binding protein. Mol Cell Biol. 2000, 20: 7463-7479. 10.1128/MCB.20.20.7463-7479.2000.
Black D: Activation of c-src neuron-specific splicing by an unusual RNA element in vivo and in vitro. Cell. 1992, 69: 795-807.
Pagani F, Buratti E, Stuani C, Romano M, Zuccato E, Niksic M, Giglio L, Faraguna D, Baralle F: Splicing factors induce cystic fibrosis transmembrane regulator exon 9 skipping through a nonevolutionary conserved intronic element. J Biol Chem. 2000, 275: 21041-21047. 10.1074/jbc.M910165199.
Shen J, Hirsh J: cis-regulatory sequences responsible for alternative splicing of the Drosophila dopa decarboxylase gene. Mol Cell Biol. 1994, 14: 7385-7393.
Tiscornia G, Mahadevan M: Myotonic dystrophy: the role of the CUG triplet repeats in splicing of a novel DMPK exon and altered cytoplasmic DMPK mRNA isoform ratios. Mol Cell. 2000, 5: 959-967.
Jin W, Huang ES-C, Bi W, Cote G: Redundant intronic repressors function to inhibit fibroblast growth factor receptor-1 α-exon recognition in glioblastoma cells. J Biol Chem. 1999, 274: 28035-28041. 10.1074/jbc.274.39.28035.
Jin W, McCutcheon I, Fuller G, Huang ES-C, Cote G: Fibroblast growth factor receptor-1 α-exon exclusion and polypyrimidine tract-binding protein in glioblastoma mutiforme tumors. Cancer Res. 2000, 60: 1221-1224.
Jin W, Bi W, Huang ES-C, Cote G: Glioblastoma cell-specific expression of fibroblast growth factor receptor-1β requires an intronic repressor of RNA splicing. Cancer Res. 1999, 59: 316-319.
del Gatto F, Breathnach R: Exon and intron sequences, respectively, repress and activate splicing of a fibroblast growth factor receptor 2 alternative exon. Mol Cell Biol. 1995, 15: 4825-4834.
del Gatto-Konczak F, Bourgeois C, Le Gunier C, Kister L, Gesnel M-C, Stévenin J, Breathnach R: The RNA-binding protein TIA-1 is a novel mammalian splicing regulator acting through intron sequences adjacent to a 5' splice site. Mol Cell Biol. 2000, 20: 6287-6299. 10.1128/MCB.20.17.6287-6299.2000.
del Gatto F, Plet A, Gesnel M-C, Fort C, Breathnach R: Multiple interdependent sequence elements control splicing of a fibroblast growth factor receptor 2 alternative exon. Mol Cell Biol. 1997, 17: 5106-5116.
Carstens R, McKeehan W, Garcia-Blanco M: An intronic sequence element mediates both activation and repression of rat fibroblast growth factor receptor 2 pre-mRNA splicing. Mol Cell Biol. 1998, 18: 2205-2217.
Carstens R, Wagner E, Garcia-Blanco M: An intronic splicing silencer causes skipping of the IIIb exon of fibroblast growth factor receptor 2 through involvement of polypyrimidine tract binding protein. Mol Cell Biol. 2000, 20: 7388-7400. 10.1128/MCB.20.19.7388-7400.2000.
Huh G, Hynes R: Elements regulating an alternatively spliced exon of the rat fibronectin gene. Mol Cell Biol. 1993, 13: 5301-5314.
Huh G, Hynes R: Regulation of alternative pre-mRNA splicing by a novel repeated hexanucleotide element. Genes Dev. 1994, 8: 1561-1574.
Ashiya M, Grabowski P: A neuron-specific splicing switch mediated by an array of pre-mRNA repressor sites: Evidence of a regulatory role for the polypyrimidine tract binding protein and a brain-specific PTB counterpart. RNA. 1997, 3: 996-1015.
Zhang L, Ashiya M, Sherman T, Grabowski P: Essential nucleotides direct neuron-specific splicing of γ2 pre-mRNA. RNA. 1996, 2: 682-698.
Fogel B, McNally M: A cellular protein, hnRNP H, binds to the negative regulator of splicing element from Rous sarcoma virus. J Biol Chem. 2000, 275: 32371-32378. 10.1074/jbc.M005000200.
McNally M, Gontarek R, Beemon K: Characterization of Rous sarcoma virus intronic sequences that negatively regulate splicing. Virology. 1991, 185: 99-108.
McNally L, McNally M: SR protein splicing factors interact with the Rous sarcoma virus negative regulator of splicing element. J Virol. 1996, 70: 1163-1172.
Jensen K, Dredge B, Stefani G, Zhong R, Buckanovich R, Okano H, Yang Y, Darnell R: Nova-1 regulates neuron-specific alternative splicing and is essential for neuronal viability. Neuron. 2000, 25: 359-371.
Blanchette M, Chabot B: Modulation of exon skipping by high-affinity hnRNP A1-binding sites and by intron elements that repress splice site utilization. EMBO J. 1999, 18: 1939-1952. 10.1093/emboj/18.7.1939.
Simard M, Chabot B: Control of hnRNP A1 alternative splicing: an intronic element represses use of the common 3' splice site. Mol Cell Biol. 2000, 20: 7353-7362. 10.1128/MCB.20.19.7353-7362.2000.
Kanopka A, Mühlemann O, Akusjärvi G: Inhibition by SR proteins of splicing of a regulated adenovirus pre-mRNA. Nature. 1996, 381: 535-538. 10.1038/381535a0.
Mühlemann O, Yue B-G, Petersen-Mahrt X, Akusjärvi G: A novel type of splicing enhancer regulating adenovirus pre-mRNA splicing. Mol Cell Biol. 2000, 20: 2317-2325. 10.1128/MCB.20.7.2317-2325.2000.
Haut D, Pintel D: Intron definition is required for excision of the minute virus of mice small intron and definition of the upstream exon. J Virol. 1998, 72: 1834-1843.
Standiford D, Sun W, Davis M, Emerson C: Positive and negative intronic regulatory elements control muscle-specific alternative exon splicing of Drosophila myosin heavy chain transcripts. Genetics. 2001, 157: 259-271.
Kawahigashi H, Harada Y, Asano A, Nakamura M: A cis-acting regulatory element that affects the alternative splicing of a muscle-specific exon in the mouse NCAM gene. Biochem Biophys Acta. 1998, 1397: 305-315. 10.1016/S0167-4781(98)00021-9.
Kawamoto S: Neuron-specific alternative splicing of nonmuscle myosin II heavy chain-B pre-mRNA requires a cis-acting intron sequence. J Biol Chem. 1996, 271: 17613-17616.
Wang J, Bell L: The Sex-lethal amino terminus mediates cooperative interactions in RNA binding and is essential for splicing regulation. Genes Dev. 1994, 8: 2072-2085.
Xie J, Black D: A CaMK IV responsive RNA element mediates depolarization-induced alternative splicing of ion channels. Nature. 2001, 410: 936-939. 10.1038/35073593.
Tange T, Damgaard C, Guth S, Valcárcel J, Kjems J: The hnRNP A1 protein regulates HIV-1 tat splicing via a novel intron silencer element. EMBO J. 2001, 20: 5748-5758. 10.1093/emboj/20.20.5748.
D'Souza I, Schellenberg G: Determinants of 4-repeat tau expression. J Biol Chem. 2000, 275: 17700-17709. 10.1074/jbc.M909470199.
D'Souza I, Schellenberg G: tau exon 10 expression involves a bipartite intron 10 regulatory sequence and weak 5' and 3' splice sites. J Biol Chem. 2002, 277: 26587-26599. 10.1074/jbc.M203794200.
Hastings M, Ingle H, Lazar M, Munroe S: Post-transcriptional regulation of thyroid hormone receptor expression by cis-acting sequences and a naturally occurring antisense RNA. J Biol Chem. 2000, 275: 11507-11513. 10.1074/jbc.275.15.11507.
Hastings M, Wilson C, Munroe S: A purine-rich intronic element enhances alternative splicing of thyroid hormone receptor mRNA. RNA. 2001, 7: 859-874. 10.1017/S1355838201002084.
Sosnowski B, Belote J, McKeown M: Sex-specific alternative splicing of RNA from the transformer gene results from sequence-dependent splice site blockage. Cell. 1989, 58: 449-459.
Valcárcel J, Singh R, Zamore P, Green M: The protein Sex-lethal antagonizes the splicing factor U2AF to regulate alternative splicing of transformer pre-mRNA. Nature. 1993, 362: 171-175. 10.1038/362171a0.
Inoue K, Hoshijima K, Sakamoto H, Shimura Y: Binding of the Drosophila Sex-lethal gene product to the alternative splice site of transformer primary transcript. Nature. 1990, 344: 461-463. 10.1038/344461a0.
Gooding C, Roberts G, Moreau G, Nadal-Ginard B, Smith C: Smooth muscle-specific switching of alpha-tropomyosin mutually exclusive exon selection by specific inhibition of the strong default exon. EMBO J. 1994, 13: 3861-3872.
Pérez I, Lin C-H, McAfee J, Patton J: Mutation of PTB binding sites causes misregulation of alternative 3' splice site selection in vivo. RNA. 1997, 3: 764-778.
Gooding C, Roberts G, Smith C: Role of an inhibitory pyrimidine element and polypyrimidine tract binding protein in repression of a regulated α-tropomyosin exon. RNA. 1998, 4: 85-100.
Gallego M, Balvay L, Brody E: cis-acting sequences involved in exon selection in the chicken beta-tropomyosin gene. Mol Cell Biol. 1992, 12: 5415-5425.
Balvay L, Libri D, Gallego M, Fiszman M: Intronic sequence with both negative and positive effects on the regulation of alternative transcripts of the chicken beta tropomyosin transcripts. Nucleic Acids Res. 1992, 20: 3987-3992.
Gallego M, Gattoni R, Stévenin J, Marie J, Expert-Bezançon A: The SR splicing factors ASF/SF2 and SC35 have antagonistic effects on intronic enhancer-dependent splicing of the β-tropomyosin alternative exon 6A. EMBO J. 1997, 16: 1772-1784. 10.1093/emboj/16.7.1772.
Expert-Bezançon A, Le Caer J, Marie J: Heterogeneous nuclear ribonucleoprotein (hnRNP) K is a component of an intronic splicing enhancer complex that activates the splicing of the alternative exon 6A from chicken β-tropomyosin pre-mRNA. J Biol Chem. 2002, 277: 16614-16623. 10.1074/jbc.M201083200.
Sirand-Pugnet P, Durosay P, Brody E, Marie J: An intronic (A/U)GGG repeat enhances the splicing of an alternative intron of the chicken beta-tropomyosin pre-mRNA. Nucleic Acids Res. 1995, 23: 3501-3507.
Lerga A, Hallier M, Delva L, Orvain C, Gallais I, Marie J, Moreau-Gachelin F: Identification of an RNA binding specificity for the potential splicing factor TLS. J Biol Chem. 2001, 276: 6807-6816. 10.1074/jbc.M008304200.
Mulligan G, Guo W, Wormsley S, Helfman D: Polypyrimidine tract binding protein interacts with sequences involved in alternative splicing of β-tropomyosin. J Biol Chem. 1992, 267: 25480-25487.
Grossman J, Meyer M, Wang Y, Mulligan G, Kobayashi R, Helfman D: The use of antibodies to the polypyrimidine tract binding protein (PTB) to analyze the protein components that assemble on alternatively spliced pre-mRNAs that use distant branch points. RNA. 1998, 4: 613-625. 10.1017/S1355838298971448.
Acknowledgements
We thank Chris Burge for helpful comments on the manuscript and for sharing his work prior to publication. The Cooper lab is supported by grants from the NIH (R01AR45653 and R01HL45565). ANL is supported by a postdoctoral NRSA fellowship from the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ladd, A.N., Cooper, T.A. Finding signals that regulate alternative splicing in the post-genomic era. Genome Biol 3, reviews0008.1 (2002). https://doi.org/10.1186/gb-2002-3-11-reviews0008
Published:
DOI: https://doi.org/10.1186/gb-2002-3-11-reviews0008