
Abstract
In sepsis, inflammation and thrombosis are both the cause and the result of interactions between circulating (for example, leukocytes and platelets), endothelial and smooth muscle cells. Microparticles are proinflammatory and procoagulant fragments originating from plasma membrane generated after cellular activation and released in body fluids. In the vessel, they constitute a pool of bioactive effectors pulled from diverse cellular origins and may act as intercellular messengers. Microparticles expose phosphatidylserine, a procoagulant phospholipid made accessible after membrane remodelling, and tissue factor, the initiator of blood coagulation at the endothelial and leukocyte surface. They constitute a secretion pathway for IL-1β and up-regulate the proinflammatory response of target cells. Microparticles circulate at low levels in healthy individuals, but undergo phenotypic and quantitative changes that could play a pathophysiological role in inflammatory diseases. Microparticles may participate in the pathogenesis of sepsis through multiple ways. They are able to regulate vascular tone and are potent vascular proinflammatory and procoagulant mediators. Microparticles' abilities are of increasing interest in deciphering the mechanisms underlying the multiple organ dysfunction of septic shock.
Similar content being viewed by others

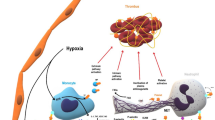

Introduction
In the 1960 s and 70 s Wolf [1] was the first to describe platelet derivatives of less than 0.1 μm as procoagulant vesicles. Later, having been given the name of 'micro-particles' (MPs), these vesicles were described as membrane-derived nano-fragments (0.05 to 1 μm) that are active in coagulation and inflammation. MPs are released in the extracellular environment through a membrane reorganization and blebbing process following cell activation or apoptosis. They constitute a storage pool of bioactive effectors with varied cellular origins and are able to act as intercellular messengers [2]. They are present in body fluids where they reflect normal tissue homeostasis, but undergo phenotypic and quantitative changes to play a pathophysiological role in several diseases, most of them associated with thrombotic disorders [3, 4] (Figure 1).

Structure of microparticles. Microparticles (MPs) are released from different cell types under physiological and pathological conditions. The plasma membrane is reorganised with active externalisation of phosphatidylserine (PhtdSer; a negatively charged phospholipid) and internalisation of phosphatidylcholine (insert). MPs bear intracytoplasmic and membrane-bound effectors from the originating cells, such as tissue factor (TF) and endothelial protein C receptor (EPCR) (endothelial cells and monocytes), CD-14 (monocytes) or glycoprotein (GP)Iba-IX-V', P-selectin or integrins (platelets).
MPs often convey tissue factor (TF) that may contribute to the dissemination of coagulopathy in sepsis [5, 6] and cytokines up-regulating deleterious inflammatory responses [7]. Circulating MPs can also provoke vascular dysfunction, and they reduce available nitric oxide (NO) and increase levels of reactive oxygen species, thereby promoting oxidative stress [8].
This review will focus on the role of MPs during sepsis, with a special emphasis on coagulation and inflammation disturbances.
Microparticles are potential intercellular messengers during sepsis
Sepsis is a syndrome characterized by excessive cellular activation involving a systemic inflammatory response to severe infection. Its most severe form may lead to septic shock. The ongoing circulatory failure is characterized by vasoplegia-related arterial hypotension and may include vasopressor resistance, and myocardial and local blood flow impairments. Inflammation plays a key role in the acute activation of the vascular wall and is associated with local thrombosis and changes in vasomotricity [9]. Thus, the endothelium-derived TF initiates the coagulation process and a proteolytic cascade [10]. The endothelial damage furthermore leads to the expression of adhesion molecules and other vasoactive factors involved in inflammation and coagulation.
Biogenesis and general features of microparticles
MPs are produced following cellular activation or apoptosis. The increase in intracellular calcium activates various cytosolic enzymes, including calpains, that cleave the cytoskeleton and facilitate the role of procaspase-3 in apoptosis [11]. As a response to stimulus, the cytoskeleton is reorganized and the asymmetric distribution of the phospholipid membrane modified with exposure of phosphatidylserine at the cell surface. Cellular blebbing then occurs, ultimately leading to the release of MPs. In addition to phosphatidylserine exposure, protein-lipid raft domains are formed and furnish the MP with its specificities and biological roles [12] (Figure 1).
The cellular origin of MPs can be determined by assessment of the antigens that they expose at their surface. However, the complete protein content of MPs remains difficult to establish. More than 300 proteins have been reported by proteomics, some of which are cytosolic and some membranous [13]. The MP phenotype is, however, known to vary according to cellular origin and parental cell response to stimulus [7, 14].
Microparticle survival and clearance
Although bearing phosphatidylserine, which is a signal for phagocytosis, MPs seem to survive longer than their parental apoptotic cell, probably because of their size, which does not allow optimal exposure of a cluster of senescence signals. Dasgupta and colleagues [15] recently described the major role of lactadherin in the removal of phosphatidylserine-expressing platelet MPs from human plasma. Lactadherin is a macrophage opsonin that mediates the clearance of apoptotic lymphocytes and knockout lactadherin (-/-) mice have increased levels of circulating platelet MPs and a hypercoagulable state; lactadherin supplementation restores the normal clearance of MPs. To date, there are no data on the effect of MP clearance on the haemostatic balance under physiological or pathological settings.
Microparticles as messengers in blood flow
As mediators of cellular communication, MPs are actors and possible mediators in the interplay between throm-bosis and inflammation, a process previously described for vascular injury in inflammatory diseases [5]. They can transfer receptors, organelles, mRNA and other proteins to target cells [16] and also comprise a secretion pathway for several cytokines, such as mature IL-1β [17]. The multiple properties of MPs and the variety of their possible cellular targets support them having a key role in cell reprogramming and tissue remodeling with physiological or pathological consequences [4]. Thus, MPs could play a major role in propagating proinflammatory and procoagulant states in sepsis. In the vascular compartment, including the arterial wall, the particular settings of sepsis and the tuning abilities of MPs point to the endothelium as a pivotal target [18, 19].
How to detect and measure microparticles?
The International Society for Thrombosis and Haemostasis (ISTH) provides information on technical procedures and recommendations for the detection and measurement of MPs. Although no standardized procedures for MP measurement are available yet, a consensus is forming on blood sampling and MP isolation by centrifugation steps that avoid exosome contamination of MP samples [20]. Several assays and phenotyping methods coexist, but these are not necessarily comparable, thus making the interpretation of results across studies difficult. MPs can be analyzed through capture techniques (using immobilized annexin V - a high affinity probe for phosphatidylserine - quantitative assessment, or insolubilized antibodies for phenotyping) combined with a functional prothrombinase assay. Flow cytometry is another method for the study of MPs. This method allows quantification and determination of cellular origin via the use of specific fluorescent antibodies and calibration beads. The protein content of MPs can also be assayed and expressed in molecular mass units [21]. Caution should be taken in the interpretation of MP analyses, taking into account the pitfalls of each method and the purpose of the experiment or clinical investigation. Furthermore, control cohorts are of prime importance in clinical investigations of MP pattern variations.
Microparticles as a player in coagulation disorders of sepsis
In the defence against pathogens, haemostasis is as fundamentally important as innate immunity and complement- mediated cell lysis. Haemostasis is activated during sepsis and septic shock, leading to thrombin and fibrin generation with dual effects: limitation of pathogen diffusion and invasion; and fibrin deposition in vessels, resulting in thrombotic microangiopathy or disseminated intravascular coagulopathy. As detailed above, MPs are efficient effectors in the haemostatic response and pathogenic markers of thrombotic disorders (Figure 2).

Microparticles and blood coagulation. (A) The plasma membrane of endothelial cells and monocytes is reorganised, with externalisation of phosphatidylserine - a negatively charged phospholipid - and encrypted tissue factor (TF) expression, allowing factor VII (FVIIa) activation and thrombin (FIIa) generation at the cell surface. Blebbing occurs, with release of microparticles (MPs) bearing TF, resulting in an increased surface for procoagulant reactions. Platelet adhesion and aggregation also occur with the release of MPs; platelets and MPs bear GPIbα', a cofactor for factor XI activation by thrombin, leading to the propagation phase with high levels of thrombin generation and fibrin formation. Endothelial TF-bearing MPs allow transfer of TF to PMNs, increasing TF dissemination and thrombotic microangiopathy or disseminated intravascular coagulopathy. (B) TF initiation of blood coagulation is quickly down-regulated by tissue factor pathway inhibitor (TFPI) on endothelial and monocytic cell surfaces, as on MPs. Endothelial protein C receptor (EPCR)-bound protein C is activated by the thrombin-thrombomodulin complex and activated protein C (APC) inhibits factor Va and factor VIIIa, limiting the propagation phase of thrombin generation. EPCR-bound APC also regulates NF-B, with cytoprotective effects on endothelial cells and monocytes. APC induces blebbing, with emission of EPCR-bearing MPs able to activate protein C, resulting in the dissemination of anticoagulant and antiapoptotic activities. LPS, lipopolysaccharide; PhtdSer, phosphatidylserine; PMN, polymorphonuclear.
Microparticles and thrombin generation
Thrombin generation requires activation of coagulation factors, which is made possible after their assembly on a catalytic surface constituted of anionic phospholipids. Cell activation constitutes the first step by furnishing exposed phosphatidylserine with a negative charge. The required remodelling of plasma membrane, resulting in phosphatidylserine translocation to the outer leaflet of the plasma membrane, occurs in platelets, endothelial cells and monocytes at sites of vascular damage or injury. Calcium ion-mediated interactions between gamma-carboxyl groups of vitamin-K-dependent factors and phosphatidylserine comprise the key step in this assembly, explaining the efficacy of anti-vitamin K treatments in hypercoagulable states [22].
At the monocyte surface a possible encrypted preformed TF would be de-encrypted by plasma membrane remodelling, thereby allowing the (auto-)activation of factor VII. Indeed, TF expression at the surface of monocyte-derived MPs has been demonstrated during in vitro endotoxin stimulation [23]. Although TF is the primary initiator of blood coagulation, whether there is a blood-borne TF (activity) is still debated, but there is growing evidence that this activity is directly tied to MPs [5, 24]. TF-bearing MPs can interact with neutrophil granulocytes by 'paracrine transfer', as demonstrated in vitro [25–27]. Circulating MPs bearing active TF have been associated with a thrombotic status in human meningococcal sepsis [28] and a primate Ebola fever model [29], pointing to their possible role in the dissemination of a procoagulant potential.
Microparticles and amplification loops in thrombin generation
TF-driven coagulation is under the control of Tissue factor pathway inhibitor (TFPI), an inhibitory complex, with factor Xa and protein S as cofactors. Although this inhibits TF-induced thrombin generation, thrombin is still generated during the propagation phase via the Josso loop: platelet-exposed factor XI (of megakaryocytic origin) is activated by the GPIbα-thrombin complex present on the surface of activated platelets. Activated platelets, and released GPIbα-FXIa bearing MPs, may, in turn, be responsible for increased thrombin generation [30–32]. In addition to blood-borne TF conveyed by MPs, polymorphonuclear (PMN)-derived MPs likely contribute to an additional amplification loop in the generation of thrombin mediated by MPs (Figure 2).
MPs could contribute to such amplification loops in sepsis. Indeed, ex vivo activation of human neutrophils by endotoxin, platelet activating factor or phorbol myristate acetate can generate MPs bearing active integrin αMβ2 (CD11b/CD18), which is able to activate GPIbα [33, 34].
Microparticles in the control of thrombin generation, cytoprotection and tissue remodelling
Interestingly, several cellular models showed that αMβ2 exposed at the MP surface can interact with other ligands, such as urokinase plasminogen activator, plasminogen and metalloproteases MMP-2 and -5, suggesting a role in fibrinolysis and in local tissue remodelling [30, 34, 35]. MPs may also display antithrombotic activities, which would be overwhelmed by procoagulant activities when MPs are released under highly thrombotic conditions, as observed during sepsis or myocardial infarction. Indeed, in purified monocyte suspensions, thrombomodulin anticoagulant activity and TF coexist at the MP surface, but when released by lipopolysaccharide treatment, the TF activity is predominant on MPs [31]. The presence of the anticoagulant endothelial protein C receptor (EPCR) at the surface of endothelial-derived MPs (mpEPCR) is another example of a cytoprotective element attached to MPs [32]; EPCR is involved in the activation of anticoagulant protein C by the thrombin-thrombomodulin complex. mpEPCR, released in response to activated protein C (APC), may switch the procoagulant properties of endothelial MPs to anticoagulant and anti-apoptotic properties. On the surface of MPs bearing mpEPCR, APC inactivates procoagulant cofactors factor Va and factor VIIIa, thereby down-regulating thrombin generation. Because a circulating soluble form of EPCR (sEPCR) has been described in sepsis, and its concentration possibly correlates with the severity of the illness, the respective contributions of mpEPCR and sEPCR is a matter of clinical relevance. sEPCR binds protein C and APC, thereby blunting their actions. The efficacy of therapeutic activated protein C (rhAPC; drotrecogin alfa (activated)) may depend on the balance between circulating sEPCR and mpEPCR [32]. Recent investigations in human endothelial cells reported that free rhAPC and rhAPC bound to mpEPCR have similar effects. rhAPC cleaves protease activated receptor-1 and induces significant changes in the activation/inhibition of genes with direct anti-apoptotic effects or indirect cell barrier protective effects, the latter requiring transactivation of KDR (vascular endothelial growth factor receptor 2/kinase insert domain receptor) via the phosphoinositide 3-kinase-Akt pathway and S1P1 (sphingosine 1-phosphate receptor) [36].
In sepsis, procoagulant MPs of endothelial, platelet, erythroid, and leukocyte origins have been reported [28, 37].
Microparticles as potential effectors in the inflammatory response of sepsis
Circulating MPs have been reported to be present in various inflammatory diseases, including sepsis [7]. MPs are a source of phospholipids, a substrate for phosphor-lipase A2, which facilitates platelet aggregation [38, 39]; they may also provoke vascular inflammation during sepsis via lysophosphatidic acid and facilitate chemo-tactic migration of platelets and/or leukocytes to the endothelium, thus playing the role of trigger in the production of monocyte cytokines (IL-1β, IL-8 and tumour necrosis factor-α) [8, 40, 41] (Figure 3).

Microparticles and inflammation in sepsis. During sepsis, microparticles (MPs) are shed from a variety of activated or apoptotic cells. MPs may be considered as both the cause and the consequence of inflammation through multiple amplification and regulatory loops affecting vascular cells and functions. Thus, MPs contribute to the spread of inflammatory and prothrombotic vascular status and they may affect the smooth muscle tissue through adhesion molecules, activation of NF-κB and the expression of inducible nitric oxide synthase and cyclooxygenase-2, with an increase in nitric oxide and vasodilator prostanoids, leading to arterial hyporeactivity. MPs form microaggregates with circulating neutrophil granulocytes and platelets and are involved in the modification of the oxidative status, markedly increasing oxidative activity. Subunits of NADPH oxidase have been identified in MPs associated with increased production of reactive oxygen species. AA, arachidonic acid; COX = cyclooxygenase; ICAM, intercellular cell adhesion molecule; iNOS, inducible NO-synthase; LPS, lipopolysaccharide; NO, nitric oxide; PAF, platelet activating factor; R, Rantes; ROS, reactive oxygen species; TF, tissue factor; VCAM, vascular cell adhesion molecule.
Microparticles targeting the endothelial function
During sepsis, the endothelial function is altered and the endothelial surface becomes proadhesive, procoagulant and antifibrinolytic [42]. The endothelium is one of the primary targets of circulating MPs, as demonstrated by Barry and colleagues [43]in vitro. Indeed, they showed that arachidonic acid exposed by platelet MPs promotes the up-regulation of cyclooxygenase-2 (COX-2) and intercellular adhesion molecules in endothelial cells. Platelet-derived MPs have been shown to modulate interactions between monocytes and endothelial cells. Released proinflammatory endothelial cytokines may themselves also contribute to the production of MPs [44], thereby amplifying the inflammatory response and the consecutive alteration of the vascular function [45]. Platelet activating factor present in endothelial cells and leukocytes is also involved in the proinflammatory effect of MPs [46].
Endothelial microparticles and inflammatory status
Circulating MPs of endothelial origin may thus vary with respect to quantity and phenotype according to the endothelial response and have been reported in inflammatory diseases and disorders [47]; the endothelial response to inflammation stimuli may be immediate, delayed or reflect a chronic endothelial activation. They were reported to participate in the regulation of arterial tone in several diseases in which oxidative stress is involved, such as human acute coronary syndromes [48] or preeclampsia [49] associated with altered NO bioavailability [50].
Sepsis induces a phenotypic change of the endothelium and the endothelial surface becomes proinflammatory, expresses cell adhesion molecules (intercellular cell adhesion molecule 1, vascular cell adhesion molecule 1) [51] and becomes prothrombotic through the increased expression of membrane TF and the inhibition of thrombomodulin and EPCR synthesis. In parallel, endothelial cells become capable of recruiting and activating platelets [52].
Microparticles contribute to the spreading of inflammatory and prothrombotic vascular status
MPs may be considered as both the cause and the consequence of inflammatory diseases through multiple amplification and regulatory loops affecting vascular cell functions. In vitro incubation of leukocyte-derived MPs with endothelial cells allowed Mesri and Altieri [53] to demonstrate their role in the secretion of inflammatory IL-6 and in the production of TF [54]. Furthermore, platelet MPs are able to deliver RANTES at the inflamed endothelium, thus promoting leukocyte recruitment and diapedesis [55]. MPs may affect the smooth muscle tissue through the activation of the transcription factor NF-κB and favour the expression of inducible NO-synthase and COX-2, resulting in an increase in NO and vasodilator prostanoids, leading to arterial hyporeactivity [45].
The interactions between platelets, leukocytes and endothelium clearly contribute to the vascular dysfunction observed in sepsis and various MPs were reported to alter the arterial wall directly or indirectly [56, 57].
Endothelial MPs may play a role in the spread of sepsis inflammatory responses leading to multiple organ dys-function [18, 58]. They may participate in the potentialisation of the procoagulant state associated with sepsis by providing renewed lipid surfaces of human endothelial cells for the generation of thrombin and by up-regulating monocyte TF expression, as demonstrated in vitro [59]. In sepsis, blockade of the human TF pathway by TFPI is very quickly overridden, clearing the way for a detrimental procoagulant state [28]. Indeed, in humans, a single endotoxin administration provokes a significant increase in endothelial-cell- or monocyte-derived MPs displaying potentiated TF [60]. This state is worsened by the exhaustion and/or faulty activation of the two other regulatory molecules, antithrombin and APC.
Several reports illustrate well the cascade of interwoven events that link cellular activation, TF up-regulation, the release of MPs presenting active TF and the triggering of disseminated intravascular coagulopathy and shock [28, 29]. With regard to vascular tone, MPs could promote the significant vasoplegia observed in septic shock [8]. Arachidonic acid transfer may up-regulate COX-2 expression and the production of prostacyclin, which is implicated in vasodilation and the inhibition of platelet activation [32].
Microparticles and oxidative stress
During sepsis, generated MPs are involved in the modification of the oxidative status; they form microaggregates with circulating neutrophil granulocytes and markedly increase oxidative activity [8]. Subunits of NADPH oxidase have been identified in endothelial- and platelet-derived MPs associated with increased production of reactive oxygen species [18, 61] (Figure 3).
Conclusion
The systemic inflammatory response that is a characteristic feature of sepsis is a major cause of cellular dysfunction that may lead to the exaggerated generation of MPs. These plasma membrane fragments are circulating markers of vascular inflammatory diseases. They also behave as pathogenic shuttles able to disseminate their deleterious proinflamatory and procoagulant potential in the systemic circulation and may be implicated in the multiple organ dysfunction characterizing sepsis and septic shock. To date, however, we have insufficient evidence to determine whether MPs are major players or bystanders in the development of the sepsis syndrome.
Abbreviations
- APC:
-
activated protein C
- COX:
-
cyclooxygenase
- EPCR:
-
endothelial protein C receptor
- IL:
-
interleukin
- MP:
-
microparticle
- MPEPCR:
-
microparticle endothelial protein C receptor
- NF:
-
nuclear factor
- NO:
-
nitric oxide
- RHAPC:
-
therapeutic activated protein C
- SEPCR:
-
soluble endothelial protein C receptor
- TF:
-
tissue factor
- TFPI:
-
Tissue factor pathway inhibitor.
References
Wolf P: The nature and significance of platelet products in human plasma. Br J Haematol 1967, 13: 269-288. 10.1111/j.1365-2141.1967.tb08741.x
Morel O, Toti F, Hugel B, Freyssinet JM: Cellular microparticles: a disseminated storage pool of bioactive vascular effectors. Curr Opin Hematol 2004, 11: 156-164. 10.1097/01.moh.0000131441.10020.87
Zwaal RF, Schroit AJ: Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood 1997, 89: 1121-1132.
Freyssinet JM: Cellular microparticles: what are they bad or good for? J Thromb Haemost 2003, 1: 1655-1662. 10.1046/j.1538-7836.2003.00309.x
Morel O, Morel N, Freyssinet JM, Toti F: Platelet microparticles and vascular cells interactions: a checkpoint between the haemostatic and thrombotic responses. Platelets 2008, 19: 9-23. 10.1080/09537100701817232
Pawlinski R, Mackman N: Cellular sources of tissue factor in endotoxemia and sepsis. Thromb Res 2010,125(Suppl 1):S70-73. 10.1016/j.thromres.2010.01.042
Distler JH, Huber LC, Gay S, Distler O, Pisetsky DS: Microparticles as mediators of cellular cross-talk in inflammatory disease. Autoimmunity 2006, 39: 683-690. 10.1080/08916930601061538
Mortaza S, Martinez MC, Baron-Menguy C, Burban M, de la Bourdonnaye M, Fizanne L, Pierrot M, Calès P, Henrion D, Andriantsitohaina R, Mercat A, Asfar P, Meziani F: Detrimental hemodynamic and inflammatory effects of microparticles originating from septic rats. Crit Care Med 2009, 37: 2045-2050. 10.1097/CCM.0b013e3181a00629
Russell JA: Management of sepsis. N Engl J Med 2006, 355: 1699-1713. 10.1056/NEJMra043632
Gando S, Kameue T, Matsuda N, Hayakawa M, Morimoto Y, Ishitani T, Kemmotsu O: Imbalances between the levels of tissue factor and tissue factor pathway inhibitor in ARDS patients. Thromb Res 2003, 109: 119-124. 10.1016/S0049-3848(03)00151-8
Cohen Z, Gonzales RF, Davis-Gorman GF, Copeland JG, McDonagh PF: Thrombin activity and platelet microparticle formation are increased in type 2 diabetic platelets: a potential correlation with caspase activation. Thromb Res 2002, 107: 217-221. 10.1016/S0049-3848(02)00334-1
Hugel B, Martinez MC, Kunzelmann C, Freyssinet JM: Membrane microparticles: two sides of the coin. Physiology (Bethesda) 2005, 20: 22-27.
Miguet L, Pacaud K, Felden C, Hugel B, Martinez MC, Freyssinet JM, Herbrecht R, Potier N, van Dorsselaer A, Mauvieux L: Proteomic analysis of malignant lymphocyte membrane microparticles using double ionization coverage optimization. Proteomics 2006, 6: 153-171. 10.1002/pmic.200500133
Miguet L, Bechade G, Fornecker L, Zink E, Felden C, Gervais C, Herbrecht R, van Dorsselaer A, Mauvieux L, Sanglier-Cianferani S: Proteomic analysis of malignant B-cell derived microparticles reveals CD148 as a potentially useful antigenic biomarker for mantle cell lymphoma diagnosis. J Proteome Res 2009, 8: 3346-3354. 10.1021/pr801102c
Dasgupta SK, Abdel-Monem H, Niravath P, Le A, Bellera RV, Langlois K, Nagata S, Rumbaut RE, Thiagarajan P: Lactadherin and clearance of platelet-derived microvesicles. Blood 2009, 113: 1332-1339. 10.1182/blood-2008-07-167148
Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ: Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia 2006, 20: 1487-1495. 10.1038/sj.leu.2404296
MacKenzie A, Wilson HL, Kiss-Toth E, Dower SK, North RA, Surprenant A: Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity 2001, 15: 825-835. 10.1016/S1074-7613(01)00229-1
Ogura H, Tanaka H, Koh T, Fujita K, Fujimi S, Nakamori Y, Hosotsubo H, Kuwagata Y, Shimazu T, Sugimoto H: Enhanced production of endothelial microparticles with increased binding to leukocytes in patients with severe systemic inflammatory response syndrome. J Trauma 2004, 56: 823-830. 10.1097/01.TA.0000084517.39244.46
Soriano AO, Jy W, Chirinos JA, Valdivia MA, Velasquez HS, Jimenez JJ, Horstman LL, Kett DH, Schein RM, Ahn YS: Levels of endothelial and platelet microparticles and their interactions with leukocytes negatively correlate with organ dysfunction and predict mortality in severe sepsis. Crit Care Med 2005, 33: 2540-2546. 10.1097/01.CCM.0000186414.86162.03
Thery C, Zitvogel L, Amigorena S: Exosomes: composition, biogenesis and function. Nat Rev Immunol 2002, 2: 569-579.
Jy W, Horstman LL, Jimenez JJ, Ahn YS, Biró E, Nieuwland R, Sturk A, Dignat-George F, Sabatier F, Camoin-Jau L, Sampol J, Hugel B, Zobairi F, Freyssinet JM, Nomura S, Shet AS, Key NS, Hebbel RP: Measuring circulating cell-derived microparticles. J Thromb Haemost 2004, 2: 1842-1851. 10.1111/j.1538-7836.2004.00936.x
Lane DA, Philippou H, Huntington JA: Directing thrombin. Blood 2005, 106: 2605-2612. 10.1182/blood-2005-04-1710
Satta N, Toti F, Feugeas O, Bohbot A, Dachary-Prigent J, Eschwege V, Hedman H, Freyssinet JM: Monocyte vesiculation is a possible mechanism for dissemination of membrane-associated procoagulant activities and adhesion molecules after stimulation by lipopolysaccharide. J Immunol 1994, 153: 3245-3255.
Key NS: Analysis of tissue factor positive microparticles. Thromb Res 2010, (125 Suppl 1:):S42-45. 10.1016/j.thromres.2010.01.035
Liu ML, Reilly MP, Casasanto P, McKenzie SE, Williams KJ: Cholesterol enrichment of human monocyte/macrophages induces surface exposure of phosphatidylserine and the release of biologically-active tissue factor-positive microvesicles. Arterioscler Thromb Vasc Biol 2007, 27: 430-435. 10.1161/01.ATV.0000254674.47693.e8
Egorina EM, Sovershaev MA, Olsen JO, Osterud B: Granulocytes do not express but acquire monocyte-derived tissue factor in whole blood: evidence for a direct transfer. Blood 2008, 111: 1208-1216. 10.1182/blood-2007-08-107698
Chou J, Mackman N, Merrill-Skoloff G, Pedersen B, Furie BC, Furie B: Hematopoietic cell-derived microparticle tissue factor contributes to fibrin formation during thrombus propagation. Blood 2004, 104: 3190-3197. 10.1182/blood-2004-03-0935
Nieuwland R, Berckmans RJ, McGregor S, Boing AN, Romijn FP, Westendorp RG, Hack CE, Sturk A: Cellular origin and procoagulant properties of microparticles in meningococcal sepsis. Blood 2000, 95: 930-935.
Geisbert TW, Young HA, Jahrling PB, Davis KJ, Kagan E, Hensley LE: Mechanisms underlying coagulation abnormalities in ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key event. J Infect Dis 2003, 188: 1618-1629. 10.1086/379724
Lacroix R, Sabatier F, Mialhe A, Basire A, Pannell R, Borghi H, Robert S, Lamy E, Plawinski L, Camoin-Jau L, Gurewich V, Angles-Cano E, Dignat-George F: Activation of plasminogen into plasmin at the surface of endothelial microparticles: a mechanism that modulates angiogenic properties of endothelial progenitor cells in vitro . Blood 2007, 110: 2432-2439. 10.1182/blood-2007-02-069997
Satta N, Freyssinet JM, Toti F: The significance of human monocyte thrombomodulin during membrane vesiculation and after stimulation by lipopolysaccharide. Br J Haematol 1997, 96: 534-542. 10.1046/j.1365-2141.1997.d01-2059.x
Perez-Casal M, Downey C, Fukudome K, Marx G, Toh CH: Activated protein C induces the release of microparticle-associated endothelial protein C receptor. Blood 2005, 105: 1515-1522. 10.1182/blood-2004-05-1896
Del Conde I, Shrimpton CN, Thiagarajan P, Lopez JA: Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 2005, 106: 1604-1611. 10.1182/blood-2004-03-1095
Pluskota E, Woody NM, Szpak D, Ballantyne CM, Soloviev DA, Simon DI, Plow EF: Expression, activation, and function of integrin alphaMbeta2 (Mac-1) on neutrophil-derived microparticles. Blood 2008, 112: 2327-2335. 10.1182/blood-2007-12-127183
Taraboletti G, D'Ascenzo S, Borsotti P, Giavazzi R, Pavan A, Dolo V: Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated components by endothelial cells. Am J Pathol 2002, 160: 673-680.
Perez-Casal M, Downey C, Cutillas-Moreno B, Zuzel M, Fukudome K, Toh CH: Microparticle-associated endothelial protein C receptor and the induction of cytoprotective and anti-inflammatory effects. Haematologica 2009, 94: 387-394. 10.3324/haematol.13547
Itakura Sumi Y, Ogura H, Tanaka H, Koh T, Fujita K, Fujimi S, Nakamori Y, Shimazu T, Sugimoto H: Paradoxical cytoskeleton and microparticle formation changes in monocytes and polymorphonuclear leukocytes in severe systemic inflammatory response syndrome patients. J Trauma 2003, 55: 1125-1132. 10.1097/01.TA.0000096663.21402.5C
Fourcade O, Simon MF, Viode C, Rugani N, Leballe F, Ragab A, Fournie B, Sarda L, Chap H: Secretory phospholipase A2 generates the novel lipid mediator lysophosphatidic acid in membrane microvesicles shed from activated cells. Cell 1995, 80: 919-927. 10.1016/0092-8674(95)90295-3
Sellam J, Proulle V, Jüngel A, Ittah M, Miceli Richard C, Gottenberg JE, Toti F, Benessiano J, Gay S, Freyssinet JM, Mariette X: Increased levels of circulating microparticles in primary Sjogren's syndrome, systemic lupus erythematosus and rheumatoid arthritis and relation with disease activity. Arthritis Res Ther 2009, 11: R156. 10.1186/ar2833
Lynch SF, Ludlam CA: Plasma microparticles and vascular disorders. Br J Haematol 2007, 137: 36-48.
Gambim MH, do Carmo Ade O, Marti L, Verissimo-Filho S, Lopes LR, Janiszewski M: Platelet-derived exosomes induce endothelial cell apoptosis through peroxynitrite generation: experimental evidence for a novel mechanism of septic vascular dysfunction. Crit Care 2007, 11: R107. 10.1186/cc6133
Sennoun N, Baron-Menguy C, Burban M, Lecompte T, Andriantsitohaina R, Henrion D, Mercat A, Asfar P, Levy B, Meziani F: Recombinant human activated protein C improves endotoxemia-induced endothelial dysfunction: a blood-free model in isolated mouse arteries. Am J Physiol Heart Circ Physiol 2009, 297: H277-282. 10.1152/ajpheart.01133.2008
Barry OP, Kazanietz MG, Pratico D, FitzGerald GA: Arachidonic acid in platelet microparticles up-regulates cyclooxygenase-2-dependent prostaglandin formation via a protein kinase C/mitogen-activated protein kinase-dependent pathway. J Biol Chem 1999, 274: 7545-7556. 10.1074/jbc.274.11.7545
Nomura S, Imamura A, Okuno M, Kamiyama Y, Fujimura Y, Ikeda Y, Fukuhara S: Platelet-derived microparticles in patients with arteriosclerosis obliterans: enhancement of high shear-induced microparticle generation by cytokines. Thromb Res 2000, 98: 257-268. 10.1016/S0049-3848(00)00186-9
Tesse A, Martinez MC, Hugel B, Chalupsky K, Muller CD, Meziani F, Mitolo-Chieppa D, Freyssinet JM, Andriantsitohaina R: Upregulation of proinflammatory proteins through NF-kappaB pathway by shed membrane microparticles results in vascular hyporeactivity. Arterioscler Thromb Vasc Biol 2005, 25: 2522-2527. 10.1161/01.ATV.0000189298.62240.5d
Wolf P, Nghiem DX, Walterscheid JP, Byrne S, Matsumura Y, Matsumura Y, Bucana C, Ananthaswamy HN, Ullrich SE: Platelet-activating factor is crucial in psoralen and ultraviolet A-induced immune suppression, inflammation, and apoptosis. Am J Pathol 2006, 169: 795-805. 10.2353/ajpath.2006.060079
VanWijk MJ, VanBavel E, Sturk A, Nieuwland R: Microparticles in cardiovascular diseases. Cardiovasc Res 2003, 59: 277-287. 10.1016/S0008-6363(03)00367-5
Boulanger CM, Scoazec A, Ebrahimian T, Henry P, Mathieu E, Tedgui A, Mallat Z: Circulating microparticles from patients with myocardial infarction cause endothelial dysfunction. Circulation 2001, 104: 2649-2652. 10.1161/hc4701.100516
Meziani F, Tesse A, David E, Martinez MC, Wangesteen R, Schneider F, Andriantsitohaina R: Shed membrane particles from preeclamptic women generate vascular wall inflammation and blunt vascular contractility. Am J Pathol 2006, 169: 1473-1483. 10.2353/ajpath.2006.051304
Brodsky SV, Zhang F, Nasjletti A, Goligorsky MS: Endothelium-derived microparticles impair endothelial function in vitro . Am J Physiol Heart Circ Physiol 2004, 286: H1910-1915. 10.1152/ajpheart.01172.2003
Lopes-Bezerra LM, Filler SG: Endothelial cells, tissue factor and infectious diseases. Braz J Med Biol Res 2003, 36: 987-991. 10.1590/S0100-879X2003000800004
Bombeli T, Schwartz BR, Harlan JM: Endothelial cells undergoing apoptosis become proadhesive for nonactivated platelets. Blood 1999, 93: 3831-3838.
Mesri M, Altieri DC: Endothelial cell activation by leukocyte microparticles. J Immunol 1998, 161: 4382-4387.
Mesri M, Altieri DC: Leukocyte microparticles stimulate endothelial cell cytokine release and tissue factor induction in a JNK1 signaling pathway. J Biol Chem 1999, 274: 23111-23118. 10.1074/jbc.274.33.23111
Mause SF, von Hundelshausen P, Zernecke A, Koenen RR, Weber C: Platelet microparticles: a transcellular delivery system for RANTES promoting monocyte recruitment on endothelium. Arterioscler Thromb Vasc Biol 2005, 25: 1512-1518. 10.1161/01.ATV.0000170133.43608.37
Morel O, Hugel B, Jesel L, Lanza F, Douchet MP, Zupan M, Chauvin M, Cazenave JP, Freyssinet JM, Toti F: Sustained elevated amounts of circulating procoagulant membrane microparticles and soluble GPV after acute myocardial infarction in diabetes mellitus. Thromb Haemost 2004, 91: 345-353.
Pfister SL: Role of platelet microparticles in the production of thromboxane by rabbit pulmonary artery. Hypertension 2004, 43: 428-433. 10.1161/01.HYP.0000110906.77479.91
Densmore JC, Signorino PR, Ou J, Hatoum OA, Rowe JJ, Shi Y, Kaul S, Jones DW, Sabina RE, Pritchard KA Jr, Guice KS, Oldham KT: Endothelium-derived microparticles induce endothelial dysfunction and acute lung injury. Shock 2006, 26: 464-471. 10.1097/01.shk.0000228791.10550.36
Brodsky SV, Malinowski K, Golightly M, Jesty J, Goligorsky MS: Plasminogen activator inhibitor-1 promotes formation of endothelial microparticles with procoagulant potential. Circulation 2002, 106: 2372-2378. 10.1161/01.CIR.0000033972.90653.AF
Aras O, Shet A, Bach RR, Hysjulien JL, Slungaard A, Hebbel RP, Escolar G, Jilma B, Key NS: Induction of microparticle- and cell-associated intravascular tissue factor in human endotoxemia. Blood 2004, 103: 4545-4553. 10.1182/blood-2003-03-0713
Janiszewski M, Do Carmo AO, Pedro MA, Silva E, Knobel E, Laurindo FR: Platelet-derived exosomes of septic individuals possess proapoptotic NAD(P)H oxidase activity: A novel vascular redox pathway. Crit Care Med 2004, 32: 818-825. 10.1097/01.CCM.0000114829.17746.19
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
All authors participated in the design of this review and in drafting the manuscript. FM and FT supervised the manuscript. All authors read and approved the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Meziani, F., Delabranche, X., Asfar, P. et al. Bench-to-bedside review: Circulating microparticles - a new player in sepsis?. Crit Care 14, 236 (2010). https://doi.org/10.1186/cc9231
Published:
DOI: https://doi.org/10.1186/cc9231