
Abstract
Continued safety assessment is essential for any newly approved therapy. Drotrecogin alfa (activated; DrotAA), which is approved for use in severe sepsis, has undergone clinical trials with corresponding safety analyses since 1995. However, the only comprehensive review of all trials is that reported in 2003 by Bernard and coworkers. This is an important review that focuses on the safety profile of DrotAA in all published studies (six randomized clinical trials and five national registry studies) and in previously unpublished data. DrotAA treatment is associated with an increased risk for bleeding (which in general is manageable). Nevertheless, the available evidence shows that any adverse effects of DrotAA are outweighed by its benefits in patients with severe sepsis who are at high risk for death. So far, more than 9,000 patients have been enrolled in clinical trials involving DrotAA, providing a valuable safety database. It is of note that although DrotAA does increase the risk of bleeding, this has not been associated with an overall increase in the rate of all severe adverse events.
Similar content being viewed by others

Introduction
Drotrecogin alfa (activated; DrotAA) is the first and thus far only compound proven to improve survival in patients with severe sepsis. The mechanisms that underlie the improved survival are not entirely clear. A number of studies have demonstrated that DrotAA exerts an anticoagulant effect through inactivation of coagulation factors Va and VIIIa [1], an anti-inflammatory effect [2] and a profibrinolytic effect [3], as well as antiapoptotic [4] and cytoprotective effects [5]. Of these, the anticoagulant property is the action associated with the only known adverse effect of treatment, namely an increased risk for bleeding.
Continued assessment of safety is essential in any newly approved therapy. DrotAA has been studied in clinical trials since 1995 (Table 1). It was approved for use in adult patients with severe sepsis at high risk of death in the USA in November 2001 [6] and in the European Union in August 2002 [7]. The European Medicines Agency indicates that DrotAA may be used in the treatment of adult patients with severe sepsis and multiple organ failure, and reaffirmed this indication as a result of the second annual licence reassessment, stating that DrotAA should be considered mainly in situations in which therapy can be initiated within 24 hours after the onset of organ failure [8].
From the very first clinical study, safety analyses have been included in each controlled trial conducted in DrotAA, and continuous safety surveillance has formed part of all studies undertaken for the purposes of registering the drug for commercial use. In this review we analyze the available literature regarding the safety of DrotAA, together with some previously unpublished data.
Safety profile in clinical trials
In the early phase I studies, begun more than 10 years ago, adverse events with a frequency of 5% or greater included headache (30.9%), ecchymosis (23.0%), and pain (5.8%), primarily at the site of venopuncture [9]. These studies were conducted in healthy volunteers mainly to evaluate pharmacokinetics, and no further safety issues were identified to prevent future clinical development [9]. The data regarding serious bleeding events are summarized in Table 2. The definition of a serious bleeding event includes any intracranial hemorrhage, any life-threatening or fatal bleed, any bleeding event requiring the administration of three or more units of packed red blood cells per day for 2 consecutive days, or any bleeding event considered serious by the investigator. A similar definition was adopted for all DrotAA studies except for ADDRESS and XPRESS, in which requirements for transfusion were not included.
The first patients with severe sepsis to receive DrotAA were those included in the EVAA (dose finding) study [10]. This was a proof-of-concept study that investigated the safety profile of DrotAA and determined the optimal dosage for future trials. Four (4%) patients receiving DrotAA and two (5%) patients receiving placebo during the 28-day period of study reported serious bleeding events. Two events, haematemesis and worsening haematoma, occurred during DrotAA infusion and the other two, worsening gastrointestinal hemorrhage and hemorrhage during laparotomy, occurred 1 and 25 days after infusion. The two gastrointestinal hemorrhages in the control group occurred 3 and 4 days after placebo infusion.
In EVAA, overall, there was no statistically significant difference between DrotAA-treated and placebo-treated patients experiencing at least one serious adverse event (39% versus 46%, respectively; P = 0.422). Skin rash occurred more frequently among DrotAA-treated than placebo-treated patients (10% versus 0%, respectively; P = 0.036). None of the rashes was classified as serious or considered by the investigators to be drug related. No significant differences in specific treatment-emergent adverse events were recorded and no safety issues were identified in central laboratory parameters or vital signs.
For 53 treated patients, samples for anti-DrotAA antibody testing were available. A low-level anti-DrotAA antibody response was reported in one patient; the antibodies were non-neutralizing with respect to the anticoagulant activity. This patient did not experience any related clinical adverse events and was negative for anti-DrotAA antibody response 1 year after exposure.
These EVAA results were sufficiently encouraging to proceed to the pivotal phase III double-blind, randomized, placebo-controlled PROWESS (Recombinant Human Activated Protein C Worldwide Evaluation in Severe Sepsis) trial.
PROWESS, ENHANCE, ADDRESS, and XPRESS
These four studies used all the same dose of DrotAA (24 μg/kg per hour), which was the one that was demonstrated to be more effective and safe in EVAA.
The pivotal phase III PROWESS trial [11] included 850 DrotAA-treated and 840 placebo-treated patients. The percentages of patients experiencing at least one serious adverse event were 12.1% in the DrotAA group and 12.5% in the placebo group (P = 0.84), whereas percentages of those experiencing at least one bleeding event were 24.9% in the DrotAA group and 17.7% in the placebo group (P < 0.05). The majority of bleeding events in both groups were ecchymosis or gastrointestinal tract bleeding and were not considered serious. The difference in the incidence of serious bleeding events between the two treatment groups occurred primarily during drug administration.
In ENHANCE (Extended Evaluation of Recombinant Human Activated Protein C, drotrecogin alfa [activated]), a single-arm, open-label trial [12], a total of 2,378 adult patients with severe sepsis received DrotAA. Given that the rate of serious bleeding events was also higher in ENHANCE during the post-infusion period, where, due to the short half-life of the drug, patients were not exposed to DrotAA, this suggests a higher baseline risk for bleeding in ENHANCE compared with PROWESS, with higher degrees of both haematologic and hepatic dysfunction, both of which are associated with increased bleeding risk. The incidences of serious bleeding events in both the infusion and post-infusion periods in the PROWESS and ENHANCE studies are summarized in Table 2.
The ADDRESS (Efficacy and Safety of Drotrecogin alfa [activated] in Adult Severe Sepsis Patients at Low Risk of Death) study [13] involved 1,317 DrotAA-treated and 1,293 placebo-treated patients. The percentages of patients experiencing at least one bleeding event in the DrotAA and placebo treatment groups were 10.9% and 6.4%, respectively (P < 0.001). The numbers of serious bleeding events during infusion (defined as study day 0 through to study day 6) were 31 (2.4%) in DrotAA-treated and 15 (1.2%) in placebo-treated patients (P = 0.02). The numbers of patients suffering central nervous system (CNS) bleeding during infusion were 4 (0.3%) and 3 (0.2%) in the DrotAA and placebo groups, respectively. Recent surgery (within 30 days before study entry) was associated with a greater risk for serious bleeding during infusion in both the DrotAA-treated and placebo-treated patients (3.6% versus 1.6% in DrotAA-treated patients with and without recent surgery, respectively; and 1.6% versus 0.9% placebo-treated patients with and without recent surgery, respectively); however, this difference between groups was not significant.
XPRESS (Xigris and Prophylactic Heparin Evaluation in Severe Sepsis) [14] was an open label study, all the patients received DrotAA and the study drug was prophylactic heparin or placebo. It included patients with severe sepsis and greater disease severity (for example, a higher rate of organ dysfunctions) than in previous studies (because it was performed in an on-label population), who were undergoing treatment with DrotAA. Serious bleeding rates were consistent with those observed in previous studies throughout the 6-day treatment period, and prophylactic heparin did not increase the risk for serious bleeding compared with placebo (2.3% and 2.5%, respectively), including CNS bleeding (0.3% in both arms). However, the group receiving prophylactic heparin had a significantly higher rate of nonserious bleeding in comparison with the placebo group (8.7% versus 5.7%, respectively; P = 0.0116).
Serious bleeding events during the 28-day study period
In PROWESS the incidence of serious bleeding events during the 28-day study period was 3.5% in DrotAA-treated and 2.0% in placebo-treated patients. The incidences of CNS bleeds during this period were 0.2% and 0.1% for DrotAA-treated and placebo-treated patients, respectively. The risk for CNS bleeding may increase with severe coagulopathy and severe thrombocytopenia [15]. In ADDRESS the corresponding results were 3.9% in DrotAA-treated and 2.2% in placebo-treated patients (P = 0.01), and 0.5% and 0.4%, respectively, for CNS bleeds.
Serious bleeding rates in XPRESS [14] were consistent with those observed in previous studies during the 28-day period of study. Prophylactic heparin did not increase the risk for serious bleeding for DrotAA compared with placebo (3.9% versus 5.2%, respectively), including CNS bleeding (1.0% versus 0.7%, respectively). In ENHANCE, 6.5% of DrotAA-treated patients had serious bleeding events during days 0 to 28, and the incidence of CNS bleeds was 1.5%.
Safety profile outside controlled clinical trials
Two years after DrotAA had been approved by the US Food and Drug Administration for use in adult patients with severe sepsis at high risk of death, Bernard and coworkers [16] reported the first post-marketing safety assessment, and at least five National Registries have been published to date [17–21].
Although all of the referenced studies [17–21] carried out a safety analysis, the findings are not entirely comparable because diverse definitions of bleeding were employed in the different registries and these were sometimes different from those used in the controlled clinical trials. Table 3 summarizes the registries that included safety findings.
Safety profile in special populations
Surgical patients
Surgical patients with severe sepsis account for 25% to 50% of patients treated in randomized clinical trials and registry studies of DrotAA. The main complication after major surgery is bleeding, and therefore this group of patients represents a special population from a safety standpoint. The main analysis of treated patients published so far concluded that although patients receiving DrotAA are at greater risk for bleeding, the risk-benefit profile still favors use of DrotAA in patients in whom it is indicated [22].
The PROWESS Trial Surgical Evaluation Committee on efficacy and safety of DrotAA in surgical patients confirmed 28% of cases as surgical. Barie et al. [22], moreover, report that for all surgical patients and those undergoing intraabdominal procedures, the absolute risk reduction for mortality was 3.2% and 9.1%, respectively. Administration of DrotAA was associated with an increased risk for serious bleeding during the infusion period in both the overall PROWESS patients (P = 0.024) and the surgical cohort (P = 0.006); however, the surgical patients did not demonstrate excessive risk of bleeding compared with nonsurgical patients who received DrotAA. Serious bleeding during the infusion and 28-day period was similar between surgical and nonsurgical patients. For the three patient populations PROWESS overall, PROWESS Surgical and PROWESS non-Surgical, respectively, the serious bleeding event rates were 2.4% versus 3.1% versus 2.1% during the infusion period, and 3.5% versus 3.5% versus 3.5% during the 28-day period. In ADDRESS, recent surgery (within 30 days prior to study entry) was associated with a numerically higher risk of serious bleeding during infusion in both the Xigris-treated and the placebo-treated patients (Xigris: 3.6% in patients with recent surgery versus 1.6% in patients without recent surgery; placebo: 1.6% versus 0.9%, respectively).
Among the small number of patients (98) enrolled in PROWESS with single organ dysfunction and recent surgery (surgery within 30 days prior to study treatment), 28-day all-cause mortality was 10/49 versus 8/49 for the Xigris and placebo patients, respectively (*P = 0.60); the in-hospital mortality was 14/48 versus 8/47 for the Xigris and placebo patients, respectively (*P = 0.16). The subset of patients (636) with single organ dysfunction and recent surgery from the randomized, placebo-controlled study (ADDRESS) of septic patients at lower risk of death (Acute Physiology and Chronic Health Evaluation [APACHE] II score <25 or single sepsis-induced organ failure at any APACHE II score) had a 28-day all-cause mortality of 67/323 versus 44/323 for Xigris and placebo patients, respectively (*P = 0.03) and an in-hospital mortality of 76/325 versus 62/314 for Xigris and placebo patients, respectively (*P = 0.26). (*Chi-square test without adjustment for multiple comparisons.)
The incidence of serious bleeding events in patients with single organ dysfunction and recent surgery reported in PROWESS during infusion was 4.2% versus 0% for Xigris and placebo patients, respectively (P = 0.16) and during the 28-day period was 4.2% versus 0% for Xigris and placebo patients, respectively (P = 0.16); in ADDRESS the incidence of serious bleeding events during infusion was 4.4% versus 1.9% for Xigris and placebo patients, respectively (P = 0.08), and for 28 days 5.0% versus 2.9% for Xigris and placebo patients, respectively (P = 0.17).
DrotAA is not indicated for patients with single organ dysfunction and in particular should not be used in patients with single organ dysfunction and recent surgery.
Recommendations for patients under interventions
When surgery or other invasive procedures are performed during the infusion of DrotAA, the recommendations for interruption of infusion should be followed to minimize the risk for bleeding. Pharmacokinetic studies show that DrotAA has a short half-life. Plasma concentrations decline rapidly, with no measurable activity within 2 hours after the infusion is stopped.
As stated in the current European Summary of Product Characteristics, 'For procedures with an inherent bleeding risk, discontinue drotrecogin alfa (activated) for 2 hours prior to the start of the procedure. Drotrecogin alfa (activated) may be restarted 12 hours after major invasive procedures or surgery if adequate haemostasis has been achieved. Drotrecogin alfa (activated) may be restarted immediately after uncomplicated less invasive procedures if adequate haemostasis has been achieved.' [8].
The decision regarding the interruption/post-procedure reinstitution of DrotAA for surgical procedures should be made by the attending clinician on a case-by-case basis after thorough assessment of benefit versus risk. Table 4[23] provides guidelines for interruption and eventual restart of DrotAA in patients treated invasively.
Pediatric patients
DrotAA is not recommended for use in persons under the age of 18 years and should not therefore be used in these patients. From a safety point of view, in the RESOLVE (REsearching severe Sepsis and Organ dysfunction in children: a gLobal perspective) study [24], a double-blind, placebo-controlled trial conducted in children with severe sepsis, the investigators attributed two deaths in the DrotAA group and five in the placebo group to bleeding events. There was a higher rate of CNS bleeding in the DrotAA group than in the placebo group. Over the infusion period (study days 0 to 6), the number of patients experiencing CNS bleeding was 5 (2.1%) in the DrotAA group compared with 1 (0.4%) in the placebo group, with four of the five events in the DrotAA group occurring in children aged 60 days or younger, or with a body weight of 3.5 kg or less. Fatal CNS bleeding events, serious bleeding events (over the infusion period and over the 28-day study period), serious adverse events, and amputations were similar in the DrotAA and placebo groups.
Interaction with other drugs
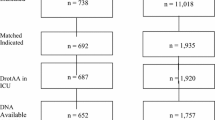
In the XPRESS trial [14] a total of 1,935 adult patients with severe sepsis treated with DrotAA were randomly assigned to receive either prophylactic heparin or placebo. Prophylactic heparin doses used in XPRESS were 10,000 U/day unfractionated heparin or 40 mg/day enoxaparin administered subcutaneously. The subgroup of patients whose baseline heparin was discontinued at randomization exhibited increased mortality and serious adverse events, including cardiac, gastrointestinal, and venous thrombotic events. Therefore, prophylactic heparin should not be discontinued before initiating treatment or during treatment with DrotAA, unless it is considered medically necessary for other reasons. Starting treatment with DrotAA does not in itself require cessation of prophylactic heparin.
In 2004, Mann and coworkers [25] reported an observation on the compatibility of DrotAA with 34 other very commonly used therapies when infused through a 'Y' site infusion device. Of the 34 test drugs, eight were defined as visually compatible with DrotAA.
Immunogenicity and re-administration
In patients with severe sepsis, the formation of anti-DrotAA antibodies was uncommon (<1%) after a single course of therapy. These antibodies were not capable of neutralizing the effect of activated protein C in the activated partial thromboplastin time assay. However, the possibility of allergic reactions to constituents of the preparation cannot be completely excluded in certain predisposed patients [8]. sick individuals, even after repeated administration. DrotAA re-administration is not contraindicated [8]; a few anecdotal reports describe repeated re-administration in a clinical setting [26].
Discussion
Clinical trials with DrotAA have now been performed for longer than 10 years and all have shown the same pattern in terms of safety. DrotAA increases the risk for bleeding without increasing general adverse events, thus resulting in similar rates of serious adverse events in treated arms compared with placebo.
Other types of adverse events (risk for superinfection, formation of blocking antibodies) were prospectively measured in randomized controlled trials and the results were consistent across trials. The only adverse event attributable to DrotAA is an increased risk for bleeding. Serious adverse bleeding events in randomized trials occurred at consistently low rates, as shown in Table 2.
The increase in the number of bleeding events compared with placebo can be explained given the mechanism of action of DrotAA. The risk for bleeding can be reduced by following simple guidelines and avoiding off-label use (use in contraindicated patients). It is important to note that the intracranial hemorrhage rate in all of the studies is in accordance with the rate of spontaneous intracranial hemorrhage in critically ill patients [27]. Moreover, some studies have suggested that the rate of intracranial hemorrhage may be much higher in patients with septic shock. In one study, autopsy examination revealed evidence of cerebral hemorrhage in six out of 23 septic shock patients (26%) [15].
In other settings, such as acute cardiac syndrome, the use of thrombolytic or antiplatelet compounds carry an inherent risk for bleeding that is even greater than that observed with DrotAA, but nevertheless this risk is considered acceptable.
It has been postulated that the anticoagulant effect of DrotAA may play a less important role in increasing survival rate in severe sepsis patients at high risk for death. Hence, development of a new form of the therapy with a reduced anticoagulant effect, while conserving its other properties, might enhance the benefit-risk ratio [28]. Nevertheless, when comparing the risk for bleeding in DrotAA-treated patients with the risk in those enrolled in trials of other natural anticoagulants, the benefit-risk ratio is greater in DrotAA patients [29, 30].
A recently published long-term follow up of ADDRESS [31] concluded that DrotAA exerts no harmful effects in patients with severe sepsis who are at low risk for death. Also, because of the drug's pharmacokinetic characteristics, any such effects would be limited to the infusion period, whereas its benefits persist in the population for which it is indicated.
Numerous registries have been reported to date. The conclusions of all of these studies recognize that DrotAA is the only compound to have demonstrated any effect on survival in septic patients and that there is no evidence that risk outweighs benefit when it used within the indicated population [32]. Efforts should be focused on education and adequate patient identification [21], especially in the setting of sepsis, where abnormal coagulation is the rule [33].
Conclusion
Although DrotAA increases the risk for bleeding, the safety profile remains acceptable for the indicated population after more than 10 years of clinical research. In other words, the benefit to patients at high risk for death is not outweighed by the adverse effects of DrotAA [34]. The increased risk for bleeding can be minimized by following simple guidelines and through appropriate patient selection.
Abbreviations
- CNS:
-
CNS = central nervous system
- DrotAA:
-
DrotAA = drotrecogin alfa (activated).
References
Marlar RA, Kleiss AJ, Griffin JH: Human protein C: inactivation of factors V and VIII in plasma by the activated molecule. Ann N Y Acad Sci 1981, 370: 303-310. 10.1111/j.1749-6632.1981.tb29743.x
Esmon CT: The anticoagulant and anti-inflammatory roles of the protein C anticoagulant pathway. J Autoimmun 2000, 15: 113-116. 10.1006/jaut.2000.0400
Macias WL, Yan SB, Williams MD, Um SL, Sandusky GE, Ballard DW, Planquois JMS: New insights into the protein C pathway: potential implications for the biological activities of drotrecogin alfa (activated). Crit Care 2005,9(Suppl 4):38-45. 10.1186/cc3747
Mosnier LO, Griffin JH: Inhibition of staurosporine-induced apoptosis of endothelial cells by activated protein C requires protease activated receptor-1 and endothelial cell protein C receptor. Biochem J 2003, 373: 65-70. 10.1042/BJ20030341
Mosnier LO, Zlokovic BV, Griffin JH: The cytoprotective protein C pathway. Blood 2007, 109: 3161-3172. 10.1182/blood-2006-09-003004
US Food and Drug Administration Anti-Infective Drugs Advisory Committee: Drotrecogin alfa (activated) [recombinant human activated protein C (rhAPC)]: Xigris, bla 125029/0. [http://www.fda.gov/ohrms/dockets/ac/01/briefing/3797b1.htm]
European Medicines Agency: Xigris: product information. [http://www.emea.europa.eu/humandocs/Humans/EPAR/xigris/Xigris.htm]
European Medicines Agency: Xigris: product information. [http://www.emea.eu.int/humandocs/PDFs/EPAR/xigris/H-396-PI-en.pdf]
Short MA, Schlichting D, Qualy R: From bench to bedside: a review of the clinical trial development of drotrecogin alfa (activated). Curr Med Res Opin 2006, 22: 2525-2540. 10.1185/030079906X154060
Bernard GR, Ely EE, Wright TJ, Fraiz J, Stasek JE Jr, Russell JA, Mayers I, Rosenfeld BA, Morris PE, Yan SB, on behalf of the rhAPC Sepsis Study Group, et al.: Safety and dose relationship of recombinant human activated protein C for coagulopathy in severe sepsis. Crit Care Med 2001, 29: 2051-2059. 10.1097/00003246-200111000-00003
Bernard GR, Vincent J-L, Laterre P-F, La Rosa SP, Dhainaut J-F, Lopez Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely W, for the Recombinant Human Activated Protein C Worldwide Evaluation in Severe Sepsis PROWESS) Study Group, et al.: Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 2001, 344: 699-709. 10.1056/NEJM200103083441001
Vincent J-L, Bernard GR, Beale R, Doig C, Putensen C, Dhainaut J-F, Artigas A, Fumagalli R, Macias WL, Wright T, for the ENHANCE Study Group, et al.: Drotrecogin alfa (activated) treatment in severe sepsis from the global open-label trial ENHANCE: further evidence for survival and safety and implications for early treatment. Crit Care Med 2005, 33: 2266-2277. 10.1097/01.CCM.0000181729.46010.83
Abraham E, Laterre P-F, Garg R, Levy H, Talwar D, Trzaskoma BL, François B, Guy JS, Brückmann M, Rea-Neto A, for the Administration of Drotrecogin Alfa (Activated) in Early Stage Severe Sepsis (ADDRESS) Study Group, et al.: Drotrecogin alfa (activated) for adults with severe sepsis and a low risk of death. N Engl J Med 2005, 353: 1332-1341. 10.1056/NEJMoa050935
Levi M, Levy M, Williams MD, Douglas I, Artigas A, Antonelli M, Wyncoll D, Janes J, Booth FV, Wang D, for the Xigris and Prophylactic HepaRin Evaluation in Severe Sepsis (XPRESS) Study Group, et al.: Prophylactic heparin in patients with severe sepsis treated with drotrecogin alfa (activated). Am J Respir Crit Care Med 2007, 176: 483-490. 10.1164/rccm.200612-1803OC
Sharshar T, Annane D, de la Grandmaison GL, Brouland JP, Hopkinson NS, Francoise G: The neuropathology of septic shock. Brain Pathol 2004, 14: 21-33.
Bernard GR, Macias WL, Joyce DE, Williams MD, Bailey J, Vincent J-L: Safety assessment of drotrecogin alfa (activated) in the treatment of adult patients with severe sepsis. Crit Care 2003, 7: 155-163. 10.1186/cc2167
Rowan K, Welch CA, North E, Harrison DA: Multicenter audit of the use of drotrecogin alfa (activated) in United Kingdom critical care units [abstract 375]. Intensive Care Med 2006,32(suppl 1):99.
Vincent JL, Laterre PF, Janes J, Nelson D, Haentjens T, Sartral M, Ermens M, Sundin D: Analysis of drotrecogin alfa (activated) use in Belgium: comparison to PROGRESS registry data [abstract 227]. Intensive Care Med 2005,31(suppl 1):62.
Kübler A, Mayzner-Zawadzka E, Durek G, Gaszyñski W, Karpel E, Mikaszewska Sokolewicz M, Majak P: Results of severe sepsis treatment program using recombinant human activated protein C in Poland. Med Sci Monit 2006, 12: CR107-CR112.
Bertolini G, Rossi C, Anghileri A, Livigni S, Addis A, Poole D: Use of drotrecogin alfa (activated) in Italian intensive care units: the results of a nationwide survey. Intensive Care Med 2007, 33: 426-434. 10.1007/s00134-007-0554-x
Kanji S, Perreault MM, Chant C, Williamson D, Burry L: Evaluating the use of Drotrecogin alfa (activated) in adult severe sepsis: a Canadian multicenter observational study. Intensive Care Med 2007, 33: 517-523. 10.1007/s00134-007-0555-9
Barie PS, Hydo LJ, Shou J, Eachempati SR: Efficacy and safety of drotrecogin alfa (activated) for the therapy of surgical patients with severe sepsis. Surg Infect (Larchmt) 2006,7(suppl 2):S77-S80.
Laterre P-F, Wittebole X: Clinical review: drotrecogin alfa (activated) as adjunctive therapy for severe sepsis – practical aspects at the bedside and patient identification. Crit Care 2003, 7: 445-450. 10.1186/cc2342
Nadel S, Goldstein B, Williams MD, Dalton H, Peters M, Macias WL, Abd-Allah SA, Levy H, Angle R, Wang D, for the REsearching severe Sepsis and Organ dysfunction in children, et al.: Drotrecogin alfa (activated) in children with severe sepsis: a multicentre phase III randomized controlled trial. Lancet 2007, 369: 836-843. 10.1016/S0140-6736(07)60411-5
Mann HJ, Demmon SL, Boelk DA, Payne CA, Effron MB, Rajagopalan N, Williams MD, Beck GM, Gopalrathnam G: Physical and chemical compatibility of drotrecogin alfa (activated) with 34 drugs during simulated Y-site administration. Am J Health Syst Pharm 2004, 61: 2664-2671.
Lee SJ, Schuster R, Bindewald M, Greaney G, Waxman K: Second course of recombinant human activated protein C delivered to a severely septic patient after recent surgery. Trauma 2006, 61: 1-3.
Oppenheim-Eden A, Glantz L, Eidelman LA, Sprung CL: Spontaneous intracerebral hemorrhage in critically ill patients: incidence over six years and associated factors. Intensive Care Med 1999, 25: 63-67. 10.1007/s001340050788
Vincent JL: Drotrecogin alfa (activated) in the treatment of severe sepsis. Expert Rev Anti Infect Ther 2006, 4: 537-547. 10.1586/14787210.4.4.537
Warren BL, Eid A, Singer P, Pillay SS, Carl P, Novak I, Chalupa P, Atherstone A, Pénzes I, Kübler A, for the KyberSept Trial Study Group, et al.: High-dose antithrombin III in severe sepsis. A randomized controlled trial. JAMA 2001, 286: 1869-1878. 10.1001/jama.286.15.1869
Abraham E, Reinhart K, Opal S, Demeyer I, Doig C, Lopez Rodriguez A, Beale R, Svoboda P, Laterre P-F, Simon S, for the OPTIMIST Trial Study Group, et al.: Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis. A randomized controlled trial. JAMA 2003, 290: 238-247. 10.1001/jama.290.2.238
Laterre PF, Abraham E, Janes J, Trzaskoma BL, Correll NL, Booth FV: ADDRESS (ADministration of Drotrecogin alfa [activated] in Early stage Severe Sepsis) long-term follow-up: one-year safety and efficacy evaluation. Crit Care Med 2007, 35: 1457-1463. 10.1097/01.CCM.0000266588.95733.63
Williams MD, Macias W, Rustige J: Safety of drotrecogin alfa (activated): a fair comparison requires consistent definitions. Intensive Care Med 2007, 33: 1487-1488. 10.1007/s00134-007-0709-9
Levi M, Opal SM: Coagulation abnormalities in critically ill patients. Crit Care 2006, 10: 222. 10.1186/cc4975
Plataki M, Vaporidi K, Georgopoulos D: Is there increasing evidence that the risks of rhAPC may outweigh its benefits? Intensive Care Med 2007, 33: 1485-1486. 10.1007/s00134-007-0707-y
Acknowledgements
This article is part of Critical Care Volume 11 Supplement 5: Severe sepsis and drotrecogin alfa (activated). The full contents of the supplement are available online at http://ccforum.com/supplements/11/S5. Publication of the supplement has been sponsored by Eli Lilly and Company.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
RF participated as Principal Investigator in Eli Lilly and Company sponsored trials, received speaker's fees from Eli Lilly and Company, and is member of their Advisory Board. He received no funds for writing this review. MAM is full-time employee of Eli Lilly and Company.
Authors' contributions
RF and MAM participated in the conception and design of the review, the acquisition of the data from the literature and the interpretation of the data, and drafted the manuscript. Both also participated in the analysis and interpretation of the data, critically revised the manuscript for important intellectual content, and gave final approval of the version to be published.
Rights and permissions
About this article
Cite this article
Fumagalli, R., Mignini, M.A. The safety profile of drotrecogin alfa (activated). Crit Care 11 (Suppl 5), S6 (2007). https://doi.org/10.1186/cc6157
Published:
DOI: https://doi.org/10.1186/cc6157