
Abstract
Impairment of the protein C pathway plays a central role in the pathogenesis of sepsis. Administration of recombinant human activated protein C (rhAPC) may correct the dysregulated anticoagulant mechanism and prevent propagation of thrombin generation and formation of microvascular thrombosis. Furthermore, it may simultaneously modulate the inflammatory response. It is likely that the beneficial effect of rhAPC observed in experimental and clinical studies of severe sepsis results from a combination of mechanisms that modulate the entangled processes of coagulation and inflammation. This review presents an analysis of the various mechanisms of action of rhAPC in sepsis.
Similar content being viewed by others

Introduction
In all patients with sepsis there is abundant activation of inflammatory pathways, which results in demonstrable circulating levels of inflammatory cytokines and chemokines, activated inflammatory cells and other markers of increased inflammatory activity. Virtually all septic patients exhibit coagulation abnormalities as well. These abnormalities range from subtle activation of coagulation, which can only be detected by sensitive markers of coagulation factor activation; to more marked activation, which may be detectable based on a small decrease in platelet count and subclinical prolongation of global clotting times; and finally to fulminant disseminated intravascular coagulation (DIC), which is characterized by simultaneous widespread microvascular thrombosis and profuse bleeding from various sites [1]. Septic patients with severe forms of DIC may present with thromboembolic disease or clinically less apparent microvascular failure that predominantly presents as multiple organ dysfunction [2, 3]. Interestingly, there is a tight, bidirectional relationship between activation of inflammation and coagulation, in which inflammatory activity results in activation of coagulation but activated coagulation proteases can also affect inflammatory pathways [4].
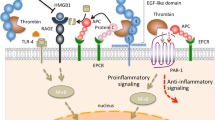
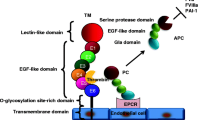
Activated protein C (APC) appears to play a central role in the pathogenesis of sepsis and associated organ dysfunction. There is ample evidence that insufficient functioning of the protein C pathway contributes to the derangement of coagulation observed in sepsis [5, 6]. The protein C system is summarized in Figure 1. The circulating zymogen protein C is activated by the endothelial cell bound thrombomodulin once this is activated by thrombin [7]. APC acts in concert with its co-factor, protein S, and can proteolytically degrade the cofactors Va and VIIIa, which are essential for coagulation; APC is therefore an effective anticoagulant. The endothelial protein C receptor (EPCR) not only accelerates activation of protein C several fold but it also serves as a receptor for APC, and binding of APC to EPCR may amplify its anticoagulant and anti-inflammatory effects [8]. A recent study [9] demonstrated that exposure of cultured endothelial cells to APC results in release of microparticles that contain EPCR, but the relevance of that observation to coagulation or inflammation is not yet clear.

The protein C system. The solid arrows indicate the mechanisms by which the protein C system is impaired in sepsis.
In patients with sepsis the APC system malfunctions at virtually all levels. First, plasma levels of the zymogen protein C are low or very low because of impaired synthesis, consumption and degradation by proteolytic enzymes such as neutrophil elastase [10–12]. Furthermore, significant downregulation of thrombomodulin caused by pro-inflammatory cytokines such as tumour necrosis factor-α and interleukin-1 has been demonstrated, resulting in diminished protein C activation [13, 14]. Low levels of free protein S may further compromise the functioning of the protein C system. In plasma, 60% of the co-factor protein S is complexed to the complement regulatory protein C4b-binding protein (C4bBP). Increased plasma levels of C4bBP, which occur as a consequence of the acute phase reaction in inflammatory disease, may result in relative protein S deficiency, which further contributes to a procoagulant state during sepsis. Although it has been shown that the β-chain of C4bBP (which mainly governs binding to protein S) is not much affected by the acute phase response [15], support for this hypothesis comes from studies conducted in baboons [16] in which infusion of C4bBP in combination with a sublethal dose of Escherichia coli resulted in a lethal response, with severe organ damage due to DIC. Finally (but importantly), in sepsis EPCR has been shown to be downregulated, which may further adversely affect the function of the protein C system [17]. Apart from these effects, sepsis may induce resistance to APC by other mechanisms that are partly dependent on a sharp increase in factor VIII levels (released from endothelial cells) and partly due to as yet unidentified mechanisms [18].
Ability of rhAPC to correct the defective protein C pathway and abnormal coagulation in sepsis
Administration of APC in a baboon model of intravenous E. coli administration resulted in survival of all animals, whereas all control animals in the same experiment died [19]. A similar beneficial effect was observed in rabbits with meningococcal endotoxin shock. In a rat model of septic shock, administration of APC prevented tumour necrosis factor-α mediated hypotension, probably caused by modulation of the nitric oxide response [20]. In patients with severe sepsis, administration of recombinant human activated protein C (rhAPC) resulted in remarkable improvement in microcirculatory perfusion [21]. Conversely, experiments conducted in baboons in which the protein C pathway was blocked with monoclonal antibodies resulted in complete lethality in an otherwise sublethal model of bacteraemia. In this same model, blockade of the EPCR also resulted in a more severe response to sublethal E. coli bacteraemia [17]. In addition to these observations in experimental sepsis models, APC was shown to have antithrombotic properties in experimental thrombosis models in dogs, rabbits and baboons [22, 23]. Interestingly, APC may also affect fibrinolysis by inhibiting plasminogen activator inhibitor type 1 (a fibrinolytic inhibitor). In a rat model of DIC, APC was shown to block activity of plasminogen activator inhibitor type 1, and other experiments demonstrated the ability of APC to enhance clot lysis in vivo [24].
More definitive proof of the beneficial effect of rhAPC in severe sepsis comes from clinical studies [25, 26]. These studies are reviewed in detail in other reviews included in this supplement [27–29] and are not discussed in detail here. However, it may be of interest to review the evidence that rhAPC acts as an anticoagulant agent in these studies.
First, it should be noted that the selected dose of rhAPC was based on the effect on D-dimer levels in a phase II clinical trial [25]. Indeed, in the pivotal PROWESS (Recombinant Human Activated Protein C Worldwide Evaluation in Severe Sepsis) trial [26], septic patients who were treated with APC exhibited a significant decrease in D-dimer levels as compared with placebo control individuals. D-dimer levels dropped by 25% after 2 days of APC administration, which was in contrast to a 10% increase in placebo-treated patients. In a more detailed analysis of coagulation activation upon administration of rhAPC, it was clearly demonstrated that markers of thrombin generation sharply dropped almost immediately after initiation of the APC infusion [30]. Second, subgroup analyses of trials including patients with severe sepsis [31, 32] demonstrated that patients with the most extreme coagulation abnormalities benefit the most from treatment with rhAPC. The relative risk reduction in mortality among patients with sepsis and DIC who received APC was 38%, as compared with a relative risk reduction of 18% observed among patients with sepsis who did not have DIC. Interestingly, the dynamics of coagulation abnormalities in the first days after intensive care unit admission for severe sepsis, including the response of the protein C system, is a strong predictor of outcome [33].
It is difficult to assess whether this anticoagulant effect of rhAPC translates into an antithrombotic effect. In the recently concluded XPRESS (Xigris [drotrecogin alfa] and Prophylactic Heparin in Severe Sepsis) study [34], all patients with severe sepsis received rhAPC but were also randomly assigned to receive prophylactic heparin or placebo. The main conclusion of this study was that heparin was not equivalent to placebo and might have a beneficial effect on 28-day mortality. Of note, this advantageous effect of heparin was completely due to a greater incidence of death and thrombotic adverse events in the patients receiving heparin but who were randomly assigned to placebo (in other words, those who stopped heparin during the trial). There was a markedly low incidence of venous thromboembolism in this study compared with previous reports (the incidence did not differ between placebo patients and patients receiving heparin), even though all patients underwent screening ultrasound for venous thrombosis at around day 6 of admission. In addition, a report of small series of patients with severe sepsis who were treated with rhAPC [35] demonstrated a lack of thrombotic obstruction of haemofiltration circuits, even in the absence of heparin or other anticoagulants.
Apart from the systemic response, there may be a differential localized effect of rhAPC on coagulation. The localized effect of APC appears to be particularly marked in the pulmonary compartment. In experiments involving unilateral instillation of endotoxin into healthy individuals, systemic administration of rhAPC resulted in a marked reduction in bronchoalveolar activation of coagulation [36]. This observation may be relevant because the vast majority of patients with severe sepsis in the various trials had a pulmonary source of infection. An interesting novel finding is the modulatory influence of alveolar epithelial cells on the protein C pathway [37]. It is likely that the underlying mechanism involves shedding of EPCR and thrombomodulin by metalloproteinases.
APC as an inflammatory mediator in sepsis
Evidence that APC acts as an important mediator in the systemic inflammatory response in sepsis comes from experiments showing that blocking the protein C pathway in septic baboons exacerbated the inflammatory response [38]. In contrast, administration of APC ameliorated the inflammatory activation that occurred upon intravenous infusion of E. coli [38]. Similar experiments in rodents yielded identical results and demonstrated a beneficial effect on inflammatory effects in various tissues [39]. Support for the notion that APC has anti-inflammatory properties comes from in vitro findings, demonstrating an APC-binding site on monocytes that may mediate downstream inflammatory processes [40, 41]. It also received support from experiments showing that APC can block nuclear factor-κB nuclear translocation, which is a prerequisite for increased levels of pro-inflammatory cytokines and adhesion molecules [42]. These in vitro findings are supported by in vivo studies in mice with targeted disruption of the protein C gene [43, 44]. In these mice with genetic deficiencies of protein C, endotoxaemia was associated with more marked increases in pro-inflammatory cytokines and other inflammatory responses than in wild-type mice.
It is likely that the effects of APC on inflammation are mediated by EPCR, which may mediate downstream inflammatory processes [45]. Binding of APC to EPCR influences gene expression profiles of cells by inhibiting endotoxin-induced calcium fluxes in the cell and by blocking nuclear factor-κB nuclear translocation [41, 42]. The EPCR-APC complex itself can translocate from the plasma membrane into the cell nucleus, which may be another mechanism of modulation of gene expression, although the relative contributions of this nuclear translocation and cell surface signalling are unclear [5]. Some studies have also suggested that EPCR binding of APC can result in activation of protease activated receptor (PAR)-1 and thereby affect cytokine responses [46]. In contrast, other experiments demonstrated that a significant physiological role for activation of PAR-1 by APC to be less probable [47]. Like APC, EPCR itself may have anti-inflammatory properties. Soluble EPCR (the extracellular domain of the cell-associated EPCR shed from the cell surface by the action of an inducible metalloproteinase [48]) can bind to proteinase 3, which is an elastase-like enzyme. The resulting complex binds to the adhesion integrin macrophage 1 antigen (Mac-1) [49]. Of considerable interest is that the crystal structure of EPCR is remarkably similar to the structure of the MHC class 1/CD1 family of proteins, the majority of which are involved in inflammation [50]. Blocking the EPCR with a specific monoclonal antibody aggravated both the coagulation and the inflammatory response to E. coli infusion [17].
Apart from the influence of APC on cytokine levels, remarkable effects of the agent on leucocyte chemotaxis and adhesion of circulating leucocytes to the activated endothelium have been demonstrated [51, 52]. This was confirmed in a hamster endotoxaemia model at concentrations of rhAPC that preclude a significant anticoagulant effect [53]. The localized effect of APC in the lung has also been shown to exhibit these anti-inflammatory properties [54]. APC was shown to inhibit the expression of platelet-derived growth factor in the lung [55], which may reflect a potential mechanism underlying this localized effect. Also, APC was shown to protect against disruption of the endothelial cell barrier in sepsis, probably by interfering with EPCR and PAR-1 on endothelial cells [56–58].
Finally, APC can inhibit endothelial cell apoptosis, which also appears to be mediated by binding of APC to EPCR and to require PAR-1 [46, 59]. Signalling through this pathway can affect Bcl-2 homologue protein, which can inhibit apoptosis, and further suppresses p53, which is a pro-apoptotic transcription factor [60, 61].
Conclusion
Inadequate functioning of the protein C system, and in particular APC, plays a central role in the pathogenesis of sepsis. Attempting to restore the function of this pathway in patients with sepsis appears to be a rational approach and is supported by the beneficial effect of APC in experimental models of sepsis and in clinical studies. Apart from its evident effect on the coagulation system and the ability of rhAPC to correct the deranged coagulation system in severe sepsis, a series of pleiotropic modulating effects of APC on inflammatory cytokines and cells as well as protective effects on disrupted endothelium have been reported. It should be noted that many of the effects on inflammatory cells and pathways, as well as the cytoprotective effect, have mostly been demonstrated in vitro and sometimes at inordinately high concentrations of APC. The relevance of these findings to the human in vivo situation and their importance to the treatment of sepsis remain to be established. Although the relative importance of the anticoagulant effect versus the inflammation-modulating effect of rhAPC is not clear, it is tempting to hypothesize that a combined effect is responsible for the benefit from rhAPC. Indeed, strategies aimed at restoring physiological pathways with less marked effects on inflammation (such as administration of antithrombin or recombinant tissue factor pathway inhibitor) were less successful. Further insight into the various mechanisms of action of rhAPC on the entangled processes of inflammation and coagulation may permit more detailed dissection of the relative importance of the various pathways that contribute to the pathogenesis of sepsis, potentially culminating in improved treatment strategies.
Abbreviations
- APC:
-
= activated protein C
- C4bBP:
-
= C4b-binding protein
- DIC:
-
= disseminated intravascular coagulation
- EPCR:
-
= endothelial protein C receptor
- PAR:
-
= protease activated receptor
- rhAPC:
-
= recombinant human activated protein C.
References
Levi M, ten Cate H: Disseminated intravascular coagulation. N Engl J Med. 1999, 341: 586-10.1056/NEJM199908193410807.
Levi M, Marder VJ: Coagulation abnormalities in sepsis. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. Edited by: Colman RW, Marder VJ, Clowes AW, George JN, Goldhaber SZ. 2006, Philadelphia, PA: Lippincott, Williams and Wilkins, 1601-1613.
Wheeler AP, Bernard GR: Treating patients with severe sepsis. N Engl J Med. 1999, 340: 207-214. 10.1056/NEJM199901213400307.
Levi M, van der Poll T, Buller HR: Bidirectional relation between inflammation and coagulation. Circulation. 2004, 109: 2698-2704. 10.1161/01.CIR.0000131660.51520.9A.
Esmon CT: Role of coagulation inhibitors in inflammation. Thromb Haemost. 2001, 86: 51-56.
Levi M, de Jonge E, van der Poll T: Rationale for restoration of physiological anticoagulant pathways in patients with sepsis and disseminated intravascular coagulation. Crit Care Med. 2001, 29 (Suppl): S90-S94. 10.1097/00003246-200107001-00028.
Esmon CT: The regulation of natural anticoagulant pathways. Science. 1987, 235: 1348-1352. 10.1126/science.3029867.
Esmon CT: The endothelial cell protein C receptor. Thromb Haemost. 2000, 83: 639-643.
Perez-Casal M, Downey C, Fukudome K, Marx G, Toh CH: Activated protein C induces the release of microparticle-associated endothelial protein C receptor. Blood. 2005, 105: 1515-1522. 10.1182/blood-2004-05-1896.
Mesters RM, Helterbrand J, Utterback BG, Yan B, Chao YB, Fernandez JA, Griffin JH, Hartman DL: Prognostic value of protein C concentrations in neutropenic patients at high risk of severe septic complications. Crit Care Med. 2000, 28: 2209-2216. 10.1097/00003246-200007000-00005.
Vary TC, Kimball SR: Regulation of hepatic protein synthesis in chronic inflammation and sepsis. Am J Physiol. 1992, 262: C445-452.
Eckle I, Seitz R, Egbring R, Kolb G, Havemann K: Protein C degradation in vitro by neutrophil elastase. Biol Chem Hoppe Seyler. 1991, 372: 1007-1013.
Nawroth PP, Stern DM: Modulation of endothelial cell hemostatic properties by tumor necrosis factor. J Exp Med. 1986, 163: 740-745. 10.1084/jem.163.3.740.
Faust SN, Levin M, Harrison OB, Goldin RD, Lockhart MS, Kondaveeti S, Laszik Z, Esmon CT, Heyderman RS: Dysfunction of endothelial protein C activation in severe meningococcal sepsis. N Engl J Med. 2001, 345: 408-416. 10.1056/NEJM200108093450603.
Garcia de Frutos P, Alim RI, Hardig Y, Zoller B, Dahlback B: Differential regulation of alpha and beta chains of C4b-binding protein during acute-phase response resulting in stable plasma levels of free anticoagulant protein S. Blood. 1994, 84: 815-822.
Taylor FBJ, Dahlback B, Chang AC, Lockhart MS, Hatanaka K, Peer G, Esmon CT: Role of free protein S and C4b binding protein in regulating the coagulant response to Escherichia coli. Blood. 1995, 86: 2642-
Taylor FBJ, Stearns-Kurosawa DJ, Kurosawa S, Ferrell G, Chang AC, Laszik Z, Kosanke S, Peer G, Esmon CT: The endothelial cell protein C receptor aids in host defense against Escherichia coli sepsis. Blood. 2000, 95: 1680-1686.
De Pont AC, Bakhtiari K, Hutten BA, de Jonge E, Vlasuk GP, Rote WE, Levi M, Buller HR, Meijers JC: Endotoxaemia induces resistance to activated protein C in healthy humans. Br J Haematol. 2006, 134: 213-219. 10.1111/j.1365-2141.2006.06127.x.
Taylor FBJ, Chang A, Esmon CT, D'Angelo A, Vigano-D'Angelo S, Blick KE: Protein C prevents the coagulopathic and lethal effects of Escherichia coli infusion in the baboon. J Clin Invest. 1987, 79: 918-925. 10.1172/JCI112902.
Isobe H, Okajima K, Uchiba M, Mizutani A, Harada N, Nagasaki A, Okabe K: Activated protein C prevents endotoxin-induced hypotension in rats by inhibiting excessive production of nitric oxide. Circulation. 2001, 104: 1171-1175. 10.1161/hc3501.093799.
De Backer D, Verdant C, Chierego M, Koch M, Gullo A, Vincent JL: Effects of drotrecogin alfa activated on microcirculatory alterations in patients with severe sepsis. Crit Care Med. 2006, 34: 1918-1924. 10.1097/01.CCM.0000220498.48773.3C.
Gruber A, Harker LA, Hanson SR, Kelly AB, Griffin JH: Antithrombotic effects of combining activated protein C and urokinase in nonhuman primates. Circulation. 1991, 84: 2454-2462.
Gruber A, Marzec UM, Bush L, Di Cera E, Fernandez JA, Berny MA, Tucker EI, McCarty OJ, Griffin JH, Hanson SR: Relative antithrombotic and antihemostatic effects of protein C activator versus low molecular weight heparin in primates. Blood. 2007, 109: 3733-3740. 10.1182/blood-2006-07-035147.
Aoki Y, Ota M, Katsuura Y, Komoriya K, Nakagaki T: Effect of activated human protein C on disseminated intravascular coagulation induced by lipopolysaccharide in rats. Arzneimittelforschung. 2000, 50: 809-815.
Bernard GR, Ely EW, Wright TJ, Fraiz J, Stasek JE, Russell JA, Mayers I, Rosenfeld BA, Morris PE, Yan SB, et al: Safety and dose relationship of recombinant human activated protein C for coagulopathy in severe sepsis. Crit Care Med. 2001, 29: 2051-2059. 10.1097/00003246-200111000-00003.
Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, et al: Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001, 344: 699-709. 10.1056/NEJM200103083441001.
Vangerow B, Shorr AF, Wyncoll D, Janes J, Nelson DR, Reinhart K: The protein C pathway: implications for the design of the RESPOND study. Crit Care. 2007, 11 (Suppl 5): S4-10.1186/cc6155.
Laterre P-F: Clinical trials in severe sepsis with drotrecogin alfa (activated). Crit Care. 2007, 11 (Suppl 5): S5-10.1186/cc6156.
Fumagalli R, Mignini MA: The safety profile of drotrecogin alfa (activated). Crit Care. 2007, 11 (Suppl 5): S6-10.1186/cc6157.
De Pont AC, Bakhtiari K, Hutten BA, de Jonge E, Vroom MB, Meijers JC, Buller HR, Levi M: Recombinant human activated protein C resets thrombin generation in patients with severe sepsis: a case control study. Crit Care. 2005, 9: R490-497. 10.1186/cc3774.
Dhainaut JF, Yan SB, Joyce DE, Pettila V, Basson BR, Brandt JT, Sundin D, Levi M: Treatment effects of drotrecogin alfa (activated) in patients with severe sepsis with or without overt disseminated intravascular coagulation. J Thromb Haemost. 2004, 2: 1924-1933. 10.1111/j.1538-7836.2004.00955.x.
Ely EW, Laterre PF, Angus DC, Helterbrand JD, Levy H, Dhainaut JF, Vincent JL, Macias WL, Bernard GR: Drotrecogin alfa (activated) administration across clinically important subgroups of patients with severe sepsis. Crit Care Med. 2003, 31: 12-19. 10.1097/00003246-200301000-00002.
Dhainaut JF, Shorr AF, Macias WL, Kollef MJ, Levi M, Reinhart K, Nelson DR: Dynamic evolution of coagulopathy in the first day of severe sepsis: Relationship with mortality and organ failure. Crit Care Med. 2005, 33: 341-348. 10.1097/01.CCM.0000153520.31562.48.
Levi M, Levy M, Williams MD, Douglas I, Artigas A, Antonelli M, Wyncoll D, Janes J, Booth FV, Wang D, et al: Prophylactic heparin in patients with severe sepsis treated with drotrecogin alfa (activated). Am J Respir Crit Care Med. 2007, 176: 483-490. 10.1164/rccm.200612-1803OC.
De Pont AC, Bouman CS, de Jonge E, Vroom MB, Buller HR, Levi M: Treatment with recombinant human activated protein C obviates additional anticoagulation during continuous venovenous hemofiltration in patients with severe sepsis. Intensive Care Med. 2003, 29: 1205-10.1007/s00134-003-1781-4.
van der Poll T, Levi M, Nick JA, Abraham E: Activated protein C inhibits local coagulation after intrapulmonary delivery of endotoxin in humans. Am J Respir Crit Care Med. 2005, 171: 1125-1128. 10.1164/rccm.200411-1483OC.
Wang L, Bastarache JA, Wickersham N, Fang X, Matthay MA, Ware LB: Novel role of the human alveolar epithelium in regulating intra-alveolar coagulation. Am J Respir Cell Mol Biol. 2007, 36: 497-503. 10.1165/rcmb.2005-0425OC.
Taylor FBJ, Chang A, Esmon CT, D'Angelo A, Vigano-D'Angelo S, Blick KE: Protein C prevents the coagulopathic and lethal effects of Escherichia coli infusion in the baboon. J Clin Invest. 1987, 79: 918-925. 10.1172/JCI112902.
Murakami K, Okajima K, Uchiba M, Johno M, Nakagaki T, Okabe H, Takatsuki K: Activated protein C attenuates endotoxin-induced pulmonary vascular injury by inhibiting activated leukocytes in rats. Blood. 1996, 87: 642-647.
Hancock WW, Tsuchida A, Hau H, Thomson NM, Salem HH: The anticoagulants protein C and protein S display potent antiin-flammatory and immunosuppressive effects relevant to transplant biology and therapy. Transplant Proc. 1992, 24: 2302-2303.
Hancock WW, Grey ST, Hau L, Akalin E, Orthner C, Sayegh MH, Salem HH: Binding of activated protein C to a specific receptor on human mononuclear phagocytes inhibits intracellular calcium signaling and monocyte-dependent proliferative responses. Transplantation. 1995, 60: 1525-1532.
White B, Schmidt M, Murphy C, Livingstone W, O'Toole D, Lawler M, O'Neill L, Kelleher D, Schwarz HP, Smith OP: Activated protein C inhibits lipopolysaccharide-induced nuclear translocation of nuclear factor kappaB (NF-kappaB) and tumour necrosis factor alpha (TNF-alpha) production in the THP-1 monocytic cell line. Br J Haematol. 2000, 110: 130-134. 10.1046/j.1365-2141.2000.02128.x.
Levi M, Dorffler-Melly J, Reitsma PH, Buller HR, Florquin S, van der Poll T, Carmeliet P: Aggravation of endotoxin-induced disseminated intravascular coagulation and cytokine activation in heterozygous protein C deficient mice. Blood. 2003, 101: 4823-4827. 10.1182/blood-2002-10-3254.
Lay AJ, Donahue D, Tsai MJ, Castellino FJ: Acute inflammation is exacerbated in mice genetically predisposed to a severe protein C deficiency. Blood. 2007, 109: 1984-1991. 10.1182/blood-2006-07-037945.
Esmon CT: New mechanisms for vascular control of inflammation mediated by natural anticoagulant proteins. J Exp Med. 2002, 196: 561-564. 10.1084/jem.20021088.
Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W: Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002, 296: 1880-1882. 10.1126/science.1071699.
Ludeman MJ, Kataoka H, Srinivasan Y, Esmon NL, Esmon CT, Coughlin SR: PAR1 cleavage and signaling in response to activated protein C and thrombin. J Biol Chem. 2005, 280: 13122-13128. 10.1074/jbc.M410381200.
Xu J, Qu D, Esmon NL, Esmon CT: Metalloproteolytic release of endothelial cell protein C receptor. J Biol Chem. 2000, 275: 6038-6044. 10.1074/jbc.275.8.6038.
Kurosawa S, Esmon CT, Stearns-Kurosawa DJ: The soluble endothelial protein C receptor binds to activated neutrophils: involvement of proteinase-3 and CD11b/CD18. J Immunol. 2000, 165: 4697-4703.
Oganesyan V, Oganesyan N, Terzyan S, Qu D, Dauter Z, Esmon NL, Esmon CT: The crystal structure of the endothelial protein C receptor and a bound phospholipid. J Biol Chem. 2002, 277: 24851-24854. 10.1074/jbc.C200163200.
Feistritzer C, Sturn DH, Kaneider NC, Djanani A, Wiedermann CJ: Endothelial protein C receptor-dependent inhibition of human eosinophil chemotaxis by protein C. J Allergy Clin Immunol. 2003, 112: 375-381. 10.1067/mai.2003.1609.
Sturn DH, Kaneider NC, Feistritzer C, Djanani A, Fukudome K, Wiedermann CJ: Expression and function of the endothelial protein C receptor in human neutrophils. Blood. 2003, 102: 1499-1505. 10.1182/blood-2002-12-3880.
Hoffmann JN, Vollmar B, Laschke MW, Inthorn D, Fertmann J, Schildberg FW, Menger MD: Microhemodynamic and cellular mechanisms of activated protein C action during endotoxemia. Crit Care Med. 2004, 32: 1011-1017. 10.1097/01.CCM.0000120058.88975.42.
Nick JA, Coldren CD, Geraci MW, Poch KR, Fouty BW, O'Brien J, Gruber M, Zarini S, Murphy RC, Kuhn K, et al: Recombinant human activated protein C reduces human endotoxin-induced pulmonary inflammation via inhibition of neutrophil chemotaxis. Blood. 2004, 104: 3878-3885. 10.1182/blood-2004-06-2140.
Shimizu S, Gabazza EC, Taguchi O, Yasui H, Taguchi Y, Hayashi T, Ido M, Shimizu T, Nakagaki T, Kobayashi H, et al: Activated protein C inhibits the expression of platelet-derived growth factor in the lung. Am J Respir Crit Care Med. 2003, 167: 1416-1426. 10.1164/rccm.200206-515OC.
Zeng W, Matter WF, Yan SB, Um SL, Vlahos CJ, Liu L: Effect of drotrecogin alfa (activated) on human endothelial cell permeability and Rho kinase signaling. Crit Care Med. 2004, 32: S302-308. 10.1097/01.CCM.0000128038.49201.8C.
Feistritzer C, Riewald M: Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood. 2005, 105: 3178-3184. 10.1182/blood-2004-10-3985.
Finigan JH, Dudek SM, Singleton PA, Chiang ET, Jacobson JR, Camp SM, Ye SQ, Garcia JG: Activated protein C mediates novel lung endothelial barrier enhancement: role of sphingosine 1-phosphate receptor transactivation. J Biol Chem. 2005, 280: 17286-17293. 10.1074/jbc.M412427200.
Cheng T, Liu D, Griffin JH, Fernandez JA, Castellino F, Rosen ED, Fukudome K, Zlokovic BV: Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med. 2003, 9: 338-342. 10.1038/nm826.
Mosnier LO, Griffin JH: Inhibition of staurosporine-induced apoptosis of endothelial cells by activated protein C requires protease activated receptor-1 and endothelial cell protein C receptor. Biochem J. 2003, 373: 65-70. 10.1042/BJ20030341.
Mosnier LO, Zlokovic BV, Griffin JH: The cytoprotective protein C pathway. Blood. 2007, 109: 3161-3172. 10.1182/blood-2006-09-003004.
Acknowledgements
This article is part of Critical Care Volume 11 Supplement 5: Severe sepsis and drotrecogin alfa (activated). The full contents of the supplement are available online at http://ccforum.com/supplements/11/S5. Publication of the supplement has been sponsored by Eli Lilly and Company.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
ML and TvdP have participated in advisory boards of Eli Lilly and Company and have participated as investigators in experimental and clinical studies with rhAPC.
Rights and permissions
About this article
Cite this article
Levi, M., van der Poll, T. Recombinant human activated protein C: current insights into its mechanism of action . Crit Care 11 (Suppl 5), S3 (2007). https://doi.org/10.1186/cc6154
Published:
DOI: https://doi.org/10.1186/cc6154