
Abstract
A progressive rise of oxidative stress due to altered reduction–oxidation (redox) homeostasis appears to be one of the hallmarks of the processes that regulate gene transcription in physiology and pathophysiology. Reactive oxygen species and reactive nitrogen species serve as signaling messengers for the evolution and perpetuation of the inflammatory process that is often associated with the condition of oxidative stress, which involves genetic regulation. Changes in the pattern of gene expression through reactive oxygen species/reactive nitrogen species-sensitive regulatory transcription factors are crucial components of the machinery that determines cellular responses to oxidative/redox conditions. The present review describes the basic components of the intracellular oxidative/redox control machinery and its crucial regulation of oxygen-sensitive and redox-sensitive transcription factors within the context of lung injury. Particularly, the review discusses mechanical ventilation and NF-κB-mediated lung injury, ischemia-reperfusion and transplantation, compromised host defense and inflammatory stimuli, and hypoxemia and the crucial role of hypoxia-inducible factor in mediating lung injury. Changes in the pattern of gene expression through regulatory transcription factors are therefore crucial components of the machinery that determines cellular responses to oxidative/redox stress.
Similar content being viewed by others


Introduction
Altering gene expression is the most fundamental and effective way for a cell to respond to extracellular signals and/or changes in its environment, in both the short term and the long term [1]. In the short term, transcription factors are involved in mediating responses to growth factors and a variety of other extracellular signals [2]. In contrast, the long-term control of gene expression induced by growth factors and the changes in gene expression, which occur during development, is generally (with few exceptions) irreversible.
During development, the expression of specific sets of genes is regulated spatially (by position/morphogenetic gradients) and temporally. Regulation of the signaling responses is governed at the genetic level by transcription factors that bind to control regions of target genes and alter their expression [1,2]. Transcription factors are endogenous substances, usually proteins, that are effective in the initiation, stimulation or termination of the genetic transcription process. While in the cytoplasm, the transcription factor is incapable of promoting transcription. A signaling event occurs, such as a change of the state of phosphorylation, which results in protein subunit translocation into the nucleus [3,4]. Transcription is a process in which one DNA strand is used as a template to synthesize a complementary RNA. Signal transduction therefore involves complex interactions of multiple cellular pathways [1,2].
In particular, reduction–oxidation/oxygen (redox)-sensitive transcription factors have gained an overwhelming backlog of interest momentum over the years, ever since the onset of the burgeoning field of free radical research and oxidative stress. The reason for this is that redox-sensitive transcription factors are often associated with the development and progression of many human disease states. Their ultimate regulation therefore bears potential therapeutic intervention for possible clinical applications [1,2,3,4].
In the present review, I will focus on elaborating a comprehensive overview of the current understanding of redox/oxidative mechanisms mediating the regulation of transcription factors. These transcription factors regulate a plethora of cellular functions that span the range from anoxia and hypoxia to oxidative stress within the context of oxidant-mediated lung injury.
Inflammatory reactions and lung injury
Mechanical ventilation and NF-κB-mediated lung injury
Some unprecedented conditions may occur during the evolution of the inflammatory process, which can eventually lead to dramatic changes in the progression of lung injury. For example, positive-pressure mechanical ventilation supports gas exchange in patients with respiratory failure but is also responsible for significant lung injury.
Pugin and colleagues, for instance, have developed an in vitro model in which isolated lung cells can be submitted to a prolonged cyclic pressure-stretching strain resembling that of conventional mechanical ventilation [5]. In this model, cells cultured on a silastic membrane were elongated up to 7% of their initial diameter, corresponding to a 12% increase in cell surface. The lung alveolar macrophage (AM) was identified as the main cellular source for critical inflammatory mediators such as tumor necrosis factor (TNF)-α, the chemokines IL-8 and IL-6, and matrix metalloproteinase-9 in this model system of mechanical ventilation. These mediators were measured in supernatants from ventilated AMs, monocyte-derived macrophages and promonocytic THP-1 cells. In addition, NF-κB was found to be activated in ventilated macrophages. Synergistic proinflammatory effects of mechanical stress and molecules such as bacterial endotoxin were observed, suggesting that mechanical ventilation might be particularly deleterious in pre-injured or infected lungs. Dexamethasone, an anti-inflammatory steroid, prevented IL-8 and TNF-α secretion in ventilated macrophages. Mechanical ventilation also induced low levels of IL-8 secretion by alveolar type II-like cells. Other lung cell types such as endothelial cells, bronchial cells and fibroblasts failed to produce IL-8 in response to a prolonged cyclic pressure-stretching load [5]. This model is of particular value for exploring physical stress-induced signaling pathways, as well as for testing the effects of novel ventilatory strategies or adjunctive substances aimed at modulating cell activation induced by mechanical ventilation.
Furthermore, alterations in AM function during sepsis-induced hypoxia may influence TNF secretion and the progression of acute lung injury. It was proposed that acute changes in partial pressure of oxygen (p O2) tension surrounding AMs alter NF-κB activation and TNF secretion in these lung cells. AM-derived TNF-α secretion and NF-κB expression were determined after acute hypoxic exposure of isolated Sprague–Dawley rat AMs. Adhered AMs (106/ml) were incubated (37°C at 5% CO2) for 2 hours with 1 μg/ml lipopolysaccharide–endotoxin (Pseudomonas aeruginosa) in normoxia (21% O2–5% CO2) or in hypoxia (1.8% O2–5% CO2). The AMs exposed to lipopolysaccharide–endotoxin in hypoxia had higher levels of TNF-α and enhanced expression of NF-κB than those in normoxia; the predominant isoforms were Rel A (p65) and c-Rel (p75). Increased mRNA bands for TNF-α, IL-1α and IL-1β were also observed in the hypoxic AMs [6]. This observation demonstrates that acute hypoxia in the lung may induce enhanced NF-κB activation in AMs, which may result in increased production and release of inflammatory cytokines.
Ischemia-reperfusion and transplantation
It has been reported that secretory leukocyte protease inhibitor (SLPI) in mice regulates local and remote organ inflammatory injury induced by hepatic ischemia-reperfusion [7,8,9]. Intravenous infusion of SLPI reduced liver and lung damage and diminished neutrophil accumulation in both organs. These effects were accompanied by reduced serum levels of TNF-α and macrophage inflammatory protein-2. SLPI also suppressed activation of NF-κB in the liver. Moreover, hepatic ischemia and reperfusion caused increased expression of SLPI mRNA and SLPI protein, which was found specifically in hepatocytes. Furthermore, treatment of mice with anti-SLPI antibodies enhanced serum levels of TNF-α and macrophage inflammatory protein-2, and it increased hepatic neutrophil accumulation and the amount of liver injury and lung injury [7,8,9,10,11,12,13]. These data indicate that SLPI has protective effects against hepatic ischemia-reperfusion injury and suggest that endogenous SLPI regulates the hepatic and remote inflammatory responses.
In concert with these observations, attenuation of lung reperfusion injury after transplantation using an inhibitor of NF-κB was achieved [14]. It was hypothesized that NF-κB is a critical early regulator of the inflammatory response in lung ischemia-reperfusion injury and that inhibition of NF-κB activation reduces this injury and improves pulmonary graft function. With the use of a porcine transplantation model, left lungs were harvested and stored in cold Euro-Collins preservation solution for 6 hours before transplantation [14]. Activation of NF-κB occurred 30 min and 1 hour after transplantation, and it declined to near baseline levels after 4 hours. Pyrrolidine dithiocarbamate, a potent inhibitor of NF-κB, given to the lung graft during organ preservation (40 mmol/l), effectively inhibited NF-κB activation and significantly improved lung function. Compared with control lungs 4 hours after transplant, pyrrolidine dithiocarbamate-treated lungs displayed significantly higher oxygenation, lower p CO2, reduced mean pulmonary arterial pressure and reduced edema and cellular infiltration [14]. This demonstrates that NF-κB is rapidly activated and is associated with poor pulmonary graft function in transplant reperfusion injury. Targeting the NF-κB pathway may therefore be a promising therapy to reduce injury and to improve lung function.
Compromised host defense
Progressive pulmonary infection may be a prominent clinical feature of lung injury, but the molecular basis for this susceptibility remains incompletely understood.
To study this problem, Sajjan et al. developed a model of chronic pneumonia by repeated instillation of a clinical isolate of Burkholderia cepacia, an opportunistic Gram-negative bacterium, from a case of cystic fibrosis (CF) into the lungs of Cftr (m1unc-/- [Cftr-/-]) and congenic Cftr+/+ controls [15]. Nine days after the last instillation, the CF transmembrane regulator knockout mice showed persistence of viable bacteria with chronic severe bronchopneumonia, while wild-type mice remained healthy. A mixed population of macrophages and neutrophils characterized the histopathological changes in the lungs of the susceptible Cftr-/- mice by infiltration of a mixed inflammatory cell population into the peribronchiolar and perivascular spaces, by Clara cell hyperplasia, by mucus hypersecretion in the airways and by exudation into alveolar airspaces. An increased proportion of neutrophils was observed in the bronchoalveolar lavage fluid from the Cftr-/- mice that, despite an increased bacterial load, demonstrated minimal evidence of activation. In addition, alveolar macrophages from Cftr-/- mice also demonstrated suboptimal activation [15].
These observations suggest that the pulmonary host defenses are compromised in lungs from animals with CF, as manifested by increased susceptibility to bacterial infection and lung injury. This murine model of chronic pneumonia thus reflects, in part, the situation in human patients and may help to elucidate the mechanisms leading to defective host defense in CF [16,17,18,19,20,21,22,23,24,25].
Summary
Acute lung injury therefore occurs as a result of a cascade of cellular events initiated by either infectious or noninfectious inflammatory stimuli. An elevated level of proinflammatory mediators combined with a decreased expression of anti-inflammatory molecules is a critical component of lung inflammation.
Expression of proinflammatory genes is regulated by transcriptional mechanisms. NF-κB is one critical transcription factor required for the expression of many cytokines involved in the pathogenesis of acute lung injury [26,27,28,29,30,31,32,33,34,35]. In acute lung injury caused by infection of bacteria, cytokine receptors play a central role in initiating the innate immune system and in activating NF-κB. Anti-inflammatory cytokines have the ability to suppress inflammatory processes via the inhibition of NF-κB, which can interact with other transcription factors, and these interactions thereby lead to greater transcriptional selectivity. Modification of transcription, and particularly of NF-κB, is likely to be a logical therapeutic target for the manipulation and treatment of acute lung injury [36,37,38,39,40,41,42].
Hypoxemia
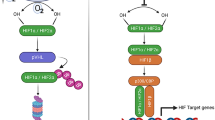
A crucial transcription factor that is a master regulatory element in sensing hypoxic conditions and in integrating an adapted response via gene expression of oxygen-sensitive and redox-sensitive enzymes and cofactors is hypoxia-inducible factor-1 (HIF-1) (Fig. 1) [43,44,45]. The signal transduction components that link the availability of oxygen to the activation of these transcription factors are poorly defined, but are broadly believed to hinge on the free abundance of oxidants.

Oxygen-sensing proposed mechanisms for the regulation of gene transcription and the involvement of hypoxia-inducible factor-1 (HIF-1) as a hypoxia-mediated transcriptional activity (see text for further details). AA, arachidonic acid; ARNT, aryl receptor hydrocarbon nuclear translocator; CREB, cAMP-responsive element binding protein; CBP, CREB-binding protein; DAG, diacyl glycerol; ECF, extracellular fluid; ICF, intracellularfluid; IP3, inositol triphosphate; MAPK, mitogen-activated protein kinase; NADP, nicotinamide dinucleotide oxidized; NADPH, nicotinamide dinucleotide reduced; PKC, protein kinase C; ROS, reactive oxygen species; SAPK, stress-activated protein kinase.
HIF-1 consists of two subunits: HIF-1α, which is unique to the oxygen response; and HIF-1β (aryl hydrocarbon receptor nuclear translocator). The stability and activity of HIF-1α, first identified as a DNA-binding activity expressed under hypoxic conditions, increase exponentially when p O2 is lowered. Whereas HIF-1β is constitutively expressed under normoxic conditions, HIF-1α is rapidly degraded by the ubiquitin–proteasome system. Under hypoxic conditions, however, HIF-1α protein stabilizes and accumulates, thus allowing the het-erodimer to translocate to the nucleus and to bind specific promoter moieties of selective genes encoding erythropoietin (EPO), vascular endothelial growth factor (VEGF), glycolytic enzymes and glucose transporters, as well as cytokines and other inflammatory mediators (Fig. 1) [44,45,46]. It is expected that any reduction of tissue oxygenation in vivo and in vitro would therefore provide a mechanistic stimulus for a graded and adaptive response mediated by hypoxia-inducible factor (Fig. 2).

Potential oxygen-sensing mechanisms and the role of the transcription factor hypoxia-inducible factor-1 (HIF-1). 6GP, 6-glucose phosphate; 6PG, 6-phosphoglycerate; FAD, flavin adenine dinucleotide oxidized; FADH, flavin adenine dinucleotide reduced; NADP, nicotinamide dinucleotide oxidized; NADPH, nicotinamide dinucleotide reduced; ROS, reactive oxygen species; VHL, von Hippel-Lindau tumor suppressor protein.
Inflammatory stimuli
The role of HIF-1α in oxidant-induced lung injury is less clear, or less prominent, than that of NF-κB. Indirect, but unprecedented and unequivocal, evidence was independently provided by Hellwig-Bürgel and colleagues [47,48,49] and by Haddad and Land [50,51], however, to indicate HIF-1 as a possible regulator of the evolution and propagation of the inflammatory process. The rate of transcription of several genes encoding proteins involved in oxygen and energy homeostasis is controlled by HIF-1. Since EPO gene expression is inhibited by the proinflammatory cytokines, such as IL-1β and TNF-α, while no such effect has been reported with respect to the VEGF gene, Hellwig-Bürgel et al. investigated the effects of these cytokines on the activation of the HIF-1 DNA-binding complex and the amount of HIF-1α protein in human hepatoma cells in culture [47]. Under normoxic conditions, both cytokines caused a moderate activation of HIF-1 DNA binding. In hypoxia, cytokines strongly increased HIF-1 activity compared with the effect of hypoxia alone. Only IL-1β increased HIF-1α protein levels. In transient transfection experiments, HIF-1-driven reporter gene expression was augmented by cytokines only under hypoxic conditions. In contrast to their effect on EPO synthesis, neither IL-1β or TNF-α decreased VEGF production. The mRNA levels of HIF-1α and VEGF were unaffected. Cytokine-induced inhibition of EPO production may thus not be mediated by impairment of HIF-1 function [47].
Hellwig-Bürgel and colleagues subsequently proposed that HIF-1 might be involved in modulating gene expression during inflammation. Furthermore, since VEGF promotes angiogenesis and inflammatory reactions, in a parallel study VEGF mRNA was found detectable in the proximal tubules of inflamed kidneys but not in normal kidneys [48]. In other organs, VEGF gene expression is induced by hypoxia and by cytokines. To identify the cellular mechanisms in control of tubular VEGF production, the effects of hypoxia and IL-1β on VEGF mRNA levels, on VEGF secretion and on activity of HIF-1 in human proximal tubular epithelial cells were assessed. The human proximal tubular epithelial cells were grown in monolayers from human kidneys, and hypoxia was induced by incubation at 3% O2. Significant amounts of VEGF mRNA and VEGF protein were measured in human proximal tubular epithelial cell extracts and culture media, respectively. Moreover, stimulation of VEGF synthesis at low p O2 tension and following IL-1β treatment was detectable at the protein level only. Nuclear HIF-1α protein levels and HIF-1 binding to DNA were also increased under these conditions [48].
VEGF induction appears to increase DNA binding of HIF-1 to hypoxia-responsive elements in the VEGF gene promoter. In inflammatory diseases of the kidney, tubular cell-derived VEGF may therefore contribute to microvascular leakage and to monocyte extravasation. Regarding the mechanisms reported, LY-294002 (an inhibitor of phosphatidylinositol 3-kinase) suppressed HIF-1 activation in a dose-dependent manner irrespective of the stimulus. With respect to target proteins controlled by HIF-1, the production of EPO was fully blocked and that of VEGF reduced following inhibition of the phosphatidylinositol 3-kinase pathway [49]. The role of mitogen-activated protein kinase kinases in this process remained ambiguous, because PD-98059 and U-0126 inhibitors did not significantly reduce HIF-1α levels at non-toxic doses [49]. It was proposed that phosphatidylinositol 3-kinase signaling is not only important in the hypoxic induction of HIF-1, but that it is also crucially involved in the response to insulin and IL-1.
Furthermore, evidence that reactive oxygen species (ROS) signaling mediates cytokine-dependent regulation of HIF-1α has been postulated by Haddad and Land [50,51]. In the airway epithelium, recombinant human IL-1β and recombinant murine TNF-α induced, in a time-dependent manner, the nuclear translocation of HIF-1α. This translocation is an effect associated with upregulating the activity of this transcription factor under normoxic conditions. In addition, analysis of the mode of action of IL-1β and TNF-α revealed a novel induction of intra-cellular ROS, including hydrogen peroxide, the superoxide anion (O2-•) and the •OH radical [50,51]. The antioxidants dimethyl sulfoxide and 1,3-dimethyl-2-thiourea, purported to be prototypical scavengers of hydrogen peroxideand •OH, attenuated cytokine-induced HIF-1α nuclear translocation and activation in a dose-dependent manner. The NADPH-oxidase inhibitor 4'-hydroxy-3'-methoxy-acetophenone, which may affect mitochondrial ROS production, attenuated cytokine-mediated nuclear translocation and activation of HIF-1α. Furthermore, inhibition of the mitochondrion complex I nicotinamide ADP-dependent oxidase by diphenylene iodonium, which blocks the conversion of ubiquinone to ubiquinol, abrogated IL-1β-dependent and TNF-α-dependent nuclear translocation and activation of HIF-1α. Similarly, interrupting the respiratory chain with potassium cyanide reversed the excitatory effect of cytokines on HIF-1α nuclear translocation and activation [50,51]. These results indicate that a nonhypoxic pathway mediates cytokine-dependent regulation of HIF-1α translocation and activation in a ROS-sensitive mechanism.
Direct evidence implicating HIF-1 in lung injury emerged with VEGF, which has been recognized as a potent mediator of endothelial barrier dysfunction and is upregulated during ischemia in many organs [43,44,45,46]. Because ventilated pulmonary ischemia causes a marked increase in pulmonary vascular permeability, it was hypothesized that VEGF would increase during ischemic lung injury.
To test this hypothesis, VEGF expression was measured by northern and western blot analysis in isolated ferret lungs after 45 or 180 min of ventilated (95% or 0% O2) ischemia [52]. Pulmonary vascular permeability, assessed by measurement of the osmotic reflection coefficient for albumin, was evaluated in the same lungs, as was expression of HIF-1α. The distribution of VEGF as a function of ischemic time and oxygen tension was also evaluated by immunohistochemical staining in separate groups of lungs. VEGF mRNA increased threefold by 180 min of ventilated ischemia, independent of oxygen tension. VEGF protein increased in parallel to VEGF mRNA. Immunohistochemical staining demonstrated the appearance of VEGF protein along alveolar septae after 180 min of hyperoxic ischemia and after 45 or 180 min of hypoxic ischemia. In addition, albumin was not altered by 45 min of hyperoxic ischemia (0.69 ± 0.09 versus 0.50 ± 0.12, respectively), but decreased significantly after 180 min of hyperoxic ischemia and after 45 and 180 min of hypoxic ischemia (0.20 ± 0.03, 0.26 ± 0.08 and 0.23 ± 0.03, respectively) [52]. HIF-1α mRNA increased during both hyperoxic and hypoxic ischemia, but HIF-1α protein increased only during hypoxic ischemia. This implicates VEGF as a potential mediator of increased pulmonary vascular permeability in this model of acute lung injury.
Further elaborating on the mechanisms involving HIF-1 in regulating the inflammatory response, Hierholzer et al. reported that hemorrhagic shock (HS) initiates an inflammatory response that includes increased expression of inducible nitric oxide synthase and production of prostaglandins [53]. Induction of inducible nitric oxide synthase during the ischemic phase of HS may involve the activation of HIF-1. Increased expression of cyclooxygenase-2 during HS contributes to prostaglandin production. The lungs of rats subjected to HS demonstrated a twofold increase in HIF-1 activation and a 7.4-fold increase in expression of cyclo-oxygenase-2 mRNA, as compared with sham controls [53]. It was concluded that the upregulation of inducible nitric oxide synthase and cyclooxygenase-2 during ischemia are two important early response genes that promote the inflammatory response and may contribute to organ damage through the rapid and exaggerated production of nitric oxide and prostaglandins.
Furthermore, in a novel study by Shoshani and colleagues, the identification and cloning of a HIF-1-responsive gene, designated RTP801, was recently reported. Strong upregula-tion of RTP801 by hypoxia was detected both in vitro and in vivo in an animal model of ischemic stroke [54]. When induced from a tetracycline-repressible promoter, RTP801 protected MCF7 and PC12 cells from hypoxia in glucose-free medium and from hydrogen peroxide-triggered apoptosis via a dramatic reduction in the generation of ROS. However, expression of RTP801 appeared toxic for nondividing neuron-like PC12 cells and increased their sensitivity to ischemic injury and oxidative stress. Furthermore, liposomal delivery of RTP801 cDNA to mouse lungs also resulted in massive cell death [54]. The biological effect of RTP801 overexpression thus depends on the cell context and may be either protecting or detrimental for cells under conditions of oxidative or ischemic stresses. Altogether, the data suggest a complex type of involvement of RTP801 in the pathogenesis of ischemic diseases.
A hypothetical schematic depicting the role of HIF-1 in lung injury is displayed in Fig. 3.

A schematic overview of the potential signaling pathways involved in cytokine-mediated regulation of hypoxia-induced hypoxia-inducible factor-1α (HIF-1α) translocation and activation. Hypoxia and inflammatory signals induce the intracellular accumulation of reactive oxygen species (ROS), which may cause changes in the phosphorylation state of target kinases, thereby mediating a specific regulatory mechanism. The mitochondrion is a potential source for cytokine-unleashed ROS, whose regulation is selectively mediated by antioxidants. ROS-mediated signaling allows HIF-1α protein stabilization, nuclear translocation and transcriptional activation.
Conclusion and future prospects
The molecular response to oxidative stress is regulated by redox-sensitive transcription factors [55,56,57,58,59,60]. The study of gene expression and regulation is critical in the development of novel gene therapies [61,62,63,64,65,66,67,68,69,70]. Recognition of reactive species and redox-mediated protein modifications as potential signals may open up a new field of cell regulation via specific and targeted genetic control of transcription factors, and can thus provide us with a novel way of controlling disease processes [71,72,73,74,75]. Dynamic variation in p O2 and redox equilibrium thus regulate gene expression, apoptosis signaling and the inflammatory process, thereby bearing potential consequences for screening emerging targets for therapeutic intervention.
Abbreviations
- AM:
-
alveolar macrophage
- CF:
-
cystic fibrosis
- EPO:
-
erythropoietin
- HIF-1 :
-
hypoxia-inducible factor-1
- HS:
-
hemorrhagic shock
- IL:
-
interleukin
- NF-κB:
-
nuclear factor-κB
- p O2:
-
partial pressure of oxygen
- redox:
-
reduction–oxidation
- ROS:
-
reactive oxygen species
- SLPI:
-
secretory leukocyte protease inhibitor
- TNF:
-
tumor necrosis factor
- VEGF:
-
vascular endothelial growth factor.
References
D'Angio CT, Finkelstein JN: Oxygen regulation of gene expression: a study in opposites. Mol Genet Metab 2000, 71: 371-380. 10.1006/mgme.2000.3074
Alder V, Yin Z, Tew KD, Ronai Z: Role of redox potential and reactive oxygen species in stress signaling. Oncogene 1999, 18: 6104-6111. 10.1038/sj.onc.1203128
Crapo JD, Harmsen AG, Sherman MP, Musson RA: Pulmonary immunobiology and inflammation in pulmonary diseases. Am J Respir Crit Care Med 2000, 162: 1983-1986.
Haddad JJ, Olver RE, Land SC: Antioxidant/pro-oxidant equilibrium regulates HIF-1α and NF-κB redox sensitivity: evidence for inhibition by glutathione oxidation in alveolar epithelial cells. J Biol Chem 2000, 275: 21130-21139. 10.1074/jbc.M000737200
Pugin J, Dunn I, Jolliet P, Tassaux D, Magnenat JL, Nicod LP, Chevrolet JC: Activation of human macrophages by mechanical ventilation in vitro . Am J Physiol 1998, 275: L1040-L1050.
Leeper-Woodford SK, Detmer K: Acute hypoxia increases alveolar macrophage tumor necrosis factor activity and alters NF-κB expression. Am J Physiol 1999, 276: L909-L916.
Lentsch AB, Yoshidome H, Warner RL, Ward PA, Edwards MJ: Secretory leukocyte protease inhibitor in mice regulates local and remote organ inflammatory injury induced by hepatic ischemia/reperfusion. Gastroenterology 1999, 117: 953-961.
Chapman WC, Debelak JP, Wright Pinson C, Washington MK, Atkinson JB, Venkatakrishnan A, Blackwell TS, Christman JW: Hepatic cryo-ablation, but not radio-frequency ablation, results in lung inflammation. Ann Surg 2000, 231: 752-761. 10.1097/00000658-200005000-00016
Chapman WC, Debelak JP, Blackwell TS, Gainer KA, Christman JW, Pinson CW, Brigham KL, Parker RE: Hepatic cryo-ablation-induced acute lung injury: pulmonary hemodynamic and permeability effects in a sheep model. Arch Surg 2000, 135: 667-672. 10.1001/archsurg.135.6.667
Basbaum C, Lemjabbar H, Longphre M, Li D, Gensch E, McNamara N: Control of mucin transcription by diverse injury-induced signaling pathways. Am J Respir Crit Care Med 1999, 160: S44-S48.
Yoshidome H, Kato A, Edwards MJ, Lentsch AB: Interleukin-10 inhibits pulmonary NF-κB activation and lung injury induced by hepatic ischemia-reperfusion. Am J Physiol 1999, 277: L919-L923.
Armstead VE, Opentanova IL, Minchenko AG, Lefer AM: Tissue factor expression in vital organs during murine traumatic shock: role of transcription factors AP-1 and NF-κB. Anesthesiology 1999, 91: 1844-1852. 10.1097/00000542-199912000-00039
Washington K, Debelak JP, Gobbell C, Sztipanovits DR, Shyr Y, Olson S, Chapman WC: Hepatic cryo-ablation-induced acute lung injury: histopathologic findings. J Surg Res 2001, 95: 1-7. 10.1006/jsre.2000.5976
Ross SD, Kron IL, Gangemi JJ, Shockey KS, Stoler M, Kern JA, Tribble CG, Laubach VE: Attenuation of lung reperfusion injury after transplantation using an inhibitor of nuclear factor-κB. Am J Physiol Lung Cell Mol Physiol 2000, 279: L528-L536.
Sajjan U, Thanassoulis G, Cherapanov V, Lu A, Sjolin C, Steer B, Wu YJ, Rotstein OD, Kent G, McKerlie C, Forstner J, Downey GP: Enhanced susceptibility to pulmonary infection with Burkholderia cepacia in Cftr-/- mice. Infect Immun 2001, 69: 5138-5150. 10.1128/IAI.69.8.5138-5150.2001
Lentsch AB, Ward PA: Regulation of experimental lung inflammation. Respir Physiol 2001, 128: 17-22. 10.1016/S0034-5687(01)00260-2
Fan J, Ye RD, Malik AB: Transcriptional mechanisms of acute lung injury. Am J Physiol Lung Cell Mol Physiol 2001, 281: L1037-L1050.
Kupfner JG, Arcaroli JJ, Yum HK, Nadler SG, Yang KY, Abraham E: Role of NF-κB in endotoxemia-induced alterations of lung neutrophil apoptosis. J Immunol 2001, 167: 7044-7051.
Park GY, Le S, Park KH, Le CT, Kim YW, Han SK, Shim YS, Yoo CG: Anti-inflammatory effect of adenovirus-mediated IκB-α overexpression in respiratory epithelial cells. Eur Respir J 2001, 18: 801-809. 10.1183/09031936.01.00099801
Semenza GL: Oxygen-regulated transcription factors and their role in pulmonary disease. Respir Res 2000, 1: 159-162. 10.1186/rr27
Cuzzocrea S, Chatterjee PK, Mazzon E, Dugo L, Serraino I, Britti D, Mazzullo G, Caputi AP, Thiemermann C: Pyrrolidine dithiocarbamate attenuates the development of acute and chronic inflammation. Br J Pharmacol 2002, 135: 496-510.
Sunil VR, Connor AJ, Guo Y, Laskin JD, Laskin DL: Activation of type II alveolar epithelial cells during acute endotoxemia. Am J Physiol Lung Cell Mol Physiol 2002, 282: L872-L880.
Haddad JJ, Safieh-Garabedian B, Saadé NE, Kanaan SA, Land SC: Chemioxyexcitation (Δ p O 2 /ROS)-dependent release of IL-1β, IL-6 and TNF-α: evidence of cytokines as oxygen-sensitive mediators in the alveolar epithelium. Cytokine 2001, 13: 138-147. 10.1006/cyto.2000.0789
Haddad JJ, Choudhary KK, Land SC: The ex vivo differential expression of apoptosis signaling cofactors in the developing perinatal lung: essential role of oxygenation during the transition from placental to pulmonary-based respiration. Biochem Biophys Res Commun 2001, 281: 311-316. 10.1006/bbrc.2001.4350
Haddad JJ, Safieh-Garabedian B, Saadé NE, Land SC: Thiol regulation of pro-inflammatory cytokines reveals a novel immunopharmacological potential of glutathione in the alveolar epithelium. J Pharmacol Exp Ther 2001, 296: 996-1005.
Haddad JJ, Collett A, Land SC, Olver RE, Wilson SM: NF-κB blockade reduces the O 2 -evoked rise in Na+ conductance in fetal alveolar cells. Biochem Biophys Res Commun 2001, 281: 987-992. 10.1006/bbrc.2001.4453
Baines DL, Ramminger SJ, Collett A, Haddad JJ, Best OG, Land SC, Olver RE, Wilson SM: Oxygen-evoked Na+ transport in rat fetal distal lung epithelial cells. J Physiol 2001, 532: 105-113.
Haddad JJ, Safieh-Garabedian B, Saadé NE, Land SC: The biphasic immunoregulation of pyrimidylpiperazine (Y-40138) is IL-10 sensitive and requires NF-κB targeting in the alveolar epithelium. Br J Pharmacol 2001, 133: 49-60.
Haddad JJ, Land SC: Nuclear factor-κB blockade attenuates but does not abrogate lipopolysaccharide-dependent tumor necrosis factor-α biosynthesis in alveolar epithelial cells. Biochem Biophys Res Commun 2001, 285: 267-272. 10.1006/bbrc.2001.5172
Haddad JJ: Glutathione depletion is associated with augmenting a proinflammatory signal: evidence for an antioxidant/pro-oxidant mechanism regulating cytokines in the alveolar epithelium. Cytokines Cell Mol Ther 2000, 6: 177-187.
Haddad JJ, Land SC: Amiloride blockades lipopolysaccharide-induced proinflammatory cytokine biosynthesis in an IκB-α/NF-κB-dependent mechanism. Evidence for the amplification of an antiinflammatory pathway in the alveolar epithelium. Am J Respir Cell Mol Biol 2002, 26: 114-126.
Haddad JJ: L-Buthionine-( S,R )-sulfoximine, an irreversible inhibitor of γ-glutamylcysteine synthetase, augments LPS-mediated pro-inflammatory cytokine biosynthesis: evidence for the implication of an IκB-α/NF-κB insensitive pathway. Eur Cytokine Netw 2001, 12: 614-624.
Haddad JJ, Saadé NE, Safieh-Garabedian B: Redox regulation of TNF-α biosynthesis: augmentation by irreversible inhibition of gamma-glutamylcysteine synthetase and the involvement of an IκB-α/NF-κB-independent pathway in alveolar epithelial cells. Cell Signal 2002, 14: 211-218. 10.1016/S0898-6568(01)00233-9
Haddad JJ, Land SC: Redox/ROS regulation of lipopolysaccharide-induced mitogen-activated protein kinase (MAPK) activation and MAPK-mediated TNF-α biosynthesis. Br J Pharmacol 2002, 135: 520-536.
Haddad JJ: The involvement of L-γ-glutamyl-L-cysteinyl-glycine (glutathione/GSH) in the mechanism of redox signaling mediating MAPKp38-dependent regulation of pro-inflammatory cytokine production. Biochem Pharmacol 2002, 63: 305-320. 10.1016/S0006-2952(01)00870-X
Haddad JJ, Fahlman CS: Nuclear factor-κB-independent regulation of lipopolysaccharide-mediated interleukin-6 biosynthesis. Biochem Biophys Res Commun 2002, 291: 1045-1051. 10.1006/bbrc.2002.6556
Rahman I, Mulier B, Gilmour PS, Watchorn T, Donaldson K, Jeffery PK, MacNee W: Oxidant-mediated lung epithelial cell tolerance: the role of intracellular glutathione and nuclear factor-κB. Biochem Pharmacol 2001, 62: 787-794. 10.1016/S0006-2952(01)00702-X
MacNee W, Rahman I: Is oxidative stress central to the pathogenesis of chronic obstructive pulmonary disease? Trends Mol Med 2001, 7: 55-62. 10.1016/S1471-4914(01)01912-8
Rahman I, MacNee W: Regulation of redox glutathione levels and gene transcription in lung inflammation: therapeutic approaches. Free Radic Biol Med 2000, 28: 1405-1420. 10.1016/S0891-5849(00)00215-X
Rahman I, van Schadewijk AA, Hiemstra PS, Stolk J, van Krieken JH, MacNee W, de Boer WI: Localization of γ-glutamylcysteine synthetase mRNA expression in lungs of smokers and patients with chronic obstructive pulmonary disease. Free Radic Biol Med 2000, 28: 920-925. 10.1016/S0891-5849(00)00179-9
Haddad JJ: Oxygen-sensitive pro-inflammatory cytokines, apoptosis signaling and redox-responsive transcription factors in development and pathophysiology. Cytokines Cell Mol Ther 2001, 7: 1-14. 10.1080/13684730216401
Haddad JJ: Oxygen homeostasis, thiol equilibrium and redox regulation of signaling transcription factors in the alveolar epithelium. Cell Signal 2002, 14: 799-810.
Semenza GL: HIF-1 and mechanisms of hypoxia sensing. Curr Opin Cell Biol 2001, 13: 167-171. 10.1016/S0955-0674(00)00194-0
Semenza GL: HIF-1 and human disease: one highly involved factor. Genes Dev 2000, 14: 1983-1991.
Semenza GL: Involvement of hypoxia-inducible factor 1 in human cancer. Intern Med 2002, 41: 79-83.
Semenza GL: HIF-1 and tumor progression: pathophysiology and therapeutics. Trends Mol Med 2002, 8: S62-S67. 10.1016/S1471-4914(02)02317-1
Hellwig-Bürgel T, Rutkowski K, Metzen E, Fandrey J, Jelkmann W: Interleukin-1β and tumor necrosis factor-alpha stimulate DNA binding of hypoxia-inducible factor-1. Blood 1999, 94: 1561-1567.
El Awad B, Kreft B, Wolber EM, Hellwig-Bürgel T, Metzen E, Fandrey J, Jelkmann W: Hypoxia and interleukin-1β stimulate vascular endothelial growth factor production in human proximal tubular cells. Kidney Int 2000, 58: 43-50. 10.1046/j.1523-1755.2000.00139.x
Stiehl DP, Jelkmann W, Wenger RH, Hellwig-Bürgel T: Normoxic induction of the hypoxia-inducible factor 1α by insulin and interleukin-1β involves the phosphatidylinositol 3-kinase pathway. FEBS Lett 2002, 512: 157-162. 10.1016/S0014-5793(02)02247-0
Haddad JJ, Land SC: A non-hypoxic, ROS-sensitive pathway mediates TNF-α-dependent regulation of HIF-1α. FEBS Lett 2001, 505: 269-274. 10.1016/S0014-5793(01)02833-2
Haddad JJ: Recombinant human interleukin (IL)-1β-mediated regulation of hypoxia-inducible factor-1α (HIF-1α) stabilization, nuclear translocation and activation requires an antioxi-dant/reactive oxygen species (ROS)-sensitive mechanism. Eur Cytokine Netw 2002, 13: 250-260.
Becker PM, Alcasabas A, Yu AY, Semenza GL, Bunton TE: Oxygen-independent upregulation of vascular endothelial growth factor and vascular barrier dysfunction during ventilated pulmonary ischemia in isolated ferret lungs. Am J Respir Cell Mol Biol 2000, 22: 272-279.
Hierholzer C, Harbrecht BG, Billiar TR, Tweardy DJ: Hypoxia-inducible factor-1 activation and cyclooxygenase-2 induction are early reperfusion-independent inflammatory events in hemorrhagic shock. Arch Orthop Trauma Surg 2001, 121: 219-222. 10.1007/s004020000211
Shoshani T, Faerman A, Mett I, Zelin E, Tenne T, Gorodin S, Moshel Y, Elbaz S, Budanov A, Chajut A, Kalinski H, Kamer I, Rozen A, Mor O, Keshet E, Leshkowitz D, Einat P, Skaliter R, Feinstein E: Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol Cell Biol 2002, 22: 2283-2293. 10.1128/MCB.22.7.2283-2293.2002
Shimaoka M, Fujino Y, Taenaka N, Hiroi T, Kiyono H, Yoshiya II: High frequency oscillatory ventilation attenuates the activation of alveolar macrophages and neutrophils in lung injury. Crit Care 1998, 2: 35-39. 10.1186/cc122
Pinsky MR: Toward a better ventilation strategy for patients with acute lung injury. Crit Care 2000, 4: 205-206. 10.1186/cc695
Wesselkamper SC, Prows DR, Biswas P, Willeke K, Bingham E, Leikauf GD: Genetic susceptibility to irritant-induced acute lung injury in mice. Am J Physiol Lung Cell Mol Physiol 2000, 279: L575-L582.
Leikauf GD, McDowell SA, Bachurski CJ, Aronow BJ, Gammon K, Wesselkamper SC, Hardie W, Wiest JS, Leikauf JE, Korfhagen TR, Prows DR: Functional genomics of oxidant-induced lung injury. Adv Exp Med Biol 2001, 500: 479-487.
Leikauf GD, McDowell SA, Wesselkamper SC, Hardie WD, Leikauf JE, Korfhagen TR, Prows DR: Acute lung injury: functional genomics and genetic susceptibility. Chest 2002, 121: 70S-75S. 10.1378/chest.121.3_suppl.70S
Pittet JF, MacKersie RC, Martin TR, Matthay MA: Biological markers of acute lung injury: prognostic and pathogenetic significance. Am J Respir Crit Care Med 1997, 155: 1187-1205.
Kaminski N, Allard JD, Pittet JF, Zuo F, Griffiths MJ, Morris D, Huang X, Sheppard D, Heller RA: Global analysis of gene expression in pulmonary fibrosis reveals distinct programs regulating lung inflammation and fibrosis. Proc Natl Acad Sci USA 2000, 97: 1778-1783. 10.1073/pnas.97.4.1778
Miyazaki H, Broaddus VC, Wiener-Kronish JP, Sawa T, Pittet JF, Kravchenko V, Mathison JC, Nishizawa H, Hattori S, Yamakawa T, Yamada H, Kudoh I: The effects of two antiinflammatory pre-treatments on bacterial-induced lung injury. Anesthesiology 1999, 90: 1650-1662. 10.1097/00000542-199906000-00022
Grimaldo DA, Wiener-Kronish JP, Jurson T, Shaughnessy TE, Curtis JR, Liu LL: A randomized, controlled trial of advanced care planning discussions during preoperative evaluations. Anesthesiology 2001, 95: 43-50. 10.1097/00000542-200107000-00012
Ernst EJ, Hashimoto S, Guglielmo J, Sawa T, Pittet JF, Kropp H, Jackson JJ, Wiener-Kronish JP: Effects of antibiotic therapy on Pseudomonas aeruginosa -induced lung injury in a rat model. Antimicrob Agents Chemother 1999, 43: 2389-2394.
Davis K Jr, Johannigman JA, Campbell RS, Marraccini A, Luchette FA, Frame SB, Branson RD: The acute effects of body position strategies and respiratory therapy in paralyzed patients with acute lung injury. Crit Care 2001, 5: 81-87. 10.1186/cc1414
Kheradmand F, Wiener-Kronish JP, Corry DB: Assessment of operative risk for patients with advanced lung disease. Clin Chest Med 1997, 18: 483-494.
Song C, Al-Mehdi AB, Fisher AB: An immediate endothelial cell signaling response to lung ischemia. Am J Physiol Lung Cell Mol Physiol 2001, 281: L993-L1000.
Merker MP, Pitt BR, Choi AM, Hassoun PM, Dawson CA, Fisher AB: Lung redox homeostasis: emerging concepts. Am J Physiol Lung Cell Mol Physiol 2000, 279: L413-L417.
Dietrich M, Block G, Hudes M, Morrow JD, Norkus EP, Traber MG, Cross CE, Packer L: Antioxidant supplementation decreases lipid peroxidation biomarker F 2 -isoprostanes in plasma of smokers. Cancer Epidemiol Biomarkers Prev 2002, 11: 7-13.
van Der Vliet A, Eiserich JP, Cross CE: Nitric oxide: a pro-inflammatory mediator in lung disease? Respir Res 2000, 1: 67-72. 10.1186/rr14
Cross CE, van der Vliet A, Eiserich JP: Peroxidases wheezing their way into asthma. Am J Respir Crit Care Med 2001, 164: 1102-1103.
Casaburi R, Mahler DA, Jones PW, Wanner A, San PG, ZuWallack RL, Menjoge SS, Serby CW, Witek T Jr: A long-term evaluation of once-daily inhaled tiotropium in chronic obstructive pulmonary disease. Eur Respir J 2002, 19: 217-224. 10.1183/09031936.02.00269802
Burrows B, Bloom JW, Traver GA, Cline MG: The course and prognosis of different forms of chronic airways obstruction in a sample from the general population. N Engl J Med 1987, 317: 1309-1314.
Kaplan LJ, Bailey H, Formosa V: Airway pressure release ventilation increases cardiac performance in patients with acute lung injury/adult respiratory distress syndrome. Crit Care 2001, 5: 221-226. 10.1186/cc1027
dos Santos CC, Zhang H, Slutsky AS: From bench to bedside: bacterial growth and cytokines. Crit Care 2002, 6: 4-6. 10.1186/cc1443
Acknowledgements
The author's own publications therein cited are, in part, financially supported by the Anonymous Trust (Scotland), the National Institute for Biological Standards and Control (England), the Tenovus Trust (Scotland), the UK Medical Research Council (MRC, London), the Wellcome Trust (London) (Stephen C. Land, Department of Child Health, University of Dundee, Scotland, UK) and the National Institutes of Health (NIH; Bethesda, USA) (Philip E. Bickler, Department of Anesthesia and Perioperative Care, University of California, San Francisco, California, USA). The work of the author was performed at the University of Dundee, Scotland, UK. This review was written at UCSF, California, USA. JJH held the Georges John Livanos prize (London, UK) under the supervision of Stephen C. Land and the NIH award fellowship (California, USA) under the supervision of Philip E. Bickler. The author also appreciatively thanks Jennifer Schuyler (Department of Anesthesia and Perioperative Care) for her excellent editing and reviewing of this manuscript. I also thank my colleagues at UCSF (San Francisco, California, USA) and the American University of Beirut (AUB, Beirut, Lebanon) who have criticised the work for enhancement and constructive purposes.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None declared.
Rights and permissions
About this article
Cite this article
Haddad, J.J. Science review: Redox and oxygen-sensitive transcription factors in the regulation of oxidant-mediated lung injury: role for hypoxia-inducible factor-1α. Crit Care 7, 47 (2002). https://doi.org/10.1186/cc1840
Published:
DOI: https://doi.org/10.1186/cc1840