
Abstract
Introduction
Mechanical ventilation (MV) could prime the lung toward an inflammatory response if exposed to another insult such as bacterial invasion. The underlying mechanisms are not so far clear. Toll-like receptors (TLRs) allow the host to recognize selectively bacterial pathogens and in turn to trigger an immune response. We therefore hypothesized that MV modulates TLR2 expression and in turn modifies responsiveness to agonists such as bacterial lipopeptide (BLP).
Method
Both in vitro and in vivo experiments were conducted. First, TLR2 expression and protein were measured in the A549 pulmonary epithelial cell line submitted to 8-hour cyclic stretch (20% elongation; 20/minute rate). After a 24-hour period of cyclic stretch, the inflammatory response of the A549 cells to the synthetic BLP, Pam3CSK4, was tested after 8 hours of exposure. In a second set of experiments, healthy anesthetized and paralyzed rabbits were submitted to 8-hour MV (tidal volume = 12 ml/kg, zero end-expiratory pressure; FIO2 = 50%; respiratory rate = 20/minute) before being sacrificed for TLR2 lung expression assessment. The lung inflammatory response to BLP was then tested in animals submitted to 24-hour MV before being sacrificed 8 hours after the tracheal instillation of Pam3CSK4.
Results
Cyclic stretch of human pulmonary epithelial cell lines increased both TLR2 mRNA and protein expression. Cells submitted to cyclic stretch also increased IL-6 and IL-8 secretion in response to Pam3CSK4, a classical TLR2 ligand. A mild-stretch MV protocol induced a 60-fold increase of TLR2 mRNA expression in lung tissue when compared with spontaneously breathing controls. Moreover, the combination of MV and airway exposure to Pam3CSK4 acted synergistically in causing lung inflammation and injury.
Conclusions
Mild-stretch MV increases lung expression of TLR2 and sensitizes the lung to bacterial TLR2 ligands. This may account for the propensity of mechanically ventilated patients to develop acute lung injury in the context of airway bacterial colonization/infection.
Similar content being viewed by others


Introduction
Bacterial superinfection of the lung is a frequent complication in critically ill patients requiring mechanical ventilation (MV) [1]. An incidence of ventilator-associated pneumonia of up to 30% has been reported in patients requiring prolonged MV. Among the bacteria causing ventilator-associated pneumonia, Staphylococcus aureus is a leading source of infection and mortality [2]. Both in vitro and in vivo experimental studies have demonstrated that MV could activate lung cells and induce a proinflammatory response. Although low-stretch MV does not lead to apparent tissue damage or inflammation, it could prime airway cells to respond massively to a second proinflammatory insult such as lipopolysaccharide (LPS) [3–6]. The subsequent release of large amounts of cytokines is probably responsible for substantial lung injury, particularly through the recruitment of neutrophils mediated by IL-8 secretion [7].
Little is known, however, about the role of Gram-positive bacterial products in this context. In several animal studies, MV has been shown to impair lung bacterial clearance, to increase lung injury and to promote pulmonary-to-systemic microbial translocation [8–11]. However, the link between these different observations remains to be established.
A prompt immune lung response is necessary to ensure microbial clearance. Early pathogen recognition via the so-called pathogen recognition receptors is considered a first and key step in this process. Among the pathogen recognition receptors, toll-like receptors (TLRs) play an indisputable and major role in the recognition of pathogen-associated microbial patterns. The ligation of TLRs by pathogen-associated molecular patterns induces the signaling of the subsequent inflammatory response. Bacterial lipopeptides (BLPs) are cell wall components of both Gram-positive and Gram-negative bacteria and have been recognized to activate TLR2 in a heterodimeric association with TLR1 or TLR6 [12, 13].
TLR2 is mainly expressed by myeloid cells, but epithelial cells from various tissues can also express the receptor [14]. In the airway, both bronchial and alveolar epithelial cells participate in the lung immune response through their ability to produce inflammatory mediators and chemokines [15]. Whereas TLR4 is essential for cell responses to Gram-negative LPS, TLR2 is implicated in innate immune responses to Gram-positive bacteria [12, 13]. For example, Tlr2-/- mice are highly susceptible to infections caused by Gram-positive bacteria such as S. aureus [16]. TLR2 expression also seems sensitive to mechanical stress, at least in the endothelium, where it has been shown to be upregulated by various types of mechanical strains [17]. Whereas the concept of lung priming was relatively well described for LPS models depending on the TLR4 pathway [3–6, 18], little is known on the possible implication of other TLRs and related bacterial ligands in MV-dependent lung injury.
To test the hypothesis that MV modulates TLR2 expression, we performed a set of in vitro and in vivo experiments to determine whether cyclic stretch of lung cells and MV modified the expression of TLR2 and to unravel the effect of MV in lung inflammatory response and injury after exposure to a classical TLR2 bacterial ligand.
Materials and methods
Cells and reagents
The human epithelial type II-like A549 cell line was purchased from the American Type Culture Collection (Rockville, MD, USA) and cultured in a 50/50 mixture of DMEM and F-12 nutrient mixture supplemented with 10% FCS, 2 mM L-glutamine, 10 mM HEPES, 50 U/ml penicillin, and 2 μg/ml gentamicin. The NF-κB inhibitor Sn50 and the p38 mitogen-activated protein kinase (MAPK) specific inhibitor SB203580 were purchased from Sigma (St Louis, MO, USA). The TLR2-specific agonist Pam3CSK4 lipopeptide was purchased from InvivoGen (San Diego, CA, USA).
Cell culture and cyclic stretch
A549 epithelial cell lines were seeded onto Bioflex® plates (Dunn Labortechnik, Asbach, Germany) at a density of 2 × 105 cells/well, and were cultured for 48 hours until 70 to 80% confluency was achieved. Mechanical stretch was then performed for different periods of time in fresh medium using the cell stretching apparatus FX-3000 Flexercell strain unit (Flexcell International, Hillsborough, N.C., U.S.A.) in a 37°C, 5% carbon dioxide incubator. The stretching rate was 20 cycles/minute with a square signal, a 1:1 stretch:relaxation ratio, and a 20% maximal equibiaxial elongation, as previously described [7]. In some experiments, Pam3CSK4 was added to the medium (final concentration 1 μg/ml) for an additional 8 hours. At the end of the experiments, conditioned supernatants were immediately collected and kept frozen at -80°C. IL-6 and IL-8 were quantified by ELISA (Endogen, Woburn, MA, USA) in thawed conditioned supernatants. Limits of detection were 45 pg/ml for IL-6 and 65 pg/ml for IL-8.
Quantitative RT-PCR
After stretching, A549 cells were harvested and kept in TriZol (Invitrogen, San Diego, CA, USA) at -70°C until the day of RNA extraction. Complementary DNA was obtained by reverse transcription using random primers, RNasin treatment, and ImProm II RT (Promega, Madison, WI, USA). Quantitative PCR was performed using the IQ5 thermocycler (Biorad, Hercules, CA, USA), and the following TaqMan probes with the universal PCR Mastermix (Applied Biosystems, Foster City, CA, USA): MD-2, Hs00209771_m1; TLR4, Hs00152939_m1; TLR2, Hs00610101_m1; CD14, Hs00169122_g1; IL-8, Hs00174103_m1; IL-6, Hs 00174131_m1; HBD1, Hs00608345_m1; HBD2, Hs00175474_m1; HBD3, Hs00218678_m1; 18S, Hs99999901_s1.
In rabbits, after sacrifice and exsanguination, the rib cage was opened and lung pieces were cut and RNA was then extracted using the RNA GenElute kit (Sigma). Quantitative PCR was performed using IQ Sybrgreen Supermix (Biorad). Melting curves were performed to ensure the presence of a single amplicon. The following primers were used: rTlr2, forward 5'-TGT CTG TCA CCG AAC CGA ATC CAC-3' and reverse 5'-TCA GGT TTT TCA GCG TCA GCA GGG-3'; rTlr4, forward 5'-GAG CAC CTG GAC CTT TCA AAT AAC-3' and reverse 5'-GAA CTT CTA AAC CAC TCA GCC CTT-3'; rGapdh, forward 5'-ATG TTT GTG ATG GGC GTG AAC C-3' and reverse 5'-CCC AGC ATC GAA GGT AGA GGA-3'; rCd14, forward 5'-TCT CTG TCC CCA CAA GTT CC-3' and reverse 5'-CAC CTG CTG CAG TCC AGT AA-3'; rMd-2, forward 5'-GGG AAC CCA AGG TTT ATT GC-3' and reverse 5'-CGT ATG CCC TTG AAG GAA AA-3'; and rIl-8, forward 5'-AAC CTT CCT GCT GCT TCT GA-3' and reverse 5'-TCT GCA CCC ACT TTT TCC TTG-3'.
The results are expressed as the fold induction using the ΔCt method since the static condition (in vitro) or the spontaneously breathing (SB) animals (in vivo) were always considered the baseline condition (that is, the '1' reference value)
Flow cytometry
A549 cells stretched or kept in static conditions for 24 hours were removed from culture plates using pH 7.4 PBS containing 0.1% ethylenediamine tetraacetic acid and were used immediately for fluorescence-activated cell sorting (FACS). For each analysis, 106 cells were washed three times with 1 ml PBS containing 0.4% BSA (Sigma) at 4°C. Cells were incubated in PBS/BSA containing 5 μg/ml mouse anti-huTLR2 mAb (T2.5 clone) antibody or an isotype control antibody (mouse IgG1; Biosource, Camarillo, CA, USA), at 4°C for 30 minutes. Cells were then washed three times with PBS and resuspended PBS containing 10 μg/ml goat anti-mouse IgG phycoerythrin-labeled antibody, at 4°C in the dark for 30 minutes. For intracellular staining, cells were pre-fixed with 1% formaldehyde and permeabilized with IC Perm Buffer (Biosource) supplemented with 4% BSA. Staining was then performed as described above. FACS was carried out using FacScan and analyzed with the CellQuest software (Becton Dickinson, San Jose, CA, USA). Results were expressed as the mean fluorescence intensity ratio, defined as the mean fluorescence intensity of cells stained with an antigen-specific antibody divided by the mean fluorescence intensity of cells incubated with the isotype control antibody.
Animal experiments
Male New Zealand White rabbits (body weight 2.7 to 3.0 kg) were obtained from the Elevage scientifique des Dombes (Romans, France). They were placed in individual cages and were fed ad libitum with water and nutriments, according to current recommendations mentioned in the Guide for the Care and Use of Laboratory Animals (National Institutes of Health No. 92-23, revised 1985). The Dijon Faculty of Medicine Ethical Committee approved the experimental protocol.
In a first set of experiments, animals were randomly allocated to the SB group (n = 4) or the MV group (n = 4) (Figure 1). Animals from the former group were immediately sacrificed by a lethal dose of penthotal, whereas the latter were orally intubated as previously described [8, 9]. The animal was then connected to a pressure-controlled ventilator (Babylog®; Drager, Lübeck, Germany). MV was performed in the supine position with continuous infusion of ketamine and pancuronium bromide. The peak inspiratory pressure was set at 12 cmH2O with zero end-expiratory pressure, a respiratory rate of 30/minute and an inspired fraction of oxygen of 0.5. The tidal volume was measured at the onset of MV by placing a pneumotachometer between the endotracheal tube and the ventilator circuit (12.6 ± 0.8 ml/kg). This corresponds to mild-stretch MV. Animals from the MV group were submitted to MV during an 8-hour period before being sacrificed. In both groups, animals were exsanguinated by venous puncture and lungs were harvested for RNA extraction. Additional samples were obtained for microscopic examination. A tissue sample of 1 cm3 was fixed in formalin and embedded in paraffin. Four-micrometer sections were obtained and colored with hematoxylin and eosin.

Experimental design description. (a) In vitro experiments. (b) In vivo experiments. FACS, fluorescence-activated cell sorting; MV, mechanical ventilation; qPCR, quantitative PCR; SB, spontaneously breathing; TLR, toll-like receptor.
In a second set of experiments, rabbits were orally intubated as described above and subjected to MV for a 24-hour period before being given intrabronchially either 500 μg Pam3CSK4 diluted in 2 ml isotonic saline or vehicle alone, through a silicon catheter introduced into the tracheal tube and pushed until one lower lobe bronchus was reached. The animals were then kept under MV during an 8-hour period before being sacrificed. An arterial catheter was inserted in most of these animals for blood sampling and blood pressure monitoring. Controls were SB animals that received 500 μg Pam3CSK4 and were allowed to go back to their cage before being sacrificed 8 hours later.
Statistical analysis
Data are presented as the mean (standard deviation). The Mann-Whitney U test or the Kruskal-Wallis test was used to compare continuous variables whether the number of groups was two or more. P ≤0.05 was considered significant.
Results
In vitro studies
Cyclic stretch induces the expression of Toll-like receptors in lung epithelial cells
A sixfold increase in epithelial cell content in TLR2 mRNA was detected by quantitative PCR in stretched A549 cells when compared with cells kept in static condition (Figure 2a). TLR4 mRNA was increased to the same extent in stretched cells but MD-2 and CD14 mRNA levels remained unchanged.

Pathogen recognition receptor expression in cells submitted to cyclic stretch or kept in static condition. (a) mRNA fold induction in A549 cells stretched for 8 hours or kept in static condition. All values are shown as the fold increase compared with control static cells normalized to 1. IL-8 mRNA levels induced by stretch served as a positive control. Both toll-like receptor (TLR)2 and TLR4 mRNA levels were markedly increased by cyclic stretch. MD-2 and CD14 mRNA levels were not significantly modified by stretch. Results are expressed as mean ± standard deviation of three distinct experiments. (b) Surface expression of TLR2, TLR4, MD-2 and CD14 by fluorescence-activated cell sorting (FACS) in A549 cells submitted to a 24-hour cyclic stretch or in cells kept in static condition. Except for CD14, none of these receptors showed significant surface expression. (c) Intracellular expression of TLR2, TLR4, MD-2 and CD14 by FACS in permeabilized A549 cells submitted to a 24-hour cyclic stretch or in cells kept in static condition. TLR2 was detected in permeabilized A549 cells after 24 hours of cyclic stretch. CD14 was detected, but not increased, by cyclic stretch. Neither TLR4 nor MD-2 proteins were detected by FACS in permeabilized cells, regardless of the experimental condition. Results are expressed as the mean fluorescence index (MFI) ± standard deviation of three distinct experiments. *P < 0.05 between static and stretched cells.
Cyclic stretch increases intracellular TLR2 in lung epithelial cells
Cells were harvested after a 24-hour cyclic stretch for FACS analysis. No TLR2 was detected in A549 cells in a static condition in nonpermeabilized cells as well as in permeabilized cells. After 24 hours of cell stretching, TLR2 became detectable in permeabilized epithelial cells and to a lesser extent in nonpermeabilized cells (Figure 2b, c). TLR4 and MD-2 remained undetectable in intact or permeabilized cells regardless of the mechanical stress. Finally, cyclic stretch did not influence CD14 expression. The promonocytic human THP1 cells expressing TLR2, TLR4, CD14 and MD-2 were used as positive controls for the mAbs used in the experiments (data not shown).
Cyclic stretch increases the inflammatory response of lung epithelial cells to bacterial lipopeptide
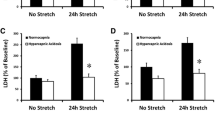
A549 cells submitted to a 24-hour stretch released higher levels of IL-6 and IL-8 in response to an additional 8-hour stimulation by the synthetic BLP Pam3CSK4 than did cells not submitted to cyclic stretch (Figure 3a). A synergistic effect between cyclic stretch and Pam3CSK4 was also observed at the IL-6 and IL-8 mRNA level (Figure 3b).

Inflammatory mediator production by A549 cells stimulated with Pam 3 CSK 4 in static and cyclic stretch conditions. (a) IL-6 and IL-8 protein concentrations in conditioned supernatants from cells stretched for 24 hours or kept in static condition, and subsequently stimulated for 8 hours with bacterial lipopeptide (BLP). Cyclic stretch increased the inflammatory response of A549 cells in response to BLP. (b) IL-6 and IL-8 mRNA levels in human type II-like A549 alveolar cells stretched for 24 hours or kept in static condition, and subsequently stimulated for 8 hours with BLP. mRNA levels were normalized to the expression of GAPDH mRNA. All values are reported as the fold increase compared with control static cells normalized to 1. For both inflammatory markers, a clear synergistic effect between cyclic stretch and BLP is observed. Results are expressed as the mean ± standard deviation of four distinct experiments. *P < 0.05 between static and stretched cells stimulated with BLP.
Cyclic stretch-induced upregulation of TLR2 depends on the p38 MAPK signaling pathway
To address the question of the signaling pathway by which cyclic stretch increased TLR2 mRNA, we tested the effects of a NF-κB inhibitor (Sn50, 18 μM), and a p38 MAPK-specific inhibitor (SB203580, 1 μM) on cyclic stretch-induced IL-8 expression. The stretch-induced TLR2 expression in A549 cells was markedly reduced by the p38 MAPK inhibitor, whereas the NF-κB blocker did not inhibit stretch-induced IL-8 (Figure 4). Notably, we checked that SB203580 did not alter cell viability using a 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide assay (MTT) test (data not shown).

Toll-like receptor 2 expression in A549 cells stretched with various inhibitors. IL-8 mRNA levels were measured as a positive control of cell activation by cyclic stretch. mRNA levels were normalized with the expression of GAPDH mRNA. All values are reported as the fold increase compared with control static cells normalized to 1. SB203580 significantly reduced the toll-like receptor (TLR)2 mRNA level induced by stretch, whereas Sn50 had no effect. The opposite was observed for IL-8 expression induced by cyclic stretch. Results are expressed as the mean ± standard error of the mean of three distinct experiments. *P < 0.05 between stretched cells with or without SB 203580. SB, spontaneously breathing.
In vivo studies
Mechanical ventilation in healthy rabbits causes a marked increase of lung TLR2 mRNA
MV induced a marked increase in lung TLR2 mRNA, much greater than those observed for TLR4, MD-2, and CD14 (Figure 5).

Pathogen recognition receptor expression in rabbits submitted to mechanical ventilation or kept spontaneously breathing. Lung IL-8 mRNA levels were measured as a positive control of lung inflammation induced by mechanical ventilation (MV). mRNA levels were normalized with the expression of GAPDH mRNA. All values are shown as the fold increase compared with spontaneously breathing (SB) rabbits normalized to 1. MV induced a strong increase of toll-like receptor (TLR)2 mRNA levels, higher than TLR4, MD-2 and CD14. Results are expressed as mean ± standard deviation. *P < 0.05 between SB and MV rabbits.
In these ventilated rabbits, no macroscopic lung abnormalities were noted. Microscopic studies, however, showed features of mild lung injury, including septal thickening and inflammatory cell infiltration together with a small increase in IL-8 mRNA (Figures 6 and 7) - features absent in lungs from SB animals.

Light photomicrographs of rabbit lungs in various conditions. (a) Lung from a spontaneously breathing (SB) animal. (b) Rabbit lung after 32-hour mild-stretch mechanical ventilation (MV). (c) Lung from a SB animal following tracheal instillation of the synthetic bacterial lipopeptide (BLP), Pam3CSK4, and sacrificed 8 hours later. (d) Lung from an animal submitted to 24-hour mild-stretch MV followed by tracheal instillation of BLP and sacrifice 8 hours later. The main abnormalities were septal thickening and inflammatory cell infiltration in rabbits submitted to MV, increased by the presence of BLP. Representative fields, original magnification, x10; hematoxylin and eosin stain.

IL-8 expression following instillation of Pam 3 CSK 4 in rabbits submitted previously, or not, to mechanical ventilation. IL-8 mRNA levels were normalized with the expression of GAPDH mRNA. Values are reported as the fold increase compared with control spontaneously breathing (SB) animals normalized to 1 (n = 4 per group). Results are expressed as the mean ± standard deviation. Lung response to bacterial lipopeptide (BLP) was markedly enhanced by previous mechanical ventilation (MV). *P < 0.05 between SB and MV stimulated rabbits.
Mechanical ventilation increases lung inflammation induced by bacterial lipopeptide
Despite MV and lung insult by Pam3CSK4, hemodynamic stability and adequate blood oxygenation were achieved in our model (Table 1).
The endobronchial instillation of the BLP Pam3CSK4 induced more lung injury in rabbits previously ventilated for 24 hours compared with those who were SB during that period of time (Figure 6). These microscopic abnormalities included more inflammatory cell infiltration and more pronounced alveolar septa thickening.
Although Pam3CSK4 alone did not induce much IL-8 mRNA, we observed a clear synergistic effect between MV and BLP on lung IL-8 gene expression (Figure 7).
Discussion
Herein, we show that cyclic stretch of lung cells and MV in rabbits upregulate TLR2. MV-dependent TLR2 upregulation was associated with a marked increase in lung cell activation and lung injury after a challenge with Pam3CSK4, a classical TLR2 agonist. These results are consistent with cyclic stretch increasing lung cell reactivity to TLR2 bacterial agonists. Prestretching type II-like human alveolar A549 cells induced a marked increase in the induction of proinflammatory mediators such as IL-6 and IL-8 in response to BLP; a synergistic effect was observed between MV and TLR2 activation by Pam3CSK4. We cannot, however, exclude that the increased expression of TLR2 by A549 cells is solely and directly caused by cyclic stretch. Actually, the release of inflammatory mediators by stretched cells might account at least in part for the observation reported herein [19].
We failed to detect TLR2 at the surface of epithelial cells by FACS. Depending on cell types, TLRs can be expressed at the surface or intracellularly. TLR4, for example, is expressed at the surface of myeloid cells but in intracellular compartments in epithelial cells such as A549, BEAS-2B cells, intestinal cells as well as endothelial cells [20, 21]. Similarly, TLR2 - which is also expressed at the plasma membrane in myeloid cells - is found intracellularly in corneal epithelial cells [22]. Using a permeabilization protocol, we showed that cyclic stretch induced intracellular TLR2 upregulation after cyclic stretch in A549 cells. TLR2 mRNA was also increased by cyclic stretch in A549 cells.
Cyclic stretch has been shown to induce proinflammatory gene transcription in lung epithelial cells via either NF-κB or MAPK pathways [7]. We therefore tested the effect of pharmacological inhibitors of the NF-κB and the p38 MAPK signalization pathways on cyclic stretch-dependent induction of TLR2. The p38 MAPK inhibitor (SB203580) abrogated cyclic stretch-induced TLR2 gene expression, whereas NF-κB had no effect. These findings are consistent with the fact that the MAPK pathway is critical in the cyclic stretch-induced cell activation. These data also indicate that TLR2 regulation is not entirely dependent on the NF-κB pathway, depending on the stimulus [23]. The involvement of the p38 MAPK pathway in TLR2 regulation by cyclic stretch is suggestive of TLR2 mRNA stabilization (post-transcriptional effect) rather than de novo TLR2 transcription, but this remains to be ascertained using TLR2 transcription assays.
We next tested whether these in vitro findings could be reproduced in an animal model. We showed that mild-stretch MV induced a marked upregulation of lung TLR2. Furthermore, the treatment with BLP of rabbits ventilated for 24 hours induced a marked IL-8 expression into the airway and significant lung injury, as compared with SB rabbits.
This is the first report showing the MV-induced upregulation of TLR2, and a synergistic effect between MV and BLP. These results are reminiscent of those found with the LPS/CD14/TLR4 pathway [3–6, 24], and highlight the increased reactivity of lung cells to Gram-positive bacterial products and bacteria induced by MV via the TLR2 pathway [25, 26]. Moreover, in contrast with previous findings, we observed that TLR2 and TLR4 could be differentially upregulated by cyclic stretch. Accordingly, the TLR2 upregulation was strikingly more pronounced, especially in the in vivo model. Interestingly, similar findings were obtained in vitro in human endothelial cells submitted to either a laminar or a turbulent flow [17]. Such data suggest that differences exist between TLRs with respect to their expression regulation by abnormal mechanical strains.
Our work adds to a growing body of literature implicating pattern-recognition receptors of the innate immunity in the development of ventilator-induced lung injury. A large tidal volume MV increased TLR4 and TLR2 expression in the lungs of preterm lambs, but the effect of the corresponding agonist was not tested [27]. Others have demonstrated that lung hyperinflation increased TLR4 expression in rats with abdominal sepsis [25]. In contrast, TLR4 knockout mice were protected against ventilator-induced lung injury due to lung hyperinflation [28]. Interestingly, it was shown that even protective MV applied to healthy mouse lungs could modify TLR expression [29].
Any clinical translation should be made cautiously, however, since the chosen ventilator settings were not in accordance with the current guidelines. The aim was to cause overdistension within healthy lungs, a goal difficult to achieve with the recommended settings (that is, 6 to 8 ml/kg and positive end-expiratory pressure). Such lung-protective settings, however, could lead to regional overdistension when applied to injured lungs [30, 31]. Another limitation is that we have not studied the relationship between lung stretch magnitude and TLR2 expression since only one degree of cell elongation as well as one tidal volume value were tested. We cannot therefore conclude definitively that TLR2 expression is stretch dependent.
Conclusions
Cyclic stretch mimicking MV in vitro induced TLR2 expression and enhanced TLR2 reactivity to BLP of epithelial human cells, most probably through a p38 MAPK-dependent effect. These findings were in part reproduced in a rabbit model, where mild-stretch MV increased lung injury in response to the instillation of a TLR2-dependent BLP. Translated into the clinical arena, our findings suggest that the increase of the TLR2 expression of the lung epithelia caused by MV may prime the lung, which in turn will overreact to the presence of colonizing or infecting Gram-positive bacteria within the airway and worsen lung injury. The potential impact on lung bacterial clearance warrants further studies.
Key messages
-
Mechanical ventilation magnifies lung inflammatory response to a Gram-positive bacterial product, and in turn worsens lung injury.
-
The stretch-dependent TLR2 upregulation could account for such findings.
-
The p38 MAPK pathway could play a critical role.
Abbreviations
- BLP:
-
bacterial lipopeptide
- BSA:
-
bovine serum albumin
- DMEM:
-
Dulbecco's modified Eagle's medium
- ELISA:
-
enzyme-linked immunosorbent assay
- FACS:
-
fluorescence-activated cell sorting
- FCS:
-
fetal calf serum
- FIO2:
-
fraction of inspired O2
- IL:
-
interleukin
- LPS:
-
lipopolysaccharide
- mAb:
-
monoclonal antibody
- MAPK:
-
mitogen-activated protein kinase
- MV:
-
mechanical ventilation
- NF:
-
nuclear factor
- PBS:
-
phosphate-buffered saline
- PCR:
-
polymerase chain reaction
- RT:
-
reverse transcriptase
- SB:
-
spontaneously breathing
- TLR:
-
toll-like receptor.
References
Chastre J, Fagon JY: Ventilator-associated pneumonia. Am J Respir Crit Care Med 2002, 165: 867-903.
Rubinstein E, Kollef MH, Nathwani D: Pneumonia caused by methicillin-resistant Staphylococcus aureus . Clin Infect Dis 2008,46(Suppl 5):S378-S385.
Murphy DB, Cregg N, Tremblay L, Engelberts BD, Laffey JG, Slutsky AS, Romaschin A, Kavanagh BP: Adverse ventilatory strategy causes pulmonary-to-systemic translocation of endotoxin. Am J Respir Crit Care Med 2000, 162: 27-33.
O'Mahony DS, Liles WC, Dhanireddy S, Frevert C, Liggit D, Martin T, Matute-Bello G: Mechanical ventilation interacts with endotoxemia to induce extrapulmonary organ dysfunction. Crit Care 2006, 10: R136. 10.1186/cc5050
Bregeon F, Delpierre S, Chetaille B, Kajikawa O, Martin TR, Autillo-Touati A, Jammes Y, Pugin J: Mechanical ventilation affects lung function and cytokine production in an experimental model of endotoxemia. Anesthesiology 2005, 102: 331-339. 10.1097/00000542-200502000-00015
Moriyama K, Ishizaka A, Nakamura M, Kotani T, Yamamoto S, Ogawa EN, Kajikawa O, Frevert CW, Kotake Y, Morisaki H, Koh H, Tasaka S, Martin TR, Takeda J: Enhancement of the endotoxin recognition pathway by ventilation with a large tidal volume in rabbits. Am J Physiol Lung Cell Mol Physiol 2004, 286: L1114-L1121. 10.1152/ajplung.00296.2003
Oudin S, Pugin J: Role of MAP kinase activation in interleukin-8 production by human BEASB-2B bronchial epithelial cells submitted to cyclic stretch. Am J Respir Cell Mol Biol 2002, 27: 107-114.
Charles PE, Piroth L, Desbiolles N, Lequeu C, Martin L, Portier H, Chavanet P: New model of ventilator-associated pneumonia in immunocompetent rabbits. Crit Care Med 2002, 30: 2278-2283. 10.1097/00003246-200210000-00016
Charles PE, Etienne M, Croisier D, Piroth L, Lequeu C, Pugin J, Portier H, Chavanet P: The impact of mechanical ventilation on the moxifloxacin treatment of experimental pneumonia caused by streptococcus pneumoniae. Crit Care Med 2005, 33: 1029-1035. 10.1097/01.CCM.0000163404.35338.72
Marquette CH, Wermert D, Wallet F, Copin MC, Tonnel AB: Characterization of an animal model of ventilator-acquired pneumonia. Chest 1999, 115: 200-209. 10.1378/chest.115.1.200
Schortgen F, Bouadma L, Joly-Guillou ML, Ricard JD, Dreyfuss D, Saumon G: Infectious and inflammatory dissemination are affected by ventilation strategy in rats with unilateral pneumonia. Intensive Care Med 2004, 30: 693-701. 10.1007/s00134-003-2147-7
Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, Klimpel GR, Godowski P, Zychlinsky A: Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science 1999, 285: 736-739. 10.1126/science.285.5428.736
Elson G, Dunn-Siegrist I, Daubeuf B, Pugin J: Contribution of toll-like receptors to the innate immune response to gram-negative and gram-positive bacteria. Blood 2007, 109: 1574-1583. 10.1182/blood-2006-06-032961
Droemann D, Goldmann T, Branscheid D, Clark R, Dalhoff K, Zabel P, Vollmer E: Toll-like receptor 2 is expressed by alveolar epithelial cells type II and macrophages in the human lung. Histochem Cell Biol 2003, 119: 103-108.
Zhang P, Summer WR, Bagby GJ, Nelson S: Innate immunity and pulmonary host defense. Immunol Rev 2000, 173: 39-51. 10.1034/j.1600-065X.2000.917306.x
Takeuchi O, Hoshino K, Akira S: Cutting edge: TLR2-deficient and Myd88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol 2000, 165: 5392-5396.
Dunzendorfer S, Lee HK, Tobias PS: Flow-dependent regulation of endothelial toll-like receptor 2 expression through inhibition of sp1 activity. Circ Res 2004, 95: 684-691. 10.1161/01.RES.0000143900.19798.47
Altemeier WA, Matute-Bello G, Frevert CW, Kawata Y, Kajikawa O, Martin TR, Glenny RW: Mechanical ventilation with moderate tidal volumes synergistically increases lung cytokine response to systemic endotoxin. Am J Physiol Lung Cell Mol Physiol 2004, 287: L533-L542. 10.1152/ajplung.00004.2004
Regueiro V, Moranta D, Capos MA, Margareto J, Garmendia J, Bengoechea JA: Klebsiella pneumoniae increases the levels of toll-like receptors 2 and 4 in human airway epithelial cells. Infect Immun 2009, 77: 714-724. 10.1128/IAI.00852-08
Guillot L, Medjane S, Le-Barillec K, Balloy V, Danel C, Chignard M, Si-Tahar M: Response of human pulmonary epithelial cells to lipopolysaccharide involves toll-like receptor 4 (TLR4)-dependent signaling pathways: evidence for an intracellular compartmentalization of TLR4. J Biol Chem 2004, 279: 2712-2718.
Dunzendorfer S, Lee HK, Soldau K, Tobias PS: Toll-like receptor 4 functions intracellularly in human coronary artery endothelial cells: roles of lbp and scd14 in mediating lps responses. Faseb J 2004, 18: 1117-1119.
Ueta M, Nochi T, Jang MH, Park EJ, Igarashi O, Hino A, Kawasaki S, Shikina T, Hiroi T, Kinoshita S, Kiyono H: Intracellularly expressed TLR2s and TLR4s contribution to an immunosilent environment at the ocular mucosal epithelium. J Immunol 2004, 173: 3337-3347.
Haehnel V, Schwarzfischer L, Fenton MJ, Rehli M: Transcriptional regulation of the human toll-like receptor 2 gene in monocytes and macrophages. J Immunol 2002, 168: 5629-5637.
Pugin J, Dunn I, Jolliet P, Tassaux D, Magnenat JL, Nicod LP, Chevrolet JC: Activation of human macrophages by mechanical ventilation in vitro. Am J Physiol 1998, 275: L1040-L1050.
Villar J, Cabrera N, Casula M, Flores C, Valladares F, Muros M, Blanch L, Slutsky AS, Kacmarek RM: Mechanical ventilation modulates toll-like receptor signaling pathway in a sepsis-induced lung injury model. Intensive Care Med 2010, 36: 1049-1057. 10.1007/s00134-010-1799-3
Charles PE, Barbar S, Toll-like receptors: A link between mechanical ventilation, innate immunity and lung injury? Intensive Care Med 2010, 36: 909-911. 10.1007/s00134-010-1804-x
Hillman NH, Moss TJ, Kallapur SG, Bachurski C, Pillo JJ, Polgase GR, Nitsos I, Kramer BW, Jobe AH: Brief, large tidal volume ventilation initiates lung injury and a systemic response in fetal sheep. Am J Respir Crit Care Med 2007, 176: 575-581. 10.1164/rccm.200701-051OC
Hu G, Malik AB, Minshall RD: Toll-like receptor 4 mediates neutrophil sequestration and lung injury induced by endotoxin and hyperinflation. Crit Care Med 2010, 38: 194-201. 10.1097/CCM.0b013e3181bc7c17
Vaneker M, Joosten LA, Heunks LM, Snijdelaar DG, Halbertsma FJ, van Egmond J, Netea MG, van der Hoeven JG, Scheffer GJ: Low-tidal-volume mechanical ventilation induces a toll-like receptor 4-dependent inflammatory response in healthy mice. Anesthesiology 2008, 109: 465-472. 10.1097/ALN.0b013e318182aef1
Terragni PP, Rosboch G, Tealdi A, Corno E, Menaldo E, Davini O, Gandini G, Herrmann P, Mascia L, Quintel M, Slutsky AS, Gattinoni L, Ranieri M: Tidal hyperinflation during low volume ventilation in acute respiratory distress syndrome. Am J Respir Crit Care Med 2007, 175: 160-166.
Sartorius A, lu Q, Vieira S, Tonnellier M, Lenaour G, Goldstein I, Rouby JJ: Mechanical ventilation and lung inflation in the genesis of air-space enlargement. Crit Care 2007, 11: R14. 10.1186/cc5680
Acknowledgements
The authors wish to thank Dr Th Fumeaux for his scientific and technical advices. JP was supported by the Swiss National Science Foundation for Scientific Research. P-EC was supported by an unrestricted grant from Wyeth.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
PEC, PT, SDB and JP designed the experiments, carried out the study, analyzed the results and drafted the manuscript. DC, IDS, JD and PC participated in the study. All of the authors read and approved the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Charles, PE., Tissières, P., Barbar, S.D. et al. Mild-stretch mechanical ventilation upregulates toll-like receptor 2 and sensitizes the lung to bacterial lipopeptide. Crit Care 15, R181 (2011). https://doi.org/10.1186/cc10330
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/cc10330