
Abstract
Background
Experimental and clinical studies on sepsis have demonstrated activation of the innate immune response following the initial host–bacterial interaction. In addition, mechanical ventilation (MV) can induce a pulmonary inflammatory response. How these two responses interact when present simultaneously remains to be elucidated. We hypothesized that MV modulates innate host response during sepsis by influencing Toll-like receptor (TLR) signaling.
Design
Prospective, randomized, controlled animal study.
Subjects
Male, septic Sprague–Dawley rats.
Interventions
Sepsis was induced by cecal ligation and perforation. At 18 h, surviving animals had the cecum removed and were randomized to spontaneous breathing or two strategies of MV for 4 h: high (20 ml/kg) tidal volume (V T) with no positive end-expiratory pressure (PEEP) versus low V T (6 ml/kg) plus 10 cmH2O PEEP.
Measurements and main results
Histological evaluation, TLR-2, TLR-4, inhibitory kappaB alpha (IκBα), interleukin-1 receptor-associated kinase-3 (IRAK-3) gene expression, protein levels and immunohistochemical lung localization, inflammatory cytokines gene expression, and protein serum concentrations were analyzed. MV with low V T plus PEEP attenuated sepsis-associated TLR-4 activation, and produced a significant decrease of IRAK-3 gene expression and protein levels, a significant increase of IκBα, and a decrease in lung gene expression and serum levels of cytokines. High-V T MV caused a significant increase of TLR-4 and IRAK-3 protein levels, lung and systemic cytokines, and mortality, and a significant decrease of IκBα.
Conclusions
Our findings suggest a novel mechanism that could partially explain how MV modulates the innate immune response in the lung by interfering with cellular signaling pathways that are activated in response to pathogens.
Similar content being viewed by others

Introduction
Mechanical ventilation (MV) is essential for the management of many patients with sepsis-induced lung injury; however, evidence from experimental and clinical studies has demonstrated that certain MV strategies can cause or worsen lung injury [1–6]. Although the Acute Respiratory Distress Syndrome (ARDS) Network trial demonstrated that lowering tidal volumes (V T) delivered to patients with acute lung injury (ALI) reduced mortality [7], the molecular mechanisms underlying this outcome remain to be elucidated.
Lung cells express a large repertoire of genes under transcriptional control of biomechanical forces [8, 9] and bacterial infections [10]. Lipopolysaccharide (LPS) has a major role in the devastating nature of sepsis, in part by binding to cell-surface receptors exerting pro-inflammatory effects. Toll-like receptors (TLRs) detect host invasion by pathogens and constitute the key link between the innate and adaptive immune responses [11, 12]. TLR signaling is regulated through a series of intracellular proteins including the interleukin (IL)-1 receptor-associated kinases (IRAK). Downstream in the TLR-4 activation, the IkappaB kinase (IKK) complex catalyzes phosphorylation of inhibitory kappaB (IκB) proteins [13]. This causes degradation of cytoplasmic inhibitors of nuclear factor-kappaB (NF-κB) and nuclear translocation of NF-κB. NF-κB is normally sequestered in the cytoplasm through association with IκBα. After cellular stimulation, IκBα is degraded, allowing NF-κB to move to the nucleus, where it binds to specific promoter sequences and initiates transcription, resulting in expression of cytokines such as IL-6 and tumor necrosis factor-alpha (TNF-α) and other gene products [14–18]. There are four distinct human genes encoding for the four IRAK proteins. IRAK-3 (also known as IRAK-M) inhibits NF-κB activation and negatively regulates TLR signaling [19]; it is a mediator of LPS-induced tolerance [20] and a selective inhibitor of the classical [19] and alternative NF-κB pathway [21].
Held et al. [22] reported that inflammatory responses induced by high-V T MV were similar to those evoked by LPS, and Moriyama et al. [23] postulated that high-V T MV enhances the LPS recognition pathway via upregulation of CD14. Therefore, we hypothesized that MV can modulate the TLR/IRAK-3 signaling pathway in lung during sepsis. We evaluated this hypothesis by comparing protective versus injurious MV in an animal model of sepsis-induced ALI.
Methods
Animal preparation
The protocol was approved by Hospital Universitario N.S. de Candelaria Research Committee and the Committee for the Use and Care of Animals, University of La Laguna (Tenerife, Spain). We studied 40 male Sprague–Dawley rats weighing 300–350 g. Animals were anesthetized by intraperitoneal injection of 50 mg/kg body weight ketamine hydrochloride and 2 mg/kg body weight xylazine. Sepsis was then induced by cecal ligation and perforation (CLP) [24]. A detailed description of this experimental model is provided elsewhere [4]. Eighteen hours after CLP, the peritoneal cavity was reopened in surviving animals, and the cecum was then excised distal to the ligature and removed. After closing the abdomen, each animal received 10 ml normal saline subcutaneously for fluid resuscitation. Then, we performed a tracheotomy using a 14-G Teflon catheter. Thereafter, animals were paralyzed with 1 mg/kg pancuronium bromide and connected to a time-cycled, volume-limited rodent ventilator (Ugo Basile, Varese, Italy).
Experimental protocol
Following surgical procedures, surviving septic rats were randomly divided into three groups: (1) control: anesthetized, spontaneously breathing, (2) low-V T group: ventilated with 6 ml/kg plus 10 cmH2O positive end-expiratory pressure (PEEP), and (3) high-V T group: ventilated with 20 ml/kg and zero PEEP. FiO2 was 60%. In pilot studies we monitored animals invasively and found comparable blood gases at the end of the 4-h ventilation period using FiO2 = 0.60 (PaO2 141 ± 21 versus 159 ± 29 mmHg and PaCO2 43 ± 4 versus 39 ± 3 mmHg, for the low-V T plus PEEP and high-V T groups, respectively; n = 5 rats/group). As such, during our experimental protocol, animals were monitored noninvasively. Respiratory rate was set to maintain constant minute ventilation in both groups. These settings were maintained for 4 h, while anesthetized and paralyzed, supine on a restraining board inclined 20° to the horizontal.
Histological examination
At the end of the 4-h ventilation period, a midline thoracotomy/laparotomy was performed in the first six surviving rats from each group and the abdominal vessels were transected. The heart and lungs were removed en bloc. The lungs were isolated from the heart, the trachea was cannulated, and the right lung was fixed by intratracheal instillation of 3 ml 10% neutral buffered formalin. After fixation, the lungs were floated in 10% formalin for 1 week. Lungs were then serially sliced from apex to base and specimens were embedded in paraffin, then cut (3 μm thickness), stained with hematoxylin–eosin, and examined under light microscopy [see Electronic Supplementary Material (ESM) for details].
RNA extraction and reverse transcription
Left lungs were excised, washed with saline, frozen in liquid nitrogen, and stored at −80°C for subsequent RNA extraction. Lungs were homogenized, and total lung tissue RNA was extracted using TRIreagent (Sigma, Hamburg, Germany) and DNase I digestion (Amersham Biosciences, Essex, UK) [25, 26]. Methods for expression levels of tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and IRAK-3 genes by real-time polymerase chain reaction (PCR) are described in the ESM.
Cytokine serum levels
At the end of the 4-h experimental period, 2 ml blood was collected from the six surviving rats in each group by cardiac puncture and centrifuged for 15 min at 3,000 rpm. Sera were divided into aliquot portions and frozen at −80°C. TNF-α and IL-6 serum protein concentrations were measured by enzyme-linked immunosorbent assay (ELISA) (see ESM for details).
Western immunoblotting
Detection of TLR-2, TLR-4, IκBα, and IRAK-3 protein expression was performed by Western blotting, as described in the ESM.
Immunohistochemistry for IRAK-3
Immunohistochemical stains were performed by applying a standard avidin–biotin complex technique. Protein was visualized using 3-amino-9-ethylcarbazole as substrate (red-pink color), and nuclei were lightly counterstained using hematoxylin (see ESM for details).
Statistical analysis
Statistical analysis was performed with the Fisher exact test and paired and unpaired Student’s t tests. Comparisons involving all groups were performed with one-way analysis of variance. If a difference was found, Student’s t test was applied. Values derived from cytokine gene expression were expressed as group median, normalized by the lowest level in the group, and tested with the Kruskall–Wallis test and the Mann–Whitney U test. Data from ELISA were analyzed by the Student–Newman–Keuls pairwise multiple-range test. Data analysis was performed using SPSS 15.0 (SPSS Inc., Chicago, IL). A value of p < 0.05 was considered significant.
Results
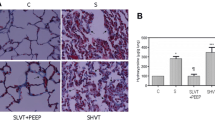
Ten rats died from sepsis after CLP; the remaining 30 animals were randomly allocated to the three study groups. After cecum removal, four animals died in the spontaneous breathing group. No animals ventilated with low V T and PEEP died during MV, but two out of ten animals ventilated with high V T died within the same experimental period (p = 0.042). Respiratory rate was 90 ± 1 cycles/min in the low-V T group and 30 ± 1 in the high-V T group. Mean peak inspiratory pressure was 28 ± 1 cmH2O in the high-V T group. Animals ventilated with high V T had histological evidence of lung injury (Fig. 1), with the highest histological scores (11.5 ± 2.0), whereas animals ventilated with low V T plus PEEP had a histological score that was lower than in septic, spontaneous breathing animals (2.2 ± 0.5 versus 4.5 ± 0.3, respectively) (p < 0.0001).

Representative histopathological features of septic lungs after 4 h of spontaneous breathing or mechanical ventilation. Left panel lung from a septic, spontaneous breathing animal showing pulmonary infiltrates and perivascular edema. Middle panel lung from a septic animal after 4 h of mechanical ventilation with low tidal volume plus 10 cmH2O PEEP showing occasional cellular infiltrates, reduced edema, and relatively normal lung architecture. Right panel lung from a septic animal after 4 h of mechanical ventilation with high tidal volume showing pulmonary infiltrates, perivascular edema, and derangement of lung architecture (hematoxylin–eosin, ×200)
Cytokine gene expression and protein levels
TNF-α and IL-6 were expressed in lung homogenates of all septic rats (Fig. 2a). High VT increased TNF-α and IL-6 gene expression compared with spontaneously breathing animals. Low VT plus PEEP caused similar IL-6 gene expression as spontaneously breathing animals but decreased TNF-α gene expression compared with unventilated animals (p < 0.001) (Fig. 2a). After 4 h of high VT, TNF-α and IL-6 serum concentrations were higher than in unventilated animals (166 ± 31 versus 89 ± 17 pg/ml, p < 0.001; 2,209 ± 153 versus 801 ± 86 pg/ml, p < 0.0001, respectively). In animals ventilated with low VT, TNF-α and IL-6 serum levels were lower than in spontaneous breathing animals (23 ± 9 versus 89 ± 17 pg/ml, p = 0.0001; and 426 ± 58 versus 801 ± 86, p < 0.0001, respectively) (Fig. 2b, c).

a Fold changes in gene expression of TNF-α and IL-6 in the lungs after 4 h of spontaneous breathing, mechanical ventilation with low VT plus 10 cmH2O PEEP, or with high VT. Bars represent the median (n = 6 rats/group). *p < 0.001 (for TNF-α between septic versus septic/low VT + PEEP, and septic/low VT + PEEP versus septic/high VT); **p < 0.01 (for IL-6 between septic/low VT + PEEP versus septic/high VT). b, c Effect of 4 h of spontaneous breathing in nonventilated, anesthetized animals and of 4 h of mechanical ventilation with low VT and high VT on systemic protein levels of TNF-α (a) and IL-6 (b). Bars represent mean values (n = 6 rats/group)
TLR-4 and TLR-2 regulation and IκBα protein levels
MV modulated TLR expression in septic lungs (Fig. 3). We did not detect changes in TLR-2 protein levels in any of the experimental groups. TLR-4 protein levels were higher in rats ventilated with high V T than in low V T plus PEEP or unventilated animals (p < 0.001). MV induced degradation of IκBα to a greater extent in septic animals ventilated with high V T compared with nonventilated or ventilated with low V T plus PEEP (p < 0.001). MV with low V T plus PEEP had no detectable effects on IκBα degradation.

Western blotting showing the effects of mechanical ventilation on TLR-4, TLR-2, and IκBα protein levels in several groups of septic animals: spontaneous breathing, ventilated with low tidal volume plus 10 cmH2O PEEP, and ventilated with high tidal volume for 4 h. Bars represent densitometric analysis [mean ± standard deviation (SD)] of six rats per group. Human lung fibroblasts IMR-90 cells were selected as positive control for TLR-2 protein levels. *p < 0.05 versus nonventilated, septic rats; ***p < 0.001 versus nonventilated, septic rats; t p < 0.001 versus septic animals ventilated with low V T plus PEEP; τ p < 0.001 versus septic animals ventilated with low V T plus PEEP
IRAK-3 gene expression and protein levels in the lungs
Levels of IRAK-3 gene expression increased slightly when septic animals were ventilated with high V T (Fig. 4a). However, MV with low V T plus PEEP caused marked downregulation of IRAK-3 gene expression compared with septic baseline (p = 0.004). IRAK-3 gene expression during low- versus high-V T MV was statistically different (p = 0.003). IRAK-3 protein levels in the lungs were downregulated in animals ventilated with low V T plus PEEP compared with those unventilated (p < 0.01). However, IRAK-3 protein levels were higher after 4 h of high V T as compared with all groups (p < 0.001) (Fig. 4b).

a Fold changes in IRAK-3 gene expression in septic lungs after 4 h of spontaneous breathing (nonventilated) or mechanical ventilation with 6 ml/kg plus 10 cmH2O PEEP and 20 ml/kg and no PEEP. Bars represent the median fold increase compared with nonventilated animals. *p < 0.004 versus nonventilated, **p < 0.003 versus ventilated with low VT plus PEEP. b Changes of IRAK-3 protein levels in lungs of septic rats after 4 h of mechanical ventilation with low or high VT. Bars represent densitometric values of IRAK-3 protein levels from all animals in each group (n = 6 animals per group). Data are reported as mean ± SD (n = 6 independent experiments). S septic, nonventilated rats; S low V T + PEEP septic rats ventilated with low VT plus 10 cmH2O PEEP; VT tidal volume; S high V T septic rats ventilated with high VT. **p < 0.01 versus septic, nonventilated rats; ***p < 0.001 versus septic, nonventilated rats; tp < 0.001 versus septic rats, ventilated with low VT plus PEEP
Immunohistochemical localization of IRAK-3
IRAK-3 was present in both the cytoplasm and nucleus. IRAK-3 was detected in the interstitium and bronchiolar epithelium of unventilated septic lungs. Positive cytoplasmic staining for IRAK-3 was found in epithelial type II cells and interstitial macrophages surrounding the alveolus in the lungs of spontaneous breathing animals and those ventilated with low V T. Positive staining of lung tissue with specific antibodies confirmed identification and localization of both cell types (see Fig. S1 of ESM). Positive staining for IRAK-3 was greatest in lungs ventilated with high V T (Fig. 5).

Representative immunohistochemical staining for IRAK-3 in lung tissues of various groups of septic animals. a Spontaneous breathing rats, b rats ventilated at low V T (6 ml/kg) plus 10 cmH2O PEEP, c rats ventilated at high V T (20 ml/kg). IRAK-3 expression is higher in lungs from septic rats ventilated at high V T (c). In the inset to b, positive type II cells located at the corner of converging alveolar septa (white arrow), and positive macrophages (black arrow). In inset to c, cytoplasmic and nuclear localization of IRAK-3 in an interstitial macrophage. IRAK-3 protein was visualized using 3-amino-9-ethylcarbazole as substrate (red-pink color), and nuclei were lightly counterstained using hematoxylin (blue/violet color). The insets are magnification of the indicated areas in b and c, respectively. Scale bars 40 μm
Discussion
Our study suggests that the TLR signaling pathway plays an important role in perpetuating pulmonary inflammatory responses which are dependent on the ventilatory strategy used during sepsis-induced ALI. Our results further support the use of low V T and moderate PEEP to minimize ventilator-induced ALI and to interfere with pro-inflammatory signaling cascades, limiting release of injurious host-derived molecules. To our knowledge, this is the first report linking the cellular response to MV with the specific inflammatory response caused by a systemic, nonpulmonary bacterial infection, and the first to provide evidence that MV regulates TLR-4/IRAK-3/IκBα levels in the lung in sepsis-induced ALI.
How mechanical forces are sensed by immune cells and converted into biochemical signals for intracellular signal transduction is unclear. Experimental [2–4, 27–31] and clinical [5] studies have shown that mechanical stretch can release proteins and mediators from a variety of lung cells. In addition, an imbalance of pro-inflammatory and anti-inflammatory cytokines predisposes to cellular injury and activation of secondary signaling pathways [32]. It might be possible that the changes in TLR-4 and IRAK-3 expression are increased by MV, as are many other genes [31, 33], but do not fully contribute to the augmentation of gene transcription and enhanced inflammation induced by MV in the septic lung. Many [4, 6, 33, 34] have reported the importance of the “two-hit model”: a previous pulmonary or systemic insult may sensitize lungs to injurious MV; however, injurious MV may also sensitize lungs to further pulmonary and systemic complications.
In an isolated perfused lung model in mice, Held et al. [22] found that MV with high V T activated NF-κB with release of cytokines in a similar manner to that of LPS. They concluded that the underlying mechanism by which overventilation activated NF-κB was unrelated to TLR-4. Moriyama et al. [23] examined the inflammatory effects in rabbits of ventilation with small (5 ml/kg) or large (20 ml/kg) V T after intratracheal LPS instillation. They postulated that the enhanced immune response to LPS resulting from ventilation with large V T may partially be controlled via upregulation of CD14. Previous studies have demonstrated increased NF-κB activation in ARDS patients [35], and that NF-κB activation in peripheral leukocytes of septic patients correlates with sepsis severity [36]. Furthermore, early alterations in neutrophil activation, particularly involving the ability to accumulate NF-κB in the nucleus, have been associated with outcome in ALI [14]. Vaneker et al. [37] reported the effects of 4-h MV using 8 ml/kg V T plus 4 cmH2O PEEP in healthy and knockout TLR-2 and TLR-4 mice and found that MV of healthy mice resulted in increased expression of endogenous TLR-4 ligands in the bronchoalveolar lavage (BAL) and enhanced TLR-4 lung gene expression associated with increased levels of TNF-α and IL-6. They found that MV increased messenger RNA (mRNA) expression of TLR-2 in lung tissue compared with nonventilated animals. However, ventilation of TLR-2 knockout mice did not result in different cytokine levels in lung tissue homogenates and plasma compared with their wild-type ventilated controls. They also found that MV did not increase cytokines plasma levels in TLR-4 knockout mice.
The modulation of TLR signaling may be a two-edged sword. On the one hand, TLRs are essential components of the innate immune response to pathogens; on the other hand, increased TLR expression and/or signaling may contribute to disease. Activation of TLRs induces expression of hundreds of genes in macrophages. TLR-induced genes follow two distinct patterns of activation on prolonged exposure to LPS or during early stages of sepsis: genes (including pro-inflammatory cytokines) are transiently silenced, which is associated with decreased inflammation, while others (including antimicrobial proteins) are activated, as an adaptation to the innate immune response [38]. Gene reprogramming that generates silencing of pro-inflammatory genes is typified by repression of mediators that initiate both systemic and local acute inflammation. This gene silencing or repression phase may develop rapidly and occurs variably in infectious and in noninfectious clinical states of severe inflammation, such as blunt trauma or hemorrhagic shock [39–41]. Sepsis causes upregulation of IRAK-3, which counteracts the TLR-4 signaling pathway leading to expression, production, and release of inflammatory mediators. Using a CLP model of sepsis in mice, Deng et al. [42] found marked upregulation of IRAK-3 in alveolar macrophages and modest induction of cytokine mRNA in the lungs at 12–24 h after intratracheal instillation of bacteria. The marked upregulation of IRAK-3 by high-V T MV in our study suggests that it could be partially responsible for mitigating the extent of inflammation and alveolar injury during the course of sepsis. By contrast, MV with low V T plus PEEP was able to reverse the upregulation of IRAK-3, a negative TLR regulator that in immunosuppression is upregulated; and this upregulation allows downregulatory TLR signaling, since the immune system needs to strike a balance constantly between activation and inhibition to avoid harmful and inappropriate inflammatory responses [43]. Many of the pro-inflammatory mediators that appear to be involved in the pathogenesis of ALI are under the regulatory control of NF-κB [14, 44]. In our experiments, injurious ventilation caused marked degradation of IκBα, resulting in higher lung gene expression and systemic levels of cytokines. It appears that modulation of TLR signaling during high-V T MV is important for maintenance of the initial LPS signal transduction that can lead to development of sepsis, multiple organ failure, and death. Studies exploring the effects of blocking the TLR-4 signaling pathway in models of ventilator-induced ALI are necessary to support this putative link. In this regard, TLRs and the IRAK proteins family might be therapeutic molecular targets which interfere with local or systemic inflammatory responses during critical illness.
Irrespective of the specific mechanism underlying IRAK-3 signaling, an interesting finding from our study is that the low-V T group not only had lower TNF-α and IRAK-3 mRNA levels than the high-V T group, but also had similar or lower levels compared with spontaneous breathing animals. These data suggest that the low-V T strategy may not only be less injurious but may in fact be therapeutic. We do not have a definitive explanation for this finding, but we postulate that ventilation of lungs with pre-existing damage using low V T and moderate PEEP levels was able to attenuate sepsis-induced ALI by a number of mechanisms: (1) animals ventilated with such a “protective” ventilation strategy were better oxygenated and ventilated than spontaneously breathing animals, (2) low V T and moderate PEEP avoids overdistension and the opening and closing of unstable lung units, and (3) in nonventilated septic animals, the failure of the alveolar epithelial barrier promotes further alveolar flooding and may increase transendothelial fluid and protein flux [45]. Septic, spontaneously breathing animals likely developed more hypoxemia, acidosis, and hemodynamic compromise, given that four of ten spontaneously breathing animals died in the 4-h experimental period. PEEP recruits flooded alveoli and improves oxygenation by redistributing extravascular lung water from alveoli to the perivascular space [46]. In addition, protective MV can modify gene expression of important components of the extracellular matrix and accelerate remodeling and repair of damaged lung tissue [47].
We acknowledge that the present study has some limitations. First, although we have shown that MV modulated TLR-4 activation in the context of sepsis-induced ALI, we did not conclusively demonstrate that TLR pathways are involved in the increased inflammation associated with high-V T MV because we did not examine the effect of disrupting these pathways. Second, we did not use IRAK-3-deficient animals to irrefutably demonstrate that IRAK-3 contributes to the pathogenesis of ventilator-induced ALI in the context of sepsis-induced ALI. However, studies from other investigators [42] have indicated that IRAK-3 is a selective regulator of different TLR downstream signaling processes. Third, a control group with nonseptic, healthy animals would have been useful for comparing the respective effects of MV and sepsis. We think that our data could have important clinical implications in the setting of sepsis-induced ALI. Both Altemeier et al. [33] and our group [4, 48] have reported that MV functions as a cofactor in development of ALI/ARDS and modulates the expression of inflammatory mediators in previously healthy animals or in response to sepsis or LPS. Our data also support the concept of TLR-4-targeted therapy in the context of sepsis-induced ALI. It has recently been reported in a mouse model of lethal peritonitis that anti-TLR-4 antibodies inhibited intracellular signaling, markedly reduced cytokine production, and protected mice from lethal endotoxic shock, when administered in a prophylactic and therapeutic manner [49].
In summary, our study offers a novel mechanism that could partially explain how MV modulates the innate immune response in the lung by interfering with cellular signaling pathways that are activated in response to pathogens. Collectively, these findings suggest a novel role for IRAK-3 in which it directly participates in NF-κB-associated transcriptional events. Our findings warrant future study regarding the selectivity and specificity of innate immunity regulation.
References
Dreyfuss D, Soler P, Basset G, Saumon G (1988) High inflation pressure pulmonary edema: respective effects of high airway pressure, high tidal volume and positive end-expiratory pressure. Am Rev Respir Dis 137:1159–1164
von Bethmann AN, Brasch F, Nüsing R, Vogt K, Volk HD, Müller KM, Wendel A, Uhlig S (1998) Hyperventilation induces release of cytokines from perfused mouse lung. Am J Respir Crit Care Med 157:263–272
Tremblay L, Valenza F, Ribeiro SP, Li J, Slutsky AS (1997) Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J Clin Invest 99:944–952
Herrera MT, Toledo C, Valladares F, Muros M, Díaz-Flores L, Flores C, Villar J (2003) Positive end-expiratory pressure modulates local and systemic inflammatory responses in a sepsis-induced lung injury model. Intensive Care Med 29:1345–1353
Ranieri VM, Suter PM, Tortorella C, De Tullio R, Dayer JM, Brienza A, Bruno F, Slutsky AS (1999) Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA 282:54–61
Fernández-Pérez ER, Keegan MT, Brown DR, Hubmayr RD, Gajic O (2006) Intraoperative tidal volume as a risk factor for respiratory failure after pneumonectomy. Anesthesiology 105:14–18
The Acute Respiratory Distress Syndrome Network (2000) Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 342:1301–1308
Dekker RJ, van Soest S, Fontijn RD, Salamanca S, de Groot PG, VanBavel E, Pannekoek H, Horrevoets AJ (2002) Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Kruppel-like factor (KLF2). Blood 100:1689–1698
Liu M, Tanswell AK, Post M (1999) Mechanical force-induced signal transduction in lung cells. Am J Physiol Lung Cell Mol Physiol 21:L667–L683
Cohen J (2002) The immunopathogenesis of sepsis. Nature 420:885–891
Aderem A, Ulevitch RJ (2000) Toll-like receptors in the induction of the innate immune response. Nature 406:782–787
Poltorak A, Ricciardi-Castagnoli P, Citterio S, Beutler B (2000) Physical contact between lipopolysaccharide and Toll-like receptor 4 revealed by genetic complementation. Proc Natl Acad Sci U S A 97:2163–2167
Arumugam TV, Okun E, Tang SC, Thundyil J, Taylor SM, Woodruff TM (2009) Toll-like receptors in ischemia–reperfusion injury. Shock 32:4–16
Yang KY, Arcaroli JJ, Abraham E (2003) Early alterations in neutrophil activation are associated with outcome in acute lung injury. Am J Respir Crit Care Med 167:1567–1574
Hotchkiss RS, Karl IE (2003) The pathophysiology and treatment of sepsis. N Engl J Med 348:138–150
Lin WJ, Yeh WC (2005) Implication of Toll-like receptor and tumor necrosis factor alpha signaling in septic shock. Shock 24:206–209
Packham G, Cleveland JL (1995) C-myc and apoptosis. Biochim Biophys Acta 1242:11–28
Ma SF, Grigoryev DN, Taylor AD, Nonas S, Sammani S, Ye SQ, García JG (2005) Bioinformatic identification of novel early stress response genes in rodent models of lung injury. Am J Physiol Lung Cell Mol Physiol 289:L468–L477
Kobayashi K, Hernandez LD, Galan JE, Janeway CA Jr, Medzhitov R, Flavell RA (2002) IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 110:191–202
Escoll P, del Fresno C, García L, Valles G, Lendinez MJ, Arnalich F, López-Collazo E (2003) Rapid up-regulation of IRAK-M expression following a second endotoxin challenge in human monocytes and in monocytes isolated from septic patients. Biochem Biophys Res Commun 311:465–472
Su J, Zhang J, Tyson J, Li L (2009) The interleukin-1 receptor-associated kinase M selectively inhibits the alternative, instead of the classical NFkB pathway. J Innate Immun 1:164–174
Held HD, Boettcher S, Hamann L, Uhlig S (2001) Ventilation-induced chemokine and cytokine release is associated with activation of nuclear factor-κB and is blocked by steroids. Am J Respir Crit Care Med 163:711–716
Moriyama K, Ishizaka A, Nakamura M, Kubo H, Kotani T, Yamamoto S, Ogawa EN, Kajikawa O, Frevert CW, Kotake Y, Morisaki H, Koh H, Tasaka S, Martin TR, Takeda J (2004) Enhancement of the endotoxin recognition pathway by ventilation with a large tidal volume in rabbits. Am J Physiol Lung Cell Mol Physiol 286:L1114–L1121
Villar J, Ribeiro SP, Mullen JBM, Kuliszewski M, Post M, Slutsky AS (1994) Induction of the heat shock response reduces mortality rate and organ damage in a sepsis-induced acute lung injury model. Crit Care Med 22:914–921
Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162:156–159
Patak E, Candenas ML, Pennefather JN, Ziccone S, Lilley A, Martín JD, Flores C, Mantecón AG, Story ME, Pinto FM (2003) Tachykinins and tachykinin receptors in human uterus. Br J Pharmacol 139:523–532
Wirtz HR, Dobbs LG (1990) Calcium mobilization and exocytosis after one mechanical stretch of lung epithelial cells. Science 250:1266–1269
Mourgeon E, Isowa N, Keshavjee S, Zhang X, Slutsky AS, Liu M (2000) Mechanical stretch stimulates macrophage inflammatory protein-2 secretion from fetal rat lung cells. Am J Physiol Lung Cell Mol Physiol 279:L699–L706
Tremblay LN, Miatto D, Hamid Q, Govindarajan A, Slutsky AS (2002) Injurious ventilation induces widespread pulmonary epithelial expression of tumor necrosis factor-alpha and interleukin-6 messenger RNA. Crit Care Med 30:1693–1700
Chiumello D, Pristine G, Slutsky AS (1999) Mechanical ventilation affects local and systemic cytokines in an animal model of acute respiratory distress syndrome. Am J Respir Crit Care Med 160:109–116
Altemeier WA, Matute-Bello G, Frevert CW, Kawata Y, Kajikawa O, Martin TR, Glenny RW (2004) Mechanical ventilation with moderate tidal volumes synergistically increases lung cytokine response to systemic endotoxin. Am J Physiol Lung Cell Mol Physiol 287:L533–L542
Shimabukuro DW, Sawa T, Gropper MA (2003) Injury and repair in lung and airways. Crit Care Med 31(Suppl 8):S524–S531
Altemeier WA, Matute-Bello G, Gharib SA, Glenny RW, Martin TR, Liles WC (2005) Modulation of lipopolysaccharide-induced gene transcription and promotion of lung injury by mechanical ventilation. J Immunol 175:3369–3376
Bouadma L, Dreyfuss D, Ricard JD, Martet G, Saumon G (2007) Mechanical ventilation and hemorrhagic shock-resuscitation interact to increase inflammatory cytokine release in rats. Crit Care Med 35:2601–2606
Schwartz MD, Moore EE, Moore FA, Shenkar R, Moine P, Haenel JB, Abraham E (1996) Nuclear factor-kappa B is activated in alveolar macrophages from patients with acute respiratory distress syndrome. Crit Care Med 24:1285–1292
Böhrer H, Qiu F, Zimmermann T, Zhang Y, Jllmer T, Mannel D, Bottiger BW, Stern DM, Waldherr R, Saeger HD, Ziegler R, Bierhaus A, Martin E, Nawroth PP (1997) Role of NFkappaB in the mortality of sepsis. J Clin Invest 100:972–985
Vaneker M, Joosten LA, Heunks LM, Snijdelaar DG, Halbertsma FJ, van Egmond J, Netea MG, van der Hoeven JG, Scheffer GJ (2008) Low-tidal-volume mechanical ventilation induces a toll-like receptor 4-dependent inflammatory response in healthy mice. Anesthesiology 109:465–472
Foster SL, Hargreaves DC, Medzhitov R (2007) Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 447:972–978
Cavaillon JM, Adib-Conquy M, Fitting C, Adrie C, Payen D (2003) Cytokine cascade in sepsis. Scand J Infect Dis 35:535–544
McCarter MD, Mack VE, Daly JM, Naama HA, Calvano SE (1998) Trauma-induced alterations in macrophage function. Surgery 123:96–101
McCall CE, Yoza BK (2007) Gene silencing in severe systemic inflammation. Am J Respir Crit Care Med 175:763–767
Deng JC, Cheng G, Newstead MW, Zeng X, Kobayashi K, Flavell RA, Standiford TJ (2006) Sepsis-induced suppression of lung innate immunity is mediated by IRAK-M. J Clin Invest 116:2532–2542
Liew FY, Xu D, Brint EK, O’Neill LA (2005) Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol 5:446–458
Moine P, McIntyre R, Schwartz MD, Kaneko D, Shenkar R, Le Tulzo Y, Moore EE, Abraham E (2000) NF-kappaB regulatory mechanisms in alveolar macrophages from patients with acute respiratory distress syndrome. Shock 13:85–91
Kuebler WM (2008) Hitting new barriers in ventilator-induced lung injury. Intensive Care Med 34:592–594
Paré PD, Warriner B, Baile EM, Hogg JC (1983) Redistribution of pulmonary extravascular water with positive end-expiratory pressure in canine pulmonary edema. Am Rev Respir Dis 127:590–593
Pelosi P, Rocco PR (2008) Effects of mechanical ventilation on the extracellular matrix. Intensive Care Med 34:631–639
Villar J, Herrera-Abreu MT, Valladares F, Muros M, Pérez-Méndez L, Flores C, Kacmarek RM (2009) Experimental ventilator-induced lung injury: exacerbation by positive end-expiratory pressure. Anesthesiology 110:1341–1347
Roger T, Froidevaux C, Le Roy D, Reymond MK, Chanson AL, Mauri D, Burns K, Riederer BM, Akira S, Calandra T (2009) Protection from lethal gram-negative bacterial sepsis by targeting Toll-like receptor 4. Proc Natl Acad Sci U S A 106:2348–2352
Acknowledgments
Supported by grants from Ministerio de Ciencia of Spain (SAF 2004-06833), FUNCIS (53/04), and by a specific agreement between Instituto de Salud Carlos III and FUNCIS (EMER07/001) under the ENCYT 2015 framework.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is discussed in the editorial available at: doi:10.1007/s00134-010-1804-x.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Villar, J., Cabrera, N., Casula, M. et al. Mechanical ventilation modulates Toll-like receptor signaling pathway in a sepsis-induced lung injury model. Intensive Care Med 36, 1049–1057 (2010). https://doi.org/10.1007/s00134-010-1799-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-010-1799-3