
Abstract
Over the past several years many advances have been made in our understanding of critical pathways involved in carcinogenesis and tumor growth. These advances have led to the investigation of small molecule inhibitors of the ErbB family of receptor tyrosine kinases across a broad spectrum of malignancies. In this article we summarize the rationale for targeting members of the ErbB family in breast cancer, and review the preclinical and clinical data for the agents that are furthest in development. In addition, we highlight directions for future research, such as exploration of the potential crosstalk between the ErbB and hormone receptor signal transduction pathways, identification of predictive markers for tumor sensitivity, and development of rational combination regimens that include the tyrosine kinase inhibitors.
Similar content being viewed by others


Introduction
The human epidermal growth factor receptor (HER) family of receptor tyrosine kinases comprises four members: epidermal growth factor receptor (EGFR; also termed HER1 or ErbB1), HER2 (also termed ErbB2 or neu), HER3 (ErbB3), and HER4 (ErbB4). Collectively, these are also referred to as the ErbB receptors. Evidence from experimental systems and from primary human breast tumors implicates the ErbB signaling network in the pathogenesis of breast cancer. In particular, amplification of HER2 is associated with an aggressive tumor phenotype that is characterized by relatively rapid tumor growth, metastatic spread to visceral sites, and drug resistance. Targeted blockade of ErbB signaling with trastuzumab, a humanized monoclonal antibody directed at the HER2 receptor, has been shown to improve survival in women with HER2-positive, advanced breast cancer. Recent data also indicate that upregulation of the ErbB receptors may mediate endocrine resistance, due to crosstalk between the ErbB and estrogen receptor (ER) signal transduction pathways.
Several orally bioavailable, low-molecular-weight tyrosine kinase inhibitors (TKIs), directed at members of the ErbB family, are now in clinical development, both as single agents and in combination with either chemotherapy or hormonal therapy. These agents may be associated with a more favorable toxicity profile than traditional cytotoxic chemotherapy.
Rationale for targeting ErbB receptors in breast cancer
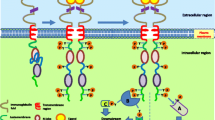
ErbB receptors are composed of an extracellular ligand binding domain, a single transmembrane domain, and an intracellular domain with tyrosine kinase activity [1]. More than 10 ligands have been identified (Fig. 1). Upon ligand binding the extracellular domain undergoes conformational change, allowing the formation of homodimers or heterodimers with other members of the ErbB family. In turn, dimerization induces tyrosine phosphorylation of specific residues in the intracellular domain that serve as docking sites for adaptor proteins and downstream effectors [2]. As a result, activation of the phosphotidylinositol 3-kinase (PI3K) and mitogen-activated protein kinase pathways occurs, leading to cell proliferation and survival (Fig. 2). Each of the ErbB receptors is thought to recruit a unique combination of effector molecules. The range of possible homodimers and heterodimers, along with the multitude of downstream effectors, is thought to account for the signaling diversity of the ErbB network.

The ErbB family of receptor tyrosine kinases. Known ligands are listed above each receptor. Human epidermal growth factor receptor (HER)2 has no known ligand. The kinase domain of HER3 is inactive. ErbB receptors can also be activated by nonconventional agonists, such as decorin and Cripto-1, which are not shown here. AR, amphiregulin; BTC, betacellulin; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; EPR, epiregulin; HB-EGF, heparin-binding epidermal growth factor; NRG, neuregulin; TGF, transforming growth factor.

The ErbB signaling pathway. Ligand binding induces dimerization, leading to activation of the intracellular tyrosine kinase. Upon auto-phosphorylation and cross-phosphorylation of the receptor complex, key downstream effectors are recruited. FasL, Fas ligand; FKHR, forkhead in rhabdomyosarcoma; Grb, growth factor receptor-bound protein; GSK, glycogen synthase kinase; MAPK, mitogen-activated protein kinase; MEK, MAPK kinase; mTOR, molecular target of rapamycin; PI3K, phosphatidylinositol 3-kinase; PTEN, phosphatase and tensin homolog deleted on chromosome 10; SOS, son-of-sevenless guanine nucleotide exchange factor.
EGFR is overexpressed in 16–48% of human breast cancers. Several groups have reported an association between EGFR expression and poor prognosis [3–5]. In addition, the constitutively active, tumorigenic EGFR vIII variant has been reported to be present in up to 78% of breast carcinomas [6]. Data from animal models also support a role for EGFR in breast cancer; for example, overexpression of the EGFR ligand transforming growth factor-α results in mammary carcinomas in mice [7].
HER2 is overexpressed in 25–30% of all human breast carcinomas; high levels of expression are generally associated with gene amplification [8]. Unlike other ErbB receptors, HER2 does not have a known ligand but instead functions as the preferred heterodimerization partner of all other ErbB receptors [2]. In preclinical models, HER2 overexpression induces the malignant transformation of NIH/3T3 cells, and transgenic mice that carry an activated HER2 oncogene develop multiple synchronous breast adenocarcinomas [9]. In the clinic, HER2 has emerged as a highly important prognostic factor for relapse and overall survival in women with primary breast cancer [8]. Trastuzumab, a monoclonal antibody directed against HER2, is active both as a single agent and in combination with chemotherapy in women with HER2-positive breast cancer [9–11]. When compared with chemotherapy alone, combinations of trastuzumab plus chemotherapy lead to higher response rates, longer time to disease progression, and an improvement in overall survival [10]. There appears to be little question that signaling through HER2 is crucial for the development, growth, and survival of breast tumors.
The roles of HER3 and HER4 in breast cancer are less well defined. HER3 is overexpressed in 15–35% of primary breast tumors and may be associated with decreased local control and overall survival [5, 12]. Although the clinical data are limited, they are consistent with the known role of HER3 as a critical partner for both EGFR (ErbB1) and HER2 in terms of signaling through the PI3K pathway [13]. In contrast, activation of HER4 appears to be both necessary and sufficient for antiproliferative and differentiation responses in human breast cancer cells in vitro, perhaps because of the decreased capacity of HER4 to activate proliferation via the Ras/Raf/mitogen-activated protein kinase signal transduction pathway [14]. Consistent with these observations, in small studies HER4 overexpression (found in approximately 10% of breast tumors) appeared to be associated with low nuclear grade, low proliferative index, and increased survival [5, 15].
Small molecule tyrosine kinase inhibitors
A number of ErbB receptor inhibitors are in clinical development (Tables 1 and 2); none have received US Food and Drug Administration approval for the treatment of breast cancer. These small molecules compete with ATP for binding to the kinase domain of the receptor. TKIs have several potential advantages over monoclonal antibodies. First, they are orally bioavailable and generally well tolerated. Second, they appear active against truncated forms of the EGFR and HER2 receptors (EGFR vIII and p95, respectively) in vitro [16, 17]. Third, their small size may allow them to penetrate sanctuary sites, such as the central nervous system. Finally, by taking advantage of the homology between kinase domains of ErbB receptors, TKIs can be developed to target more than one member of the receptor family simultaneously.
Epidermal growth factor receptor inhibitors
The reversible EGFR inhibitors gefitinib (Iressa™; AstraZeneca, London, UK) and erlotinib (Tarceva™; Genentech, South San Francisco, CA, USA) are furthest in the course of development. Gefitinib is approved in the USA for the treatment of non-small-cell lung cancer (NSCLC), based on clinical trials that have demonstrated activity in a relatively small percentage of patients [18]. Both gefitinib and erlotinib have activity against multiple breast cancer cell lines in vitro and in xenograft models. In clinical trials the most commonly reported adverse effects were grade 1 or 2 rash, diarrhea, asthenia, nausea, and vomiting. However, neither gefitinib nor erlotinib has demonstrated significant single-agent activity against refractory metastatic breast cancer. Across three phase II studies of gefitinib, only three partial responses and eight patients with stable disease lasting 6 months or longer were observed among a total of 113 patients [19–21]. Two phase II studies of erlotinib have also been disappointing, with only one partial response seen among 87 patients treated [22, 23]. It is unclear whether response rates would have been higher in a less heavily pretreated patient population. Of interest, there was no correlation between EGFR expression and response, but with such a small number of responses, any conclusion about the relationship between response and expression is quite limited.
EKB-569 is an irreversible EGFR inhibitor that is in phase I trials for advanced solid tumors [24]. Response data in breast cancer have not been published to date. Given the experience with gefitinib and erlotinib, it seems unlikely that monotherapy with EKB-569 will have a substantial degree of activity in an unselected and heavily pretreated group of women with advanced breast cancer.
Dual kinase inhibitors
Promiscuous heterodimerization of ErbB receptors allows for signaling through a variety of receptor–receptor combinations. Thus, one potential mechanism of resistance to agents targeted against a single ErbB receptor (e.g. gefitinib or trastuzumab) is upregulation of other members of the receptor family. Consequently, dual kinase inhibitors against EGFR and HER2 could theoretically have activity in resistant tumors, as well as in tumors that have not previously been exposed to an EGFR or HER2 inhibitor. Indeed, in animal models simultaneous blockade of both receptors has been shown to result in increased cell kill compared with blockade of either receptor alone.
Two dual TKIs have been evaluated in phase I trials: GW572016 and PKI-166. The safety, tolerability, and pharmacokinetics of both drugs have been characterized [25, 26]. Among patients with advanced breast cancer treated with GW572016, there have been seven partial responses reported thus far in women with trastuzumab-refractory disease [25, 27]. Of patients treated with GW572016, 25% developed grade 1 or 2 rash. Mild gastrointestinal symptoms were also seen, with 21% having grade 1 or 2 nausea and vomiting and 27% having grade 1 or 2 diarrhea. Phase II trials of GW572016 in patients with metastatic breast cancer are ongoing. PKI-166 is no longer under development because of toxicity concerns. However, AEE788, a compound that targets EGFR, HER2, and the vascular endothelial growth factor receptor, is currently under evaluation in phase I trials of advanced solid tumors, colorectal cancer, and glioblastoma multiforme. Based on its mechanism of action, this agent could have promise in the treatment of breast cancer.
Pan-ErbB Inhibitors
CI-1033 is an irreversible pan-erbB inhibitor that covalently binds to the ATP binding site of the intracellular kinase domain. In mouse xenograft models, a variety of irreversible inhibitors have demonstrated superior antitumor activity in comparison with reversible inhibitors. Whether irreversible inhibitors will offer any advantages in humans is unknown. CI-1033 has been studied in phase I trials of heavily pretreated patients with a variety of solid tumors, including more than 10 patients with breast cancer. One patient has achieved stable disease for more than 25 weeks, but objective responses have not yet been reported [16, 28–30]. Like the other TKIs, CI-1033 is associated with grade 1–2 diarrhea, rash, nausea, and vomiting. However, it has also been associated with a 28% rate of thrombocytopenia. Rarely, hypersensitivity reactions have been observed at doses in excess of 500 mg. A phase II trial of CI-1033 in patients with metastatic breast cancer recently reached its accrual goal.
Future directions
Targeting 'targeted therapy'
Although the clinical results with EGFR inhibitors in patients with breast cancer have been disappointing to date, it has been argued that trastuzumab would have been considered an inactive drug if studied in unselected populations of patients with breast cancer. Like trastuzumab, it is possible that TKIs will be highly active in selected patient populations. However, it is unlikely that prediction of sensitivity to TKIs will be as straightforward as was the case for trastuzumab; thus far, the data suggest that the presence of receptor overexpression does not adequately predict response. Recent studies have demonstrated that many patients with NSCLC who benefit from gefitinib have somatic mutations in the kinase domain of the EGFR [31, 32]. It is unknown whether breast tumors contain similar mutations, or whether such mutations predict response to EGFR inhibitors. Other groups have focused on predictors of response to dual and pan-ErbB TKIs. There is preliminary evidence that response to these agents is associated with high levels of EGFR, HER2, pAkt, and pErk at baseline, whereas disease progression is associated with persistently high levels of pAkt and pErk despite treatment. Of interest, in one study [33] high levels of pAkt and pErk at baseline predicted response to TKIs but not to trastuzumab.
Furthermore, although monoclonal antibodies and TKIs ostensibly inhibit the same targets, there appear to be substantial differences in their clinical activity [34]. For example, cetuximab, a monoclonal antibody directed against EGFR, has single-agent activity against colorectal cancer, whereas gefitinib and erlotinib do not [34, 35]. Moreover, although combinations of TKIs with chemotherapy have not yet been demonstrated to improve outcomes, trastuzumab-based regimens are associated with improved survival in metastatic breast cancer [10]. In the setting of advanced colorectal cancer, the addition of cetuximab to irinotecan is associated with increased response rates and time to progression compared with irinotecan alone [36]. It is not yet clear what accounts for these observed differences, but the differences do raise questions as to the mechanisms by which monoclonal antibodies and TKIs function to inhibit tumor growth and/or result in cell death.
Crosstalk between signal transduction pathways
Importantly, data from in vitro models and from retrospective series in women with breast cancer suggest that both EGFR and HER2 overexpression confer resistance to hormonal therapy [2]. Crosstalk between ER and ErbB pathways has been postulated to occur via multiple mechanisms, including upregulation of EGFR and HER2 expression by tamoxifen, ligand-independent signaling of ER via PI3K, and modulation of coactivators of ER via downstream effectors of the ErbB pathway [37]. In preclinical models, co-blockade of the EGFR and ER pathways with gefitinib and either tamoxifen or fulvestrant resulted in restoration of tamoxifen sensitivity and delay of resistance to estrogen deprivation in HER2-over-expressing breast tumors [38]. Based on these data, several phase II trials of gefitinib with hormonal therapy, and a phase III trial of letrozole ± the dual inhibitor GW572016 are ongoing in women with ER-positive metastatic breast cancer.
Combining tyrosine kinase inhibitors with chemotherapy, radiotherapy, and other targeted agents
Enthusiasm for adding EGFR inhibitors to chemotherapy has waned slightly since the results of two randomized studies in NSCLC (Iressa NSCLC Trials Assessing Combination Therapy [INTACT] 1 and 2) were reported [18]. In these trials, the addition of gefitinib to cytotoxic chemotherapy did not improve progression-free or overall survival. Nevertheless, given the success of combination regimens with trastuzumab and chemotherapy in metastatic breast cancer, and the preclinical data showing synergy between TKIs and chemotherapy, it is still reasonable to study chemotherapy-based regimens. Several ongoing phase II and phase III trials in metastatic breast cancer utilize gefitinib, erlotinib, or GW572016 in combination with chemotherapy. Alternatively, combining antibody-based therapy (e.g. C225 or trastuzumab) with TKIs may allow for more complete blockade of ErbB-mediated signal transduction pathways, and thereby delay or overcome drug resistance. This strategy is being employed in a phase II study of trastuzumab and gefitinib.
TKIs are also potent radiosensitizers [39]. Their small size may allow for improved penetration into tumors, which, combined with concurrent radiation, could result in improved cell kill. There are no trials in breast cancer utilizing this approach, but it is being studied in phase I/II trials in primary brain tumors.
Chemoprevention
There has been interest in developing novel chemoprevention strategies that have an impact upon the incidence of hormone receptor negative tumors. Lu and coworkers [40] recently reported that gefitinib delayed the development of ER-negative tumors in transgenic mice engineered to overexpress HER2. In women with ductal carcinoma in situ, gefitinib reduced proliferation, as measured by Ki67 [41]. It is too early to know whether these results will translate into improved outcomes in women at high risk for developing breast cancer. Although the toxicity profile of the existing TKIs may be reasonable in the setting of metastatic disease, it is unclear whether protracted use of these agents would be tolerated by otherwise healthy women.
Conclusion
The ErbB receptor family represents an attractive target for cancer therapy. A number of TKIs that inhibit EGFR, HER2, HER4, or combinations thereof are in clinical development. The challenge that lies ahead is to identify those patients who will benefit from targeting of the ErbB receptor family, and to determine how best to integrate these compounds into the armamentarium of both standard and novel treatments for breast cancer. It may be most fruitful to combine agents that target multiple neoplastic pathways, with the goal of producing highly effective and minimally toxic regimens.
Note
This article is the first in a review series on New targets for therapy in breast cancer, edited by Stephen RD Johnston.
Other articles in the series can be found at http://breast-cancer-research.com/articles/review-series.asp?series=bcr_NewTargets
Abbreviations
- EGFR:
-
epidermal growth factor receptor
- ER:
-
estrogen receptor
- HER:
-
human epidermal growth factor receptor
- NSCLC:
-
non-small-cell lung cancer
- PI3K:
-
phosphatidylinositol 3-kinase
- TKI:
-
tyrosine kinase inhibitor.
References
Schlessinger J: Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell. 2002, 110: 669-672. 10.1016/S0092-8674(02)00966-2.
Atalay G, Cardoso F, Awada A, Piccart MJ: Novel therapeutic strategies targeting the epidermal growth factor receptor (EGFR) family and its downstream effectors in breast cancer. Ann Oncol. 2003, 14: 1346-1363. 10.1093/annonc/mdg365.
Klijn JG, Berns PM, Schmitz PI, Foekens JA: The clinical significance of epidermal growth factor receptor (EGF-R) in human breast cancer: a review on 5232 patients. Endocr Rev. 1992, 13: 3-17. 10.1210/er.13.1.3.
Tsutsui S, Ohno S, Murakami S, Hachitanda Y, Oda S: Prognostic value of epidermal growth factor receptor (EGFR) and its relationship to the estrogen receptor status in 1029 patients with breast cancer. Breast Cancer Res Treat. 2002, 71: 67-75. 10.1023/A:1013397232011.
Witton CJ, Reeves JR, Going JJ, Cooke TG, Bartlett JM: Expression of the HER1-4 family of receptor tyrosine kinases in breast cancer. J Pathol. 2003, 200: 290-297. 10.1002/path.1370.
Moscatello DK, Holgado-Madruga M, Godwin AK, Ramirez G, Gunn G, Zoltick PW, Biegel JA, Hayes RL, Wong AJ: Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res. 1995, 55: 5536-5539.
Matsui Y, Halter SA, Holt JT, Hogan BL, Coffey RJ: Development of mammary hyperplasia and neoplasia in MMTV-TGF alpha transgenic mice. Cell. 1990, 61: 1147-1155. 10.1016/0092-8674(90)90077-R.
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL: Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987, 235: 177-182.
Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, Wolter JM, Paton V, Shak S, Lieberman G, et al: Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999, 17: 2639-2648.
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, et al: Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001, 344: 783-792. 10.1056/NEJM200103153441101.
Burstein HJ, Harris LN, Marcom PK, Lambert-Falls R, Havlin K, Overmoyer B, Friedlander RJ, Gargiulo J, Strenger R, Vogel CL, et al: Trastuzumab and vinorelbine as first-line therapy for HER2-overexpressing metastatic breast cancer: multicenter phase II trial with clinical outcomes, analysis of serum tumor markers as predictive factors, and cardiac surveillance algorithms. J Clin Oncol. 2003, 21: 2889-2895. 10.1200/JCO.2003.02.018.
Travis A, Pinder SE, Robertson JF, Bell JA, Wencyk P, Gullick WJ, Nicholson RI, Poller DN, Blamey RW, Elston CW, et al: C-erbB-3 in human breast carcinoma: expression and relation to prognosis and established prognostic indicators. Br J Cancer. 1996, 74: 229-233.
Soltoff SP, Carraway KLIII, Prigent SA, Gullick WG, Cantley LC: ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol Cell Biol. 1994, 14: 3550-3558.
Sartor CI, Zhou H, Kozlowska E, Guttridge K, Kawata E, Caskey L, Harrelson J, Hynes N, Ethier S, Calvo B, et al: HER4 mediates ligand-dependent antiproliferative and differentiation responses in human breast cancer cell. Mol Cell Biol. 2001, 21: 4265-4275. 10.1128/MCB.21.13.4265-4275.2001.
Tovey SM, Witton CJ, Bartlett JM, Stanton PD, Reeves JR, Cooke TG: Outcome and human epidermal growth factor receptor (HER) 1–4 status in invasive breast carcinomas with proliferation indices evaluated by bromodeoxyuridine labeling. Breast Cancer Res. 2004, 6: R246-R251. 10.1186/bcr783.
Allen LF, Lenehan PF, Eiseman IA, Elliott WL, Fry DW: Potential benefits of the irreversible pan-erbB inhibitor, CI-1033, in the treatment of breast cancer. Semin Oncol. 2002, Suppl 11: 11-21. 10.1053/sonc.2002.34049.
Xia W, Liu L-H, Ho P, Spector NL: Truncated ErbB2 receptor (p95ErbB2) is regulated by heregulin through heterodimer formation with ErbB3 yet remains sensitive to the dual EGFR/ErbB2 kinase inhibitor GW572016. Oncogene. 2004, 23: 646-653. 10.1038/sj.onc.1207166.
Cohen MH, Williams GA, Sridhara R, Chen G, Pazdur R: FDA drug approval summary: gefitinib (ZD1839) (Iressa) tablets. Oncologist. 2003, 8: 303-306.
Albain K, Elledge R, Gradishar WJ, Hayes DF, Rowinsky E, Hudis C, Pusztai L, Tripathy D, Modi S, Rubi S: Open-label, phase II, multicenter trial of ZD1839 ('Iressa') in patients with advanced breast cancer [abstract 20]. Breast Cancer Res Treat. 2002, 76 (Suppl 1): s33-
Robertson JFR, Gutteridge E, Cheung KL, Owers R, Koehler M, Hamilton L, Gee J, Nicholson RI: Gefitinib (ZD1839) is active in acquired tamoxifen (TAM)-resistant oestrogen receptor (ER)-positive and ER-negative breast cancer: results from a phase II study [abstract 23]. Proc Am Soc Clin Oncol. 2003, 22: 7-
Baselga J, Albanell J, Ruiz A, Lluch A, Gascon P, Gonzalez S, Guillen V, Sauleda S, Averbuch S, Rojo F: Phase II and tumor pharmacodynamic study of gefitinib (ZD1839) in patients with advanced breast cancer [abstract 24]. Proc Am Soc Clin Oncol. 2003, 22: 7-
Winer E, Cobleigh M, Dickler M, Miller K, Fehrenbacher L, Jones C, Justice R: Phase II multicenter study to evaluate the efficacy and safety of Tarceva (erlotinib, OSI-774) in women with previously treated locally advanced or metastatic breast cancer [abstract 445]. Breast Cancer Res Treat. 2002, 76 (Suppl 1): s115-
Tan AR, Yang X, Berman A, Lepper ER, Cox MC, Parr AL, Hewitt SM, Steinberg SM, Lipkowitz S, Swain SM: Evaluation of epidermal growth factor receptor signaling in tumor and skin biopsies after treatment with OSI-774 in patients with metastatic breast cancer [abstract 784]. Proc Am Soc Clin Oncol. 2003, 22: 196-
Hidalgo M, Erlichman C, Rowinsky EK, Koepp-Norris J, Jensen K, Boni J, Korth-Bradley J, Quinn S, Zacharchuk C: Phase I trial of EKB0569, an irreversible inhibitor of the epidermal growth factor receptor (EGFR), in patients with advanced solid tumors [abstract 65]. Proc Am Soc Clin Oncol. 2002, 21: 17a-
Dees EC, Burris H, Hurwitz H, Dowlati A, Smith D, Koch K, Stead A, Mangum S, Harris J, Spector N: Clinical summary of 67 heavily pre-treated patients with metastatic breast carcinomas treated with GW572016 in a phase Ib study [abstract 3188]. Proc Am Soc Clin Oncol. 2004, 23: 241-
Murren JR, Papadimitrakopoulou VA, Sizer KC, Vaidyanathan S, Ravera C, Abbruzzese JL: A phase I dose-escalating study to evaluate the biological activity and pharmacokinetics of PKI166, a novel tyrosine kinase inhibitor, in patients with advanced cancers [abstract 377]. Proc Am Soc Clin Oncol. 2002, 21: 95a-
Blackwell KL, Kaplan EH, Franco SX, Marcom PK, Maleski JE, Sorensen MJ, Berger MS: A phase II, open-label, multicenter study of GW572016 in patients with trastuzumab-refractory metastatic breast cancer [abstract 3006]. Proc Am Soc Clin Oncol. 2004, 23: 196-
Nemunaitis JJ, Eiseman I, Cunningham C, Lenehan P, Olson S, Bycott P, Schlict M, Zentgraff R, Shin D, Zinner R: A phase I trial of CI-1033, a pan-erbB tyrosine kinase inhibitor, given daily for 14 days every 3 weeks, in patients with advanced solid tumors [abstract 974]. Proc Am Soc Clin Oncol. 2003, 22: 243-
Zinner RG, Donato NJ, Nemunaitis JJ, Cunningham CC, Shin HJ, Zentgraf RE, Ayers GD, Glisson BS, Khuri FR, Kies MS, et al: Biomarker modulation in tumor and skin biopsy samples from patients with solid tumors following treatment with the pan-erbB tyrosine kinase inhibitor, CI-1033 [abstract 58]. Proc Am Soc Clin Oncol. 2002, 21: 15a-
Garrison MA, Tolcher A, McCreery H, Rowinsky EK, Schott A, Mace J, Drengler R, Patnaik A, Denis L, Lenehan P, et al: A phase I and pharmacokinetic study of CI-1033, a pan-ErbB tyrosine kinase inhibitor, given orally on days 1, 8, and 15 every 28 days to patients with solid tumors [abstract 283]. Proc Am Soc Clin Oncol. 2001, 20: a283-10.1023/A:1011817205673.
Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et al: EGFR mutations in lung cancer: correlation with clinical response to gefi-tinib therapy. Science. 2004, 304: 1497-1500. 10.1126/science.1099314.
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al: Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004, 350: 2129-2139. 10.1056/NEJMoa040938.
Bacus SS, Beresford PJ, Yarden Y, Spector N, Smith B: The use of predicting factors and surrogate markers in patients' cancer biopsies treated with targeted antibodies to erbB receptors and erbB tyrosine kinase inhibitors [abstract 3408]. Proc Am Soc Clin Oncol. 2003, 22: 848-
Mendelsohn J, Baselga J: Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J Clin Oncol. 2003, 21: 2787-2799. 10.1200/JCO.2003.01.504.
Saltz LB, Meropol NJ, Loehrer PJ, Needle MN, Kopit J, Mayer RJ: Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004, 22: 1201-1208. 10.1200/JCO.2004.10.182.
Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Van Cutsem E: Cetuximab (C225) alone or in combination with irinotecan (CPT-11) in patients with epidermal growth factor receptor (EGFR)-positive, irinotecan-refractory metastatic colorectal cancer (MCRC) [abstract 1012]. Proc Am Soc Clin Oncol. 2003, 22: 252-
Johnston SR, Head J, Pancholi S, Detre S, Martin LA, Smith IE, Dowsett M: Integration of signal transduction inhibitors with endocrine therapy: an approach to overcoming hormone resistance in breast cancer. Clin Cancer Res. 2003, 9: 524S-532S.
Massarweh S, Shou J, Mohsin SK: Inhibition of epidermal growth factor/HER2 receptor signaling using ZD1839 ('Iressa') restores tamoxifen sensitivity and delays resistance to estrogen deprivation in HER2-overexpressing breast tumors [abstract 130]. Proc Am Soc Clin Oncol. 2002, 21: 33a-
May BBY, Bristow RB, Kim J, Siu LL: Combined-modality treatment of solid tumors using radiotherapy and molecular targeted agents. J Clin Oncol. 2003, 21: 2760-2776. 10.1200/JCO.2003.10.044.
Lu C, Speers C, Zhang Y, Xu X, Hill J, Steinbis E, Celestino J, Shen Q, Kim H, Hilsenbeck S, et al: Effect of epidermal growth factor receptor inhibitor on development of estrogen receptor-negative mammary tumors. J Natl Cancer Inst. 2003, 17: 1825-1833.
Bundred NJ, Anderson EA, Gee J, Barr L, Baildam A, Zeiton A, Byrne G, Grassby S, Nicholson RI, Koehler M: Randomised, double-blind, placebo-controlled trial of short-term effects of gefitinib ('Iressa', ZD1839) on ductal carcinoma in situ [abstract 14]. Breast Cancer Res Treat. 2003, 82: s10-
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None declared.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Lin, N.U., Winer, E.P. New targets for therapy in breast cancer: Small molecule tyrosine kinase inhibitors. Breast Cancer Res 6, 204 (2004). https://doi.org/10.1186/bcr919
Published:
DOI: https://doi.org/10.1186/bcr919