
Abstract
Growth and progression of breast cancers are accompanied by increased neovascularization (angiogenesis). A variety of factors, including hypoxia and genetic changes in the tumor cells, contribute to increased production of angiogenic factors. Furthermore, cells within the activated tumor stroma also contribute to the increase in production of vascular endothelial growth factor and other angiogenic factors, including basic fibroblast growth factor and platelet-derived growth factor. The contribution of the microenvironment to tumor-induced angiogenesis is underscored by findings that breast tumors implanted into different tissue sites show marked differences in the extent and nature of the angiogenic response. These findings have important implications for designing anti-angiogenic therapies.
Similar content being viewed by others


Introduction
Like most solid tumors, breast cancers require new blood vessel growth (neovascularization) if they are to grow beyond a few millimeters in diameter [1]. The new vessels not only help to meet the growing metabolic demands of the tumor by supplying additional nutrients, but also provide potential routes for tumor dissemination and metastasis.
In breast cancers, tumor-induced angiogenesis is first evident at the pre-invasive stage of high-grade ductal carcinoma in situ. In this instance, a characteristic rim of microvessels is formed around the ducts that are filled with proliferating epithelial cells [2]. As the tumor continues to progress, so does the degree of neovascularization. Not surprisingly, poor breast cancer prognosis has been shown to correlate with increasing microvascular density or production and with factors that stimulate new vessel growth [3, 4]. Accordingly, an extensive body of research has focused on identifying the factors in the tumor microenvironment that promote and support angiogenesis, with the hope of limiting neovascularization and ultimately tumor growth and metastasis. Moreover, anti-angiogenic therapy is particularly attractive, because, unlike the tumor cells, which are genetically unstable and can rapidly acquire resistance to many therapeutic agents, the normal vascular endothelium does not harbor mutations that would facilitate acquisition of drug resistance. Both strands of research are reviewed in this article.
The angiogenic cycle
In normal, quiescent capillaries, the endothelial cells are in contact with a laminin-rich basement membrane and a 1- to 2-cell-thick layer of supporting pericytes. During angiogenesis, the connections between the adjacent pericytes must be weakened and the surrounding basement membrane must be degraded. Endothelial cells re-enter the cell cycle and invade the surrounding stromal matrix; this invasion is facilitated by the integrin adhesion receptors [5]. The endothelial cells begin to resynthesize a basement membrane, which promotes their acquisition of capillary-like morphology and assists in withdrawal from the cell cycle [6]. Pericytes are subsequently recruited to the newly formed capillaries to help stabilize the maturing vessels. In the tumor microenvironment, chronic exposure to angiogenic factors that either support proteolysis of the basement membrane and/or antagonize endothelial–pericyte interactions results in creation of a relatively unstable, highly permeable network of vessels which do not fully mature, but nonetheless are capable of supplying nutrients to meet the increasing metabolic demands of the tumor. In fact, the increased permeability of these vessels is often considered to facilitate extravasation and ultimately metastasis of the tumor cells.
Factors that promote angiogenesis
Hypoxia
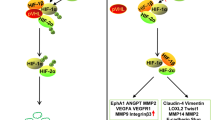
One of the features within the tumor microenvironment that has long been suspected to act as a potent angiogenic stimulus is hypoxia. The low tissue oxygen tension arises from masses of tightly packed, rapidly growing cells that lack access to an adequate supply of nutrients. Significant progress has been made in recent years toward understanding the biochemical and molecular responses to hypoxia and how the tissue senses the low oxygen tension. Most notably has been the discovery of the hypoxia-inducible factor (HIF), a heterodimeric transcription factor consisting of the hypoxic response factor (HIF-1α) and the constitutively expressed aryl hydrocarbon receptor nuclear translocator (ARNT or HIF-1β) [7]. Under oxygenated conditions, HIF-1α is bound to von Hippel–Lindau (VHL) protein, which leads to ubiquitination and rapid degradation of HIF-1α. In contrast, in hypoxic conditions, this factor is stabilized: it cannot interact with VHL protein, because prolyl hydroxylase, an enzyme that normally modifies HIF-1α to promote its interactions with VHL protein, is not active in hypoxic conditions [8]. Thus, prolyl hydroxylase has been suggested to serve as the oxygen sensor.
Strong evidence shows that HIF-1α plays a significant role in experimental tumor growth and tumor-associated angiogenesis; mice deficient in this factor have markedly reduced angiogenic responses [9, 10]. In humans, HIF-1α is overexpressed in ductal carcinomas but not in benign tumors, which lack extensive angiogenesis [11]. In the hypoxic tumor microenvironment, stabilized HIF-1α directly induces expression of many proangiogenic mediators, most notably vascular endothelial growth factor (VEGF), as well as one of its receptors, VEGF receptor 1 (VEGFR1) [12, 13].
Vascular endothelial growth factor
As its name implies, VEGF is a potent and selective endothelial mitogen that can induce a rapid and complete angiogenic response. VEGF consists of a family of related glycoproteins (VEGF-A, -B, -C and -D) identified by homology with VEGF (VEGF-A), which has been the most extensively studied and has been implicated in tumor-induced angiogenesis. VEGF-C and -D primarily influence lymphatic endothelium [14].
VEGF is produced and secreted by a number of normal cell types and its expression is markedly increased in tumor cells, including a variety of breast tumors, and is also highly upregulated in reactive breast tumor stromal cells [15]. However, in contrast to other cytokines produced by tumor cells, VEGF acts almost exclusively on endothelial cells, since expression of the major VEGF receptor, VEGFR2, is restricted to such cells. Thus, interfering with VEGF or VEGFR2 provides a means to selectively target tumor endothelium. In contrast, VEGFR1 is expressed by endothelial cells as well as monocytes and macrophages and until recently the role of VEGFR1 has been more enigmatic [16, 17]. Once bound to its receptor, VEGF initiates an intracellular signaling cascade that leads to changes in expression of the gene that promote endothelial cell migration and proliferation. In addition, VEGF not only is a potent mitogen for endothelium, but also produces a marked increase in permeability of capillaries [18], and perhaps it is not surprising that a major distinguishing feature between normal capillaries and tumor vessels is the leakiness of the tumor vessels [18, 19].
VEGF and breast tumor angiogenesis
As mentioned already, angiogenesis induced by breast tumors has been linked to an increased production of VEGF both by the tumor cells and by cells within the tumor stroma [15]. Furthermore, an increase in expression of VEGFR2 has also been noted in the adjacent breast tumor endothelial cells. In fact, an increased expression of VEGF corresponds with the earliest visible breast-tumor-induced angiogenesis that is evident in pre-invasive high-grade ductal carcinoma in situ [20].
A number of factors are believed to contribute to the increased expression of VEGF in the breast tumor environment. Clearly, hypoxia and HIF-1α play a prominent role [11]. The findings that there are higher levels of VEGF expression in premenopausal than in postmenopausal women suggested that steroid hormones may also increase VEGF expression. [21]. It has long been recognized that estradiol is angiogenic, and evidence suggests that estrogen effects may be mediated by induction of VEGF [22]. For example, VEGF is upregulated by estrogens and decreased by progestins in certain breast cancer cell lines [23, 24]. More recently, the estrogen receptor inhibitor tamoxifen was shown to suppress VEGF transcription [25, 26]. However, it remains to be established whether estrogen receptor expression directly correlates with VEGF expression and vascular density.
Other changes within the tumor environment also positively influence VEGF production. For example, many tumor cells, including human breast tumors, often secrete high levels of matrix metalloproteinases. One member of this family that has received considerable attention is matrix metalloproteinase (MMP)-9, which is released by tumor cells and is expressed at high levels in human breast tumors [27]. MMP-9 was shown to be capable of proteolyzing the adjacent extracellular matrix and releasing sequestered VEGF, thus increasing its bioavailability [28].
Another important change in breast tumors is expression of HER2. HER2 is a member of the epidermal growth factor receptor family of tyrosine kinase receptors encoded by the ERB2 gene and signals in the absence of any known ligand. Moreover, HER2 overexpression or heregulin stimulation leads to an increase in VEGF mRNA [29, 30], while treatment of breast cancers with a neutralizing antibody against HER2 results in a dose-dependent inhibition of VEGF production [31]. Furthermore, HER2 was shown to increase the rate of HIF-1α protein synthesis, not by preventing degradation as is observed during hypoxia, but in a novel, rapamycin-dependent manner [32].
Other changes in epithelial gene expression associated with increased tumorigenicity can also enhance VEGF production. The α6β4 integrin, which normally promotes interactions of breast epithelium with basement membrane, is increased in breast tumor cells and is also mislocalized and consequently promotes invasiveness of tumor cells. Recent studies show that α6β4 signaling leads to an inactivation of eIF-4E, a translational repressor, which in turn increases VEGF translation and ultimately increases survival of these tumor cells [33]. Similar to the HER2-mediated increases in HIF-1α and VEGF, the α6β4 signaling pathway that promotes translation of VEGF also converges on a rapamycin-sensitive pathway. Also importantly, the increase in VEGF production by the tumor cells has been suggested to function in an autocrine manner to directly promote epithelial cell survival [33].
Host microenvironment and angiogenesis
It has long been suspected that the tumor microenvironment affects the angiogenic response of the tumor cells. In studies of the degree of vascularity of primary breast tumors and their axiliary lymph node metastasis, marked variations were found in the level of vascularity and angiogenesis in individual tumors and their metastatic clones within the same patient, raising the possibility that different microenvironments affected the tumor's angiogenic response [34]. In more recent studies, the same estrogen-dependent breast tumor line implanted either into the mammary gland fat pad or into the cranium displayed different angiogenic responses. Tumors implanted in the mammary fat pad displayed increased expression of VEGF and increased vascular permeability. The adjacent endothelial cells also displayed greater expression of VEGFR1 and VEGFR2 than endothelial cells adjacent to tumors in the cranial site (a common metastatic route) [35]. However, in the cranial tumors there was an overall increase in angiogenesis.
These results clearly show that the tumor microenvironment is a major determinant of the angiogenic response. They also indicate that experimental models designed to study breast tumor angiogenesis should carefully consider the site of tumor implantation. To date, most models of breast cancer angiogenesis have relied on subcutaneous injection into mice or implantation of tumors in human skin grafted onto immunocompromised mice, the latter providing the benefit that the response of human vessels can be evaluated. One recent study showed, in fact, that when a mammary carcinoma cell line derived from mice infected with the mouse mammary tumor virus (MMTV) was subcutaneously injected into syngeneic mice, the resulting tumor vasculature was highly sensitive to inhibitors including interleukin 12 and interferon gamma. In contrast, in tumors that developed naturally in the mammary gland of mice carrying the MMTV oncogene, the resulting tumor vasculature did not respond to the same agents and the vessels associated with the tumor appeared to be relatively more mature than those developing in the subcutaneous environment [36]. Again, these findings emphasize the need to consider the appropriate environment when evaluating breast-cancer-induced angiogenesis. In addition to the MMTV mice, a variety of other transgenic models is currently available that would facilitate direct evaluation of tumor-induced angiogenesis in the mammary gland [37]. In many of these mouse models, breast cancers can be selectively induced by using mammary-gland-specific promoters to drive expression of a variety of tumor-promoting genes.
Anti-VEGF treatment and breast tumor angiogenesis
Given the large body of work implicating VEGF and its receptors as prime culprits in facilitating breast tumor angiogenesis, several therapies designed to counteract the effects of VEGF are currently undergoing advanced clinical trials. Notably, soluble antagonists of the VEGF receptors, specific tyrosine kinase inhibitors (Su5416) and monoclonal antibodies against VEGF (bevacizumab, avastin) have been developed to target the tumor endothelium specifically. Recent reports, however, indicate that although avastin showed initial promise in combating breast tumor angiogenesis, follow-up studies showed that the long-term prognosis in treated patients was not improved.
The underlying reasons for the lack of long-term effectiveness using anti-VEGF or perhaps other anti-angiogenic therapies are not known, yet it raises several potentially informative issues regarding sustained angiogenic responses by tumor cells. First of all, no evidence has been provided to suggest whether endothelial cells eventually acquire resistance to these or other anti-angiogenic treatments. Secondly, given that the tumor microenvironment retains many of the features that enhance VEGF expression, such as hypoxia, it is unlikely that tumor cells simply stop producing this factor. However, it is possible that tumors may evolve in a manner such that they no longer require angiogenesis or VEGF for survival. Recent studies also showed that many tumors that have mutations in the p53 gene have consequently adapted to chronic hypoxic environments and in fact are not significantly affected by anti-angiogenic therapies [38]. Thus, failure of long-term anti-VEGF treatment to improve patient survival might indicate that certain tumors have acquired p53 mutations and thus are no longer as susceptible to death due to a limited blood supply. Since many breast tumors and commonly used breast cancer cell lines, such as MDA231, have nonfunctional p53 [4], these findings should be taken into consideration when designing studies to evaluate breast tumor angiogenesis.
Another possible explanation for the lack of long-term effects of anti-VEGF therapy in breast cancer may be that the genetic instability of tumor cells allows them to acquire the capability of expressing other angiogenic factors to supplant their dependency on VEGF. For example, in addition to expressing high levels of VEGF, many solid tumors, including breast tumors, secrete high levels of other angiogenic factors, including basic fibroblast growth factor (bFGF) and platelet-derived growth factor (PDGF) [39]. In fact, studies have shown that although VEGF is required for the initial growth of human breast tumor carcinoma cells, after the tumor had reached a certain size, they were no longer dependent on VEGF but instead showed increased expression of bFGF [40]. Given a tumor's notorious ability to acquire resistance to a number of therapeutic agents, it may well be that tumors change the profile of angiogenic factors they produce in response to therapeutic selection pressures. The major drawback to targeting bFGF or PDGF receptors, however, is that these receptors are expressed on cells other than the endothelium, a fact which may increase the potential for adverse side effects.
Integrins and angiogenesis
Endothelial cells must also be enabled to adhere to the activated tumor stroma to effectively migrate and invade adjacent tissue. Invasion and adhesion to the tumor stroma requires an increase in the expression of specialized extracellular matrix receptors, the integrins, which regulate adhesion to and migration through extracellular matrices [5]. To date, no endothelium-specific integrins have been discovered, but there are several endothelial-cell integrins that show a limited tissue distribution and, more importantly, are not expressed on quiescent endothelium but are upregulated in response to the tumor microenvironment. One of these is αvβ3 integrin, which is not expressed on resting quiescent endothelial cells but is markedly upregulated in endothelial cells at sites of angiogenesis, including wound repair and angiogenesis induced in response to human breast tumor cells [41]. This integrin binds to fibrinogen, tenascin, fibronectin and proteolyzed collagen, which are highly expressed in the breast tumor stroma [15]. Binding to these ligands by αvβ3 allows endothelial cells to migrate into the adjacent stroma, and blocking αvβ3 not only prevents endothelial cell migration and angiogenesis but also leads to apoptosis of endothelial cells [5, 41].
A humanized antibody against αvβ3 integrin has been developed and is now entering clinical trials, but its effectiveness in treating breast cancer remains to be established. Nonetheless, the finding that αvβ3 is largely restricted to angiogenic endothelium has provided an additional strategy for targeting vascular endothelial cells by coupling αvβ3-specific antibodies to nanoparticles capable of delivering more generalized inhibitors of signaling pathways activated during angiogenesis [42].
Earlier findings showed that the tumor microenvironment is a critical determinant of the nature and extent of the angiogenic response [35, 36]. It is not clear how integrin expression levels or the nature of the integrin heterodimers expressed by the angiogenic endothelium correlates with the different tumor environments.
Another integrin that is upregulated in angiogenic vessels associated with human breast tumors is the fibronectin receptor α5β1. Blocking this integrin with specific antibodies can impair angiogenesis induced by bFGF but not VEGF in the chick chorioallantoic membrane assay [43]. In addition to α5β1, the collagen receptor α2β1 can also mediate angiogenesis in response to VEGF [44].
It is possible that the specific integrin used for angiogenesis by a particular tumor type may be dictated by the nature of the factors the tumor secretes or other properties of the tumor microenvironment. For example, although α2β1 is upregulated by VEGF, αvβ3 and α5β1 are not, but instead are induced by bFGF, tumor necrosis factor alpha and other angiogenic cytokines [43–45]. Therefore if the tumor changes its production of angiogenic factors, this may also change the relative expression levels of various integrins on the corresponding tumor endothelium. It would therefore seem necessary to compare different stages of tumors (VEGF-dependent vs other angiogenic factors) in order to fully appreciate the types of integrins that may contribute to tumor-induced angiogenesis.
Recent studies also showed that mice lacking both β3 and integrins could still undergo a significant degree of β5 angiogenesis in response to melanoma or Lewis lung carcinoma tumors injected subcutaneously and showed a normal angiogenic response to hypoxia, suggesting that β3 or β5 integrins were not essential for tumor angiogenesis [46]. Again, it is not clear whether tumors that do not rely on VEGF would do equally well in these mice, or whether the same tumor would elicit strong angiogenic responses in other microenvironments such as the brain or mammary fat pad.
Proteolyzed fragments of extracellular matrix as inhibitors of angiogenesis
In addition to their role in facilitating tumor invasion and angiogenesis, matrix metalloproteinases have recently been assigned a role in helping block tumor progression and angiogenesis. One of the first naturally occurring inhibitors of angiogenesis identified was angiostatin, a 38-kDa internal cleavage production of plasminogen, generated by tumor cell MMP-2 [47]. Subsequently another naturally occurring, potent inhibitor of angiogenesis, endostatin, was discovered; it is a product of proteolyzed collagen XVIII [48]. More recently, two groups have reported that various proteolytic fragments of the basement membrane component type IV collagen can inhibit angiogenesis [49, 50]. One fragment, named tumstatin because of its ability to impair tumor growth and angiogenesis, initially binds to αvβ3 and subsequently leads to enhanced binding of 4EBP1 to eIF-4E to block protein translation through a rapamycin-sensitive pathway. Moreover, this aspect of tumstatin's action was specific to endothelial cells [50]. In contrast, the collagen-XVIII fragment, endostatin, does not influence protein synthesis in endothelial cells.
Recent studies, however, have shed light on the mechanism of endostatin's antiproliferative and anti-angiogenic action. Endostatin can interfere with the Wnt signaling pathway and block β-catenin/TCF-mediated transcription of the cell cycle mediators cyclin D and c-Myc [51]. Not clear, however, is the relative abundance of these inhibitors in the tumor environment. Do dormant or less aggressive tumors produce relatively more of these inhibitory fragments or do different tumor microenvironments also influence the levels produced? Since generation of inhibitory fragments requires matrix-degrading proteinases, it is not clear whether the disappointing results seen using protease inhibitors as antimetastastic and anti-angiogenic agents is related to interfering with the production of these inhibitory fragments.
Endothelial progenitor cells and tumor-induced angiogenesis
One of the most exciting developments in the area of tumor-induced angiogenesis is the finding that endothelial progenitor cells (EPCs) can be recruited into the circulation, become incorporated into new tumor-associated vasculature and differentiate into endothelial cells [52]. Although endothelial-cell progenitors were initially isolated from peripheral blood, EPCs originate from a renewing population of hematopoietic stem cells (HSCs) residing in the bone marrow [53, 54]. In an elegant series of experiments, Lyden et al. [52] showed that deletion of the mouse Id-1 and Id-3 genes, which encode transcription factors, impaired the mobilization of HSCs. Furthermore, inhibition of HSC and EPC mobilization prevented xenografted tumors from inducing an initial angiogenic response in these animals. However, when wild-type HSCs were grafted into the marrow of the Id-1- and Id-3-null mice, the tumors consequently produced a robust angiogenic response and tumor growth was increased. Thus, the tumors were capable of producing angiogenic factors that mobilize and recruit HSCs to the areas of neovascularization. Although the Id-null mice are heavily dependent on recruitment of HSCs for establishing any measurable tumor vasculature, it is not clear to what extent the recruitment of HSCs contributes to tumor angiogenesis in other settings.
Not surprisingly, VEGF and VEGFR2, expressed on HSCs, are believed to be critical for maintenance, expansion and recruitment of HSC populations, since mice lacking VEGF or VEGFR2 are deficient in angiogenesis as well as hematopoiesis [55]. Recent studies have helped to establish an essential role for VEGFR1 in this process. Specific inhibition of VEGFR1 blocked cycling of HSCs as well as repopulation of the bone marrow after suppression [17] and could also block tumor-induced angiogenesis [16]. Furthermore, addition of placental growth factor, a member of the VEGF family that acts exclusively on VEGFR1, could restore hematopoiesis. A corresponding increase in expression of MMP-9 leads to proteolysis and release of c-kit ligand from the marrow matrix, which in turn stimulates expansion of the HSC population [56].
Although recruitment of EPCs into tumor vasculature has so far been observed only in experimental models of tumor angiogenesis, it is worth noting that studies in humans have identified a renewable source of EPCs in bone marrow, and circulating endothelial progenitor cells have been detected in inflammatory breast cancers [54, 57]. Furthermore, human HSC progenitors transplanted into nude mice can be recruited to the tumor vasculature [58]. Therefore, the likelihood that EPC recruitment and incorporation will emerge as an important component of tumor angiogenesis in human disease is strong. At the moment, however, it is not clear whether selective inhibition of VEGFR1 to block recruitment of these cells will also have a negative effect on other aspects of bone marrow function.
Vascular mimicry
Another example of a tumor cell's survival strategy in the absence of classic angiogenesis is the phenomenon of vascular mimicry, where bona fide capillaries are not present, yet conduits for the transport of nutrients are observed. A controversial study [59] showed that certain nonvascular tumors contained channels that were devoid of endothelial cells but lined by tumor cells and displayed many properties of a circulatory system. In this instance of vascular mimicry, channels between tightly packed uveal melanoma cells appear to express many of the genes normally associated with endothelial cells. It is not clear whether there was evidence of blood flow through the channels formed between the uveal melanoma cells. Nonetheless, this finding raises the interesting question of whether the ability to form a pseudo-vasculature may be another example of an elaborate survival strategy arising from heterogeneous, genetically pliable tumors and present an additional complication for conventional anti-angiogenic therapies.
The future of anti-angiogenic treatment and breast cancer
Despite the variety of means that can be adopted by the tumor cells to ensure an angiogenic response and despite their ability to overcome the need for conventional angiogenesis to survive, anti-angiogenic therapy still holds great promise. In particular, agents that target both the growth of breast tumor cells and their ability to produce angiogenic factors, such as HER2-neutralizing antibodies, should prove to be particularly effective. In addition, since tumor cells can rapidly acquire resistance to many chemotherapeutic agents, direct targeting of the vascular endothelium using particles coupled to αvβ3 or VEGFR2 to deliver potent inhibitors of essential signaling pathways also provides an additional strategy to keep tumor growth and metastasis in check. It will also be important to characterize the nature of angiogenic factors produced by increasingly aggressive tumors as well as consider means to combat the potential recruitment of endothelial progenitor cells to sites of tumor growth. By studying the angiogenic responses to tumors generated within the breast microenvironment, it may be possible to determine the relative contribution of each of these factors and design the appropriate means to control breast-tumor-induced angiogenesis.
Abbreviations
- bFGF =:
-
basic fibroblast growth factor
- EPC =:
-
endothelial progenitor cell
- HIF =:
-
hypoxia-inducible factor
- HIF-1α =:
-
hypoxia-inducible factor 1α
- HSC =:
-
hematopoietic stem cell
- MMP =:
-
matrix metalloproteinase
- MMTV =:
-
mouse mammary tumor virus
- PDGF =:
-
platelet-derived growth factor
- VEGF =:
-
vascular endothelial growth factor
- VEGFR =:
-
VEGF receptor
- VHL =:
-
von Hippel-Lindau protein.
References
Folkman J: Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995, 1: 27-31.
Engels K, Fox SB, Whitehouse RM, Gatter KC, Harris AL: Distinct angiogenic patterns are associated with high-grade in situ ductal carcinomas of the breast. J Pathol. 1997, 181: 207-212. 10.1002/(SICI)1096-9896(199702)181:2<207::AID-PATH758>3.0.CO;2-4.
Gasparini G, Harris AL: Clinical importance of the determination of tumor angiogenesis in breast carcinoma: much more than a new prognostic tool. J Clin Oncol. 1995, 13: 765-782.
Linderholm B, Lindh B, Tavelin B, Grankvist K, Henriksson R: p53 and vascular endothelial growth factor expression predicts outcome in 833 patients with primary breast carconoma. Int J Cancer. 2000, 89: 51-62. 10.1002/(SICI)1097-0215(20000120)89:1<51::AID-IJC9>3.0.CO;2-8.
Elcieri BP, Cheresh DA: Adhesion events in angiogenesis. Curr Opin Cell Biol. 2001, 13: 563-568. 10.1016/S0955-0674(00)00252-0.
Kubota Y, Kleinman HK, Martin GR, Lawley TJ: Role of laminin and basement membrane in the morphological differentiation of human endothelial cells into capillary-like structures. J Cell Biol. 1988, 107: 1589-1596.
Wang GL, Jiang BH, Rue EA, Semenza GL: Hypoxia inducible factor is a basic helix-loop-helix PAS heterodimer regulated by cellular oxygen tension. Proc Natl Acad Sci USA. 1995, 92: 5510-5514.
Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ: Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 2001, 20: 5197-5206. 10.1093/emboj/20.18.5197.
Maxwell PH, Dachs GU, Gleadle JM, Nicholls LG, Harris AL, Stratford IJ, Hankinson O, Pugh CW, Ratcliffe PJ: Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci USA. 1997, 94: 8104-8109. 10.1073/pnas.94.15.8104.
Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshert E, Keshet E: Role of Hif-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998, 394: 485-490. 10.1038/28867.
Bos R, Zhong H, Hanrahan CF, Mommers EC, Semenza GL, Pinedo HM, Abeloff MD, Simons JW, van Diest PJ, van der Wall E: Levels of hypoxia-inducible factor-1 alpha during breast carcinogenesis. J Natl Cancer Inst. 2001, 93: 309-314. 10.1093/jnci/93.4.309.
Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL: Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996, 16: 4604-4613.
Gerber HP, Condorelli F, Park J, Ferrara N: Differential transcriptional regulation of the two vascular endothelial growth factor receptor genes. Flt-1, but not Flk-1/KDR, is up-regulated by hypoxia. J Biol Chem. 1997, 272: 23659-23667. 10.1074/jbc.272.38.23659.
Olofsson B, Jeltsch M, Eriksson U, Alitalo K: Current biology of Vegf-B and Vegf-C. Curr Opin Biotechnol. 1999, 10: 528-535. 10.1016/S0958-1669(99)00024-5.
Brown LF, Guidi AJ, Schnitt SJ, Van De Water L, Iruela-Arispe ML, Yeo TK, Tognazzi K, Dvorak HF: Vascular stroma formation in carcinoma in situ, invasive carcinoma, and metastatic carcinoma of the breast. Clin Cancer Res. 1999, 5: 1041-1056.
Hattori K, Heissig B, Wu Y, Dias S, Tejada R, Ferris B, Hicklin DJ, Zhu Z, Bohlen P, Witte L, Hendrikx J, Hackett NR, Crystal RG, Moore MA, Werb Z, Lyden D, Rafii S: Placental growth factor reconstitutes hematopoiesis by recruiting VEGFR1(+) stem cells from bone-marrow microenvironment. Nat Med. 2002, 8: 841-849.
Luttun A, Tjwa M, Moons L, Wu Y, Angelillo-Scherrer A, Liao F, Nagy JA, Hooper A, Priller J, De Klerck B, Compernolle V, Daci E, Bohlen P, Dewerchin M, Herbert JM, Fava R, Matthys P, Carmeliet G, Collen D, Dvorak HF, Hicklin DJ, Carmeliet P: Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat Med. 2002, 8: 831-840.
Dvorak HF, Sioussat TM, Brown LF, Berse B, Nagy JA, Sotrel A, Manseau EJ, Van de Water L, Senger DR: Distribution of vascular permeability factor (vascular endothelial growth factor) in tumors: concentration in tumor blood vessels. J Exp Med. 1991, 174: 1275-1278.
Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, Jain RK, McDonald DM: Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000, 156: 1363-1380.
Guidi AJ, Schnitt SJ, Fischer L, Tognazzi K, Harris JR, Dvorak HF, Brown LF: Vascular permeability factor (vascular endothelial growth factor) expression and angiogenesis in patients with ductal carcinoma in situ of the breast. Cancer. 1997, 80: 1945-1953. 10.1002/(SICI)1097-0142(19971115)80:10<1945::AID-CNCR11>3.0.CO;2-Y.
Greb RR, Maier I, Wallwiener D, Kiesel L: Vascular endothelial growth factor a (Vegf-a) mRNA expression levels decrease after menopause in normal breast tissue but not in breast cancer lesions. Br J Cancer. 1999, 81: 225-231. 10.1038/sj.bjc.6690681.
Losordo DW, Isner JM: Estrogen and angiogenesis: A review. Arterioscler Thromb Vasc Biol. 2001, 21: 6-12.
Hyder SM, Murthy L, Stancel GM: Progestin regulation of vascular endothelial growth factor in human breast cancer cells. Cancer Res. 1998, 58: 392-395.
Ruohola JK, Valve EM, Karkkainen MJ, Joukov V, Alitalo K, Harkonen PL: Vascular endothelial growth factors are differentially regulated by steroid hormones and antiestrogens in breast cancer cells. Mol Cell Endocrinol. 1999, 149: 29-40. 10.1016/S0303-7207(99)00003-9.
Buteau-Lozano H, Ancelin M, Lardeux B, Milanini J, Perrot-Applanat M: Transcriptional regulation of vascular endothelial growth factor by estradiol and tamoxifen in breast cancer cells: a complex interplay between estrogen receptors alpha and beta. Cancer Res. 2002, 62: 4977-4984.
Takei H, Lee ES, Jordan CV: In vitro regulation of vascular endothelial growth factor by estrogens and antiestrogens in estrogen-receptor positive breast cancer. Breast Cancer. 2002, 9: 39-42.
Scorilas A, Karameris A, Arnogiannaki N, Ardavanis A, Bassilopoulos P, Trangas T, Talieri M: Overexpression of matrix-metalloproteinase-9 in human breast cancer: a potential favourable indicator in node-negative patients. Br J Cancer. 2001, 84: 1488-1496. 10.1054/bjoc.2001.1810.
Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D: Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000, 2: 737-744. 10.1038/35036374.
Bagheri-Yarmand R, Vadlamudi RK, Wang RA, Mendelsohn J, Kumar R: Vascular endothelial growth factor up-regulation via p21-activated kinase-1 signaling regulates heregulin-beta1-mediated angiogenesis. J Biol Chem. 2000, 275: 39451-39457. 10.1074/jbc.M006150200.
Yen L, You XL, Al Moustafa AE, Batist G, Hynes NE, Mader S, Meloche S, Alaoui-Jamali MA: Heregulin selectively upregulates vascular endothelial growth factor secretion in cancer cells and stimulates angiogenesis. Oncogene. 2000, 19: 3460-3469. 10.1038/sj.onc.1203685.
Petit AM, Rak J, Hung MC, Rockwell P, Goldstein N, Fendly B, Kerbel RS: Neutralizing antibodies against epidermal growth factor and ErbB-2/neu receptor tyrosine kinases down-regulate vascular endothelial growth factor production by tumor cells in vitro and in vivo: angiogenic implications for signal transduction therapy of solid tumors. Am J Pathol. 1997, 151: 1523-1530.
Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL: HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001, 21: 3995-4004. 10.1128/MCB.21.12.3995-4004.2001.
Chung J, Bachelder RE, Lipscomb EA, Shaw LM, Mercurio AM: Integrin (alpha 6 beta 4) regulation of eIF-4E activity and VEGF translation: a survival mechanism for carcinoma cells. J Cell Biol. 2002, 158: 165-174. 10.1083/jcb.200112015.
Edel MJ, Harvey JM, Papadimitriou JM: Comparison of vascularity and angiogenesis in primary invasive mammary carcinomas and in their respective axillary lymph node metastases. Clin Exp Metastasis. 2000, 18: 695-702. 10.1023/A:1013139022051.
Monsky WL, Mouta Carreira C, Tsuzuki Y, Gohongi T, Fukumura D, Jain RK: Role of host microenvironment in angiogenesis and microvascular functions in human breast cancer xenografts: mammary fat pad versus cranial tumors. Clin Cancer Res. 2002, 8: 1008-1013.
Lee JC, Kim DC, Gee MS, Saunders HM, Sehgal CM, Feldman MD, Ross SR, Lee WM: Interleukin-12 inhibits angiogenesis and growth of transplanted but not in situ mouse mammary tumor virus-induced mammary carcinomas. Cancer Res. 2002, 62: 747-755.
Animal models to study mammary gland development, physiology and tumorigenesis. [http://mammary.nih.gov/models/index.html]
Yu JL, Rak JW, Coomber BL, Hicklin DJ, Kerbel RS: Effect of p53 status on tumor response to antiangiogenic therapy. Science. 2002, 295: 1526-1528. 10.1126/science.1068327.
de Jong JS, van Diest PJ, van der Valk P, Baak JP: Expression of growth factors, growth-inhibiting factors, and their receptors in invasive breast cancer. II: Correlations with proliferation and angiogenesis. J Pathol. 1998, 184: 53-57. 10.1002/(SICI)1096-9896(199801)184:1<53::AID-PATH6>3.0.CO;2-7.
Yoshiji H, Harris SR, Thorgeirsson UP: Vascular endothelial growth factor is essential for initial but not continued in vivo growth of human breast carcinoma cells. Cancer Res. 1997, 57: 3924-3928.
Brooks PC, Stromblad S, Klemke R, Visscher D, Sarkar FH, Cheresh DA: Anti-integrin alpha v beta 3 blocks human breast cancer growth and angiogenesis in human skin. J Clin Invest. 1995, 96: 1815-1822.
Hood JD, Bednarski M, Frausto R, Guccione S, Reisfeld RA, Xiang R, Cheresh DA: Tumor regression by targeted gene delivery to the neovasculature. Science. 2002, 296: 2404-2407. 10.1126/science.1070200.
Kim S, Bell K, Mousa SA, Varner JA: Regulation of angiogenesis in vivo by ligation of integrin alpha5beta1 with the central cell-binding domain of fibronectin. Am J Pathol. 2000, 156: 1345-1362.
Senger DR, Claffey KP, Benes JE, Perruzzi CA, Sergiou AP, Detmar M: Angiogenesis promoted by vascular endothelial growth factor: regulation through alpha1beta1 and alpha2beta1 integrins. Proc Natl Acad Sci USA. 1994, 94: 13612-13617. 10.1073/pnas.94.25.13612.
Boudreau N, Andrews C, Srebrow A, Ravanpay A, Cheresh DA: Induction of the angiogenic phenotype by Hox D3. J Cell Biol. 1997, 139: 257-264. 10.1083/jcb.139.1.257.
Reynolds LE, Wyder L, Lively JC, Taverna D, Robinson SD, Huang X, Sheppard D, Hynes RO, Hodivala-Dilke KM: Enhanced pathological angiogenesis in mice lacking beta3 integrin or beta3 and beta5 integrins. Nat Med. 2002, 8: 27-34. 10.1038/nm0102-27.
O'Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, Lane WS, Cao Y, Sage EH, Folkman J: Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994, 79: 315-328.
O'Reilly MS, Wiederschain D, Stetler-Stevenson WG, Folkman J, Moses MA: Regulation of angiostatin production by matrix metalloproteinase-2 in a model of concomitant resistance. J Biol Chem. 1999, 274: 29568-29571. 10.1074/jbc.274.41.29568.
Petitclerc E, Boutaud A, Prestayko A, Xu J, Sado Y, Ninomiya Y, Sarras MP, Hudson BG, Brooks PC: New functions for non-collagenous domains of human collagen type IV. Novel integrin ligands inhibiting angiogenesis and tumor growth in vivo. J Biol Chem. 2000, 275: 8051-8061. 10.1074/jbc.275.11.8051.
Maeshima Y, Sudhakar A, Lively JC, Ueki K, Kharbanda S, Kahn CR, Sonenberg N, Hynes RO, Kalluri R: Tumstatin, an endothelial cell-specific inhibitor of protein synthesis. Science. 2002, 295: 140-143. 10.1126/science.1065298.
Hanai J, Gloy J, Karumanchi SA, Kale S, Tang J, Hu G, Chan B, Ramchandran R, Jha V, Sukhatme VP, Sokol S: Endostatin is a potential inhibitor of Wnt signaling. J Cell Biol. 2002, 158: 529-539. 10.1083/jcb.200203064.
Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L, Chadburn A, Heissig B, Marks W, Witte L, Wu Y, Hicklin D, Zhu Z, Hackett NR, Crystal RG, Moore MA, Hajjar KA, Manova K, Benezra R, Rafii S: Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001, 7: 1194-1201. 10.1038/nm1101-1194.
Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM: Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997, 275: 964-967. 10.1126/science.275.5302.964.
Reyes M, Dudek A, Jahagirdar B, Koodie L, Marker PH, Verfaillie CM: Origin of endothelial progenitors in human postnatal bone marrow. J Clin Invest. 2002, 109: 337-346. 10.1172/JCI200214327.
Gerber HP, Malik AK, Solar GP, Sherman D, Liang XH, Meng G, Hong K, Marsters JC, Ferrara N: VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature. 2002, 417: 954-958. 10.1038/nature00821.
Heissig B, Hattori K, Dias S, Friedrich M, Ferris B, Hackett NR, Crystal RG, Besmer P, Lyden D, Moore MA, Werb Z, Rafii S: Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell. 2002, 109: 625-637.
Shirakawa K, Shibuya M, Heike Y, Takashima S, Watanabe I, Konishi F, Kasumi F, Goldman CK, Thomas KA, Bett A, Terada M, Wakasugi H: Tumor-infiltrating endothelial cells and endothelial precursor cells in inflammatory breast cancer. Int J Cancer. 2002, 99: 344-351. 10.1002/ijc.10336.
de Bont ES, Guikema JE, Scherpen F, Meeuwsen T, Kamps WA, Vellenga E, Bos NA: Mobilized human CD34+ hematopoietic stem cells enhance tumor growth in a nonobese diabetic/ severe combined immunodeficient mouse model of human non-Hodgkin's lymphoma. Cancer Res. 2001, 61: 7654-7659.
Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe'er J, Trent JM, Meltzer PS, Hendrix MJ: Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999, 155: 739-752.
Acknowledgements
NB is supported by NIH grants CA- 85249-and -P50 GM27345.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None declared.
Rights and permissions
About this article
Cite this article
Boudreau, N., Myers, C. Breast cancer-induced angiogenesis: multiple mechanisms and the role of the microenvironment. Breast Cancer Res 5, 140 (2003). https://doi.org/10.1186/bcr589
Published:
DOI: https://doi.org/10.1186/bcr589