
Abstract
The insulin-like growth factor (IGF) system exerts pleiotropic effects on mammalian cells. This review focuses on type I IGF receptor (IGF1R)-mediated signal transduction and its relevance in breast cancer. Upon activation by the IGFs, IGF1R, a transmembrane tyrosine kinase receptor, undergoes autophosphorylation, and then binds and phosphorylates additional signaling molecules. These intermediates initiate a series of downstream signaling events that are involved in multiple physiologic processes for cells. Recent data demonstrate that the IGF receptor system actively interacts with the estrogen receptor and integrin receptor systems. Cross-talk among these pathways regulates breast cancer proliferation, protection from cell death, and metastasis. Better understanding of IGF biochemical signaling pathways is of utmost importance for developing therapies for breast cancer.
Similar content being viewed by others


Introduction
The IGF system is composed of IGF ligands, receptors, and binding proteins. These system components form a highly regulated network of interactions both among themselves and between other biologic signaling pathways.
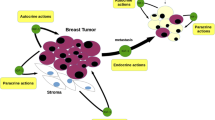
The two well-characterized ligands, IGF-1 and IGF-II, are mitogenic peptides that are highly homologous to each other and to insulin [1]. Whereas insulin is composed of two chains (A and B) of 21 and 30 amino acids, respectively, the IGFs are single-chain molecules that retain the equivalent of the connecting (C)-peptide of proinsulin between the A and B domains. IGF-1 and IGF-II are thought to have autocrine, endocrine, and paracrine roles in normal mammary development and in the etiology of breast cancer [2,3,4,5].
Unlike insulin, circulating IGFs are found complexed to high-affinity binding proteins known as IGF-binding proteins (IGFBPs). Six distinct species have been cloned. An additional family of structurally homologous proteins has been identified and named IGFBP-related proteins, because their affinity for the IGFs is significantly lower than that of the IGFBPs [6,7]. Cleavage of IGFBPs by specific proteases modulates levels of free IGFs and IGFBPs, and thereby their actions. In addition, IGFBPs may also have effects that are completely independent of their role in modulating the action of IGF [8].
The cellular actions of IGFs are mediated by type I and type II receptors, insulin receptor, and insulin receptor-IGF1R hybrids. The type II IGF receptor (IGF2R) is a multifunctional nontyrosine kinase receptor [9,10,11] that is also known as the cation-independent mannose-6-phosphate receptor, and its function in regulating the action of IGF-II has been controversial. IGF1R is a glycosylated heterotetramer that is composed of two extracellular α-subunits and two transmembrane β-subunits that have intrinsic tyrosine kinase activity [12,13]. This review focuses on IGF1R-mediated signaling and its relevance in breast cancer.
Type I insulin-like growth factor receptor signaling
Activation of the IGF1R by IGFs results in its oligomerization, autophosphorylation, and activation of the intrinsic tyrosine kinase [12,13,14,15]. The IGF1R tyrosine kinase further directly phosphorylates various intracellular substrates. Several substrates of the IGF1R have been identified, including insulin receptor substrates (IRSs) 1,2, and 4 [16,17,18,19], src-homology 2/collagen-α proteins (Shc) [20,21], phosphatidyl inositol-3 kinase (PI-3K) [22], growth factor receptor-binding protein 10 [23], focal adhesion kinase (FAK) [24*], and carboxyl-terminal src kinase (CSK) [25*].
IRS-1 is a well-characterized IGF1R-signaling molecule that has multiple sites for tyrosine phosphorylation and acts as a 'docking protein' for other signaling molecules [26,27]. Upon activation of IGF1R, IRS-1 binds and becomes rapidly tyrosine phosphorylated, allowing docking sites for SH2 domain-containing proteins. IRS-1 phosphorylation results in the activation of many downstream signaling pathways; many of these pathways are implicated in mitogenesis and protection from apoptosis. For instance, the following are all known to be stimulated through IRS-1: PI-3K pathway through the association with the p85 regulatory subunit of PI-3K [28]; Ras-mitogen-activated protein kinase (MAPK) cascade through Grb-2/Sos [29]; Syp phosphatase [30]; as well as other pathways involving adapters Nck and Crk [31,32]. Upon tyrosine phosphorylation by the activated IGF1R, Shc (a common substrate of most tyrosine kinase receptors) also recruits Grb-2-Sos complexes and activates the Ras-MAPK pathway.
Additional pathways may be affected by IGF1R activation. For example, the cytoplasmic tyrosine kinase c-Src can phosphorylate IGF1R on the same sites as the IGF-induced autophosphorylation sites [33]. CSK, a negative regulator of Src activity, associates with activated IGF1R, and therefore may play a role in the decrease in Src activity after IGF-1 stimulation. Other substrates of Src are almost exclusively proteins that regulate actin cytoskeleton dynamics, such as FAK, p130 Crk-associated substrate, cortactin, and p190RhoGAP. IGF-1 through its receptors has been shown to positively or negatively modulate tyrosine phosphorylation of focal adhesion proteins such as FAK, p130 Crk-associated substrate, and paxillin [24*,34,35]. Thus, activation of IGF1R, via its interaction with Src, could influence aspects of cytoskeletal organization and cell adhesion.
Insulin-like growth factors and insulin-like growth factor receptor signaling in breast cancer
Expression of IGF-1 and IGF-II has been measured in normal and breast tumor tissues by in situ hybridization and immunohistochemistry. IGF-1 is found mainly in stromal cells that are adjacent to normal breast cells [2]. IGF-II is also mainly expressed in the stroma [5], but may occasionally be found in malignant epithelial cells [4]. Increased IGF-II expression is seen in stromal cells that are adjacent to malignant epithelial cells, whereas levels are lower in stroma that are adjacent to benign and normal breast epithelium [36,37]. Furthermore, malignant breast epithelial cells can induce IGF-II expression in breast stroma in vitro [38]. High IGF-II expression is reported to be associated with poor prognostic features in breast cancer [39].
Endocrine levels of IGF-I have been implicated in breast cancer. Breast cancer patients have higher serum IGF-I levels than do matched control individuals [40]. Higher IGF-I levels have also been associated with an increased risk of developing breast cancer [41].
IGF1R has been found to be both significantly overexpressed [42,43,44] and highly activated [45**] in cancer cells with respect to its status in normal or benign breast tissues. Recent reports [46*] have suggested that insulin receptor may mediate the IGF-II response in breast cancer cells. In addition, insulin receptor-IGF1R hybrids are overexpressed in breast cancer, and these receptors can also mediate IGF responsiveness [47*]. Although the IGF2R does not appear to function in a signaling pathway, there is significant loss of heterozygosity at the IGF2R locus in breast cancer, suggesting that IGF2R may represent a breast tumor suppressor gene [48]. Mutation in the IGF-II binding domain of the remaining IGF2R allele has been identified in cancer cells [49*]. These observations suggest that IGF2R loses the ability to bind IGF-II in some cancer cells. This would allow enhanced interaction of IGF-II with the tyrosine kinase receptors and, perhaps, tumor promotion.
Expression of the IGF downstream signaling molecule IRS-1 is also detected in breast cancer. Increased levels of expression were found to correlate with estrogen receptor (ER) status in 200 node-negative patients, and identified patients with a decreased disease-free survival in a subset of patients with small tumors [50*]. Taken together, these studies show that the IGFs are freely available to the malignant epithelial cells from endocrine or paracrine sources. Furthermore, IGF receptors are present on breast cancer cells to mediate the biologic effects of the IGFs.
Consequence of insulin-like growth factor activation in breast cancer
Activation of the IGF system is known to have substantial pleiotropic effects on mammalian cells. Mitogenesis, transformation, and antiapoptosis induced by IGF1R stimulation could account for many aspects of the malignant phenotype. Both IGF-1 and IGF-II stimulate ER-positive breast cancer cell proliferation at picomolar to nanomolar concentrations [51]. Once IGFs interact with receptors, we found that IRS-1 is the predominant signaling molecule activated in ER-positive human breast cancer cells [52*].
There is also accumulating evidence that IGF action influences breast cancer cell responsiveness to estrogen. It is well established that estrogens stimulate the growth of ER-positive breast cancer cells. ER acts as a ligand-activated transcription factor. Two forms of ER have been cloned, ERα and ERβ [53,54,55]. ERα contains a hormone-binding domain, a DNA-binding domain, and two transcriptional activation domains (AF-1 and AF-2). Estradiol binding to ERα results in dimerization and subsequent binding of the hormone-receptor complex to specific DNA palindromic sequences (estrogen response elements) to initiate gene transcription, and therefore induce the expression of growth promoting genes. To date, a similar role for ERβ has not been found. Antiestrogens, such as tamoxifen, influence ERα function by blocking the initiation of transcription from estrogen response elements without interfering with the binding of ligand-receptor complex to DNA.
Estrogen induces the expression of several members of the IGF family, including IGF-II [3,56,57], IGF1R [58], IGF2R [59], IGFBPs [60], and IRS-1 and IRS-2 [61*]. The increased expression of IGF1R and IRS-1 results in an enhanced response to IGF-1 that is manifested in greater downstream signaling through MAPK. Removal of estrogen results in a dramatic decrease in IRS-1 expression and MAPK activity. Antiestrogens may inhibit IGF action by increasing IGFBP-3 [62,63], affecting phosphorylation of IGF1R or IRS-1 [64*,65,66], downregulating expression of IGF1R and IRS-1 [58,67], and inhibiting ligand-independent activation of the ER by IGF-1 [68,69,70,71]. Thus, several members of the IGF family could be the growth-promoting genes that are regulated by estrogen.
On the other hand, IGF-1 also directly increases the transcriptional activity of the ER and increases expression of estrogen-inducible genes, such as the progesterone receptor gene [71]. Furthermore, IGFBP-1, an inhibitor of IGF-1 action, not only inhibited IGF-mediated activation of the ER, but also had a significant inhibitory effect upon estrogen-mediated activation of the ER. Although the mechanisms that account for this cross-talk are not clear, it is obvious that both signaling pathways can positively influence each other, resulting in reinforcement of biologic effects for both estrogen and IGFs.
Many model systems have shown that IGF1R activation protects cells from programmed cell death. The PI-3K pathway and its substrate AKT1 probably mediate this effect. It has been reported [72] that AKT1 is highly expressed in several human breast carcinoma cell lines, and its activity in MCF-7 cells is modulated by estradiol and IGF-1. Overexpression and activation of AKT1 produces estrogen and IGF-1 independent proliferation and controls an antiapoptotic pathway. IGF-1 reduces apoptosis in doxorubicin-treated and paclitaxel-treated MCF-7 cells [73]. Detailed studies indicate that IGF-I rescue of MCF-7 cells from chemotherapy-induced cell death involves at least two mechanisms: inhibition of apoptosis through PI-3K and induction of proliferation through both PI-3K and MAPK cascades. In clinical specimens, high levels of IGF1R may protect cells from radiation therapy-induced apoptosis [74*].
Several reports have documented the interaction of IGF and integrin signaling pathways. The direct interaction of the two pathways was demonstrated through a physical association between αvβ3 integrin and IRS-1 [75**]. Later reports [24*] also showed that FAK, a downstream signaling molecule of integrins, is a substrate for the insulin receptor and IGF1R. In vascular smooth muscle cells, ligand occupancy of αvβ3 integrin is required for full activation of the IGF1R β-subunit and IRS-1 by IGF-1 stimulation [76,77*]. IGFs are chemoattractants for breast cancer cells, perhaps due to the ability of IGF to affect the integrins [78]. Activation of integrin signaling pathways have been reported [79] to inhibit the mitogenic effect of IGF-1 in human breast cancer cell lines. Recently, IGF1R activation was shown to induce rapid and transient tyrosine dephosphorylation of FAK, p130 Crk-associated substrate, and paxillin in MCF-7 breast cancer epithelial cells [80]. Finally, IGFs may be involved in cell migration and invasion, because dominant-negative IGF1R constructs inhibit invasion and metastasis of MDA-435 breast cancer cells in vitro and in vivo [81*].
Conclusion
Breast cancer is a lethal disease because the transformed epithelial cells proliferate, metastasize, and are protected from programmed cell death. The pathways responsible for each of these phenotypes are only now becoming understood. Despite the multiple accumulated genetic abnormalities that cause malignant transformation, however, it is evident that some of the transformed cells can still respond to signals from their external environment. Notably, the inhibition of ER function has proven to be a powerful weapon in breast cancer treatment.
There now is a large body of evidence showing that IGF activation is involved in these malignant processes; clearly some fully transformed cells can still respond to these cues. It is also evident that the signaling pathways that are activated by the IGFs are not simple or linear. Multiple divergent and convergent biochemical signaling pathways are stimulated after receptor activation, which then impinge upon multiple other pathways that are known to be important in breast cancer biology. We are now just beginning to understand how the IGFs affect breast cancer biology. The next challenge will be to untangle the web of signal cascades initiated by these factors. By doing so, we will be better positioned to develop therapies based on interruption of the key signaling pathways that are responsible for the malignant phenotype.
References
Daughaday WH, Rotwein P: Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr Rev. 1989, 10: 68-91.
Yee D, Paik S, Lebovic GS, et al: Analysis of insulin-like growth factor I gene expression in malignancy: evidence for a paracrine role in human breast cancer. Mol Endocrinol. 1989, 3: 509-517.
Yee D, Cullen KJ, Paik S, et al: Insulin-like growth factor II mRNA expression in human breast cancer. Cancer Res. 1988, 48: 6691-6696.
Paik S: Expression of IGF-I and IGF-II mRNA in breast tissue. Breast Cancer Res Treat. 1992, 22: 31-38.
Giani C, Cullen KJ, Campani D, et al: IGF-II mRNA and protein are expressed in the stroma of invasive breast cancers: an in situ hybridization and immunohistochemistry study. Breast Cancer Res Treat. 1996, 41: 43-50.
Baxter RC, Binoux MA, Clemmons DR, et al: Recommendations for nomenclature of the insulin-like growth factor binding protein superfamily. J Clin Endocrinol Metab. 1998, 83: 3213-3213. 10.1210/jc.83.9.3213.
Ferry RJ, Cerri RW, Cohen P: Insulin-like growth factor binding proteins: new proteins, new functions. Horm Res. 1999, 51: 53-67. 10.1159/000023315.
Gucev ZS, Oh Y, Kelley KM, et al: Insulin-like growth factor binding protein 3 mediates retinoic acid- and transforming growth factor beta 2-induced growth inhibition in human breast cancer cells. Cancer Res. 1996, 56: 1545-1550.
Morgan DO, Edman JC, Standring DN, et al: Insulin-like growth factor II receptor as a multifunctional binding protein. Nature. 1988, 329: 301-307. 10.1038/329301a0.
Blanchard F, Raher S, Duplomb L, et al: The mannose 6-phosphate/insulin-like growth factor II receptor is a nanomolar affinity receptor for glycosylated human leukemia inhibitory factor. J Biol Chem. 1998, 273: 20886-20893. 10.1074/jbc.273.33.20886.
Kang JX, Li Y, Leaf A: Mannose-6-phosphate/insulin-like growth factor-II receptor is a receptor for retinoic acid. Proc Natl Acad Sci USA. 1997, 94: 13671-13676. 10.1073/pnas.94.25.13671.
Kato H, Faria TN, Stannard B, et al: Role of tyrosine kinase activity in signal transduction by the insulin-like growth factor-I (IGF-I) receptor. Characterization of kinase-deficient IGF-I receptors and the action of an IGF-I-mimetic antibody (alpha IR-3). J Biol Chem. 1993, 268: 2655-2661.
Ullrich A, Gray A, Tam AW, et al: Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 1986, 5: 2503-2512.
Frattali AL, Pessin JE: Relationship between alpha subunit ligand occupancy and beta subunit autophosphorylation in insulin/insulin-like growth factor-1 hybrid receptors. J Biol Chem. 1993, 268: 7393-7400.
LeRoith D, Werner H, Beitner-Johnson D, et al: Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev. 1995, 16: 143-163. 10.1210/er.16.2.143.
Myers MG, Sun XJ, White MF: The IRS-1 signaling system. Trends Biochem Sci. 1994, 19: 289-293. 10.1016/0968-0004(94)90007-8.
He W, Craparo A, Zhu Y, et al: Interaction of insulin receptor substrate-2 (IRS-2) with the insulin and insulin-like growth factor I receptors. Evidence for two distinct phosphotyrosine-dependent interaction domains with IRS-2. J Biol Chem. 1996, 271: 11641-11645. 10.1074/jbc.271.20.11641.
Qu BH, Karas M, Koval A, et al: Insulin receptor substrate-4 enhances insulin-like growth factor-I-induced cell proliferation [In Process Citation]. J Biol Chem. 1999, 274: 31179-31184. 10.1074/jbc.274.44.31179.
Fantin VR, Sparling JD, Slot JW, et al: Characterization of insulin receptor substrate 4 in human embryonic kidney 293 cells. J Biol Chem. 1998, 273: 10726-10732. 10.1074/jbc.273.17.10726.
Pelicci G, Lanfrancone L, Grignani F, et al: A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell. 1992, 70: 93-104.
Giorgetti S, Pelicci PG, Pelicci G, et al: Involvement of Src-homology/collagen (SHC) proteins in signaling through the insulin receptor and the insulin-like-growth-factor-I-receptor. Eur J Biochem. 1994, 223: 195-202.
Lamothe B, Bucchini D, Jami J, et al: Interaction of p85 subunit of PI 3-kinase with insulin and IGF-1 receptors analysed by using the two-hybrid system. FEBS Lett. 1995, 37: 51-55. 10.1016/0014-5793(95)01011-3.
Morrione A, Valentinis B, Li S, et al: Grb10: a new substrate of the insulin-like growth factor I receptor. Cancer Res. 1996, 56: 3165-3167.
Baron V, Calleja V, Ferrari P, et al: p125Fak focal adhesion kinase is a substrate for the insulin and insulin-like growth factor-I tyrosine kinase receptors. J Biol Chem. 1998, 273: 7162-7168. 10.1074/jbc.273.12.7162. Data from this paper provide evidence that insulin and IGF-I are able to induce p125FAK (an important molecule involved in integrin and other growth factor signaling) phosphorylation and activation. Therefore, the importance of the interaction of IGF system and adhesion-dependent signaling system is emphasized.
Arbet-Engels C, Tartare-Deckert S, Eckhart W: C-terminal Src kinase associates with ligand-stimulated insulin-like growth factor-I receptor. J Biol Chem. 1999, 274: 5422-5428. 10.1074/jbc.274.9.5422. The authors demonstrate that CSK can bind to activated IGF1R and the insulin receptor. The results suggest that c-Src and CSK are involved in IGF1R and insulin receptor signaling and that the interaction of CSK with the IGF1R may play a role in the decrease in c-Src activity after IGF-I stimulation
Myers MG, Sun XJ, Cheatham B, et al: IRS-1 is a common element in insulin and insulin-like growth factor-I signaling to the phosphatidylinositol 3'-kinase. Endocrinology. 1993, 132: 1421-1430. 10.1210/en.132.4.1421.
Sun XJ, Crimmins DL, Myers MG, et al: Pleiotropic insulin signals are engaged by multisite phosphorylation of IRS-1. Mol Cell Biol. 1993, 13: 7418-7428.
Myers MG, Wang LM, Sun XJ, et al: Role of IRS-1-GRB-2 complexes in insulin signaling. Mol Cell Biol. 1994, 14: 3577-3587.
Skolnik EY, Lee CH, Batzer A, et al: The SH2/SH3 domain-containing protein GRB2 interacts with tyrosine-phosphorylated IRS1 and Shc: implications for insulin control of ras signalling. EMBO J. 1993, 12: 1929-1936.
Kuhne MR, Pawson T, Lienhard GE, et al: The insulin receptor substrate 1 associates with the SH2-containing phosphotyrosine phosphatase Syp. J Biol Chem. 1993, 268: 11479-11481.
Lee CH, Li W, Nishimura R, et al: Nck associates with the SH2 domain-docking protein IRS-1 insulin-stimulated cells. Proc Natl Acad Sci USA. 1993, 90: 11713-11717.
Beitner-Johnson D, Blakesley VA, Shen-Orr Z, et al: The proto-oncogene product c-Crk associates with insulin receptor substrate-1 and 4PS. Modulation by insulin growth factor-I (IGF) and enhanced IGF-I signaling. J Biol Chem. 1996, 271: 9287-9290. 10.1074/jbc.271.16.9287.
Peterson JE, Kulik G, Jelinek T, et al: Src phosphorylates the insulin-like growth factor type I receptor on the autophosphorylation sites. Requirement for transformation by src. J Biol Chem. 1996, 271: 31562-31571. 10.1074/jbc.271.49.31562.
Leventhal PS, Shelden EA, Kim B, et al: Tyrosine phosphorylation of paxillin and focal adhesion kinase during insulin-like growth factor-I-stimulated lamellipodial advance. J Biol Chem. 1997, 272: 5214-5218. 10.1074/jbc.272.8.5214.
Konstantopoulos N, Clark S: Insulin and insulin-like growth factor-1 stimulate dephosphorylation of paxillin in parallel with focal adhesion kinase. Biochem J. 1996, 314: 387-390.
Giani C, Cullen KJ, Campani D, et al: IGF-II mRNA and protein are expressed in the stroma of invasive breast cancers: an in situ hybridization and immunohistochemistry study. Breast Cancer Res Treat. 1996, 41: 43-50.
Giani C, Pinchera A, Rasmussen A, et al: Stromal IGF-II messenger RNA in breast cancer: relationship with progesterone receptor expressed by malignant epithelial cells. J Endocrinol Invest. 1998, 21: 160-165.
Singer C, Rasmussen A, Smith HS, et al: Malignant breast epithelium selects for insulin-like growth factor II expression in breast stroma: evidence for paracrine function. Cancer Res. 1995, 55: 2448-2454.
Yu H, Levesque MA, Khosravi MJ, et al: Associations between insulin-like growth factors and their binding proteins and other prognostic indicators in breast cancer. Br J Cancer. 1996, 74: 1242-1247.
Pollak MN: Endocrine effects of IGF-I on normal and transformed breast epithelial cells: potential relevance to strategies for breast cancer treatment and prevention. Breast Cancer Res Treat. 1998, 47: 209-217. 10.1023/A:1005950916707.
Hankinson SE, Willett WC, Colditz GA, et al: Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet. 1998, 351: 1393-1396. 10.1016/S0140-6736(97)10384-1.
Peyrat JP, Bonneterre J: Type 1 IGF receptor in human breast diseases. Breast Cancer Res Treat. 1992, 22: 59-67.
Papa V, Gliozzo B, Clark GM, et al: Insulin-like growth factor-I receptors are overexpressed and predict a low risk in human breast cancer. Cancer Res. 1993, 53: 3736-3740.
Pezzino V, Papa V, Milazzo G, et al: Insulin-like growth factor-I (IGF-I) receptors in breast cancer. Ann N Y Acad Sci. 1996, 784: 189-201.
Resnik JL, Reichart DB, Huey K, et al: Elevated insulin-like growth factor I receptor autophosphorylation and kinase activity in human breast cancer. Cancer Res. 1998, 58: 1159-1164.
Sciacca L, Costantino A, Pandini G, et al: Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene. 1999, 18: 2471-2479. 10.1038/sj/onc/1202600. In addition to IGF1R, these authors suggest that insulin receptor could have a function in mediating breast cancer response to IGF-II.
Pandini G, Vigneri R, Costantino A, et al: Insulin and insulin-like growth factor-I (IGF-I) receptor overexpression in breast cancers leads to insulin/IGF-I hybrid receptor overexpression: evidence for a second mechanism of IGF-I signaling. Clin Cancer Res. 1999, 5: 1935-1944.
Hankins GR, De Souza AT, Bentley RC, et al: M6P/IGF2 receptor: a candidate breast tumor suppressor gene. Oncogene. 1996, 12: 2003-2009.
Byrd JC, Devi GR, De Souza AT, et al: Disruption of ligand binding to the insulin-like growth factor II/mannose 6-phosphate receptor by cancer-associated missense mutations. J Biol Chem. 1999, 274: 24408-24416. 10.1074/jbc.274.34.24408. A potential mechanism for IGF2R as a tumor suppressor gene in breast cancer is described
Rocha RL, Hilsenbeck SG, Jackson JG, et al: Insulin-like growth factor binding protein-3 and insulin receptor substrate-1 in breast cancer: correlation with clinical parameters and disease-free survival. Clin Cancer Res. 1997, 3: 103-109. A demonstration is provided that the effectors of IGF action (IGFBP-3 and IRS-1) are associated with outcome in lymph node negative breast cancer.
Karey KP, Sirbasku DA: Differential responsiveness of human breast cancer cell lines MCF-7 and T47D to growth factors and 17 beta-estradiol. Cancer Res. 1998, 48: 4083-4092.
Jackson JG, White MF, Yee D: Insulin receptor substrate-1 is the predominant signaling molecule activated by insulin-like growth factor-I, insulin, and interleukin-4 in estrogen receptor-positive human breast cancer cells. J Biol Chem. 1998, 273: 9994-10003. 10.1074/jbc.273.16.9994.
Green S, Walter P, Kumar V, et al: Human oestrogen receptor cDNA: sequence, expression and homology to v-erb-A. Nature. 1986, 320: 134-139.
Green S, Walter P, Greene G, et al: Cloning of the human oestrogen receptor cDNA. J Steroid Biochem. 1986, 24: 77-83. 10.1016/0022-4731(86)90035-X.
Kuiper GG, Enmark E, Pelto-Huikko M, et al: Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA. 1996, 93: 5925-5930. 10.1073/pnas.93.12.5925.
Lee AV, Darbre P, King RJ: Processing of insulin-like growth factor-II (IGF-II) by human breast cancer cells. Mol Cell Endocrinol. 1994, 99: 211-220. 10.1016/0303-7207(94)90010-8.
Osborne CK, Coronado EB, Kitten LJ, et al: Insulin-like growth factor-II (IGF-II): a potential autocrine/paracrine growth factor for human breast cancer acting via the IGF-I receptor. Mol Endocrinol. 1989, 3: 1701-1709.
Stewart AJ, Westley BR, May FE: Modulation of the proliferative response of breast cancer cells to growth factors by oestrogen. Br J Cancer. 1992, 66: 640-648.
Mathieu M, Vignon F, Capony F, et al: Estradiol down-regulates the mannose-6-phosphate/insulin-like growth factor-II receptor gene and induces cathepsin-D in breast cancer cells: a receptor saturation mechanism to increase the secretion of lysosomal proenzymes. Mol Endocrinol. 1991, 5: 815-822.
McGuire WL, Jackson JG, Figueroa JA, et al: Regulation of insulin-like growth factor-binding protein (IGFBP) expression by breast cancer cells: use of IGFBP-1 as an inhibitor of insulin-like growth factor action. J Natl Cancer Inst. 1992, 84: 1336-1341.
Lee AV, Jackson JG, Gooch JL, et al: Enhancement of insulin-like growth factor signaling in human breast cancer: estrogen regulation of insulin receptor substrate-1 expression in vitro and in vivo. Mol Endocrinol. 1999, 13: 787-796. 10.1210/me.13.5.787. This paper provides evidence that IGF1R, IRS-1 and IRS-2 are estrogenregulated proteins. These data reinforce the concept of cross-talk between IGF and ER signaling pathways
Huynh H, Yang X, Pollak M: Estradiol and antiestrogens regulate a growth inhibitory insulin-like growth factor binding protein 3 autocrine loop in human breast cancer cells. J Biol Chem. 1996, 271: 1016-1021. 10.1074/jbc.271.2.1016.
Figueroa JA, Jackson JG, McGuire WL, et al: Expression of insulin-like growth factor binding proteins in human breast cancer correlates with estrogen receptor status. J Cell Biochem. 1993, 52: 196-205.
Freiss G, Puech C, Vignon F: Extinction of insulin-like growth factor-I mitogenic signaling by antiestrogen-stimulated Fas-associated protein tyrosine phosphatase-1 in human breast cancer cells. Mol Endocrinol. 1998, 12: 568-579. 10.1210/me.12.4.568. Antiestrogens can inhibit IGF signaling in breast cancer cells by inducing increased phosphatase activity.
Freiss G, Rochefort H, Vignon F: Mechanisms of 4-hydroxytamoxifen anti-growth factor activity in breast cancer cells: alterations of growth factor receptor binding sites and tyrosine kinase activity. Biochem Biophys Res Commun. 1990, 173: 919-926.
Guvakova MA, Surmacz E: Tamoxifen interferes with the insulin-like growth factor I receptor (IGF-IR) signaling pathway in breast cancer cells. Cancer Res. 1997, 57: 2606-2610.
Salerno M, Sisci D, Mauro L, et al: Insulin receptor substrate 1 is a target for the pure antiestrogen ICI 182,780 in breast cancer cells. Int J Cancer. 1999, 81: 299-304. 10.1002/(SICI)1097-0215(19990412)81:2<299::AID-IJC21>3.0.CO;2-8.
Ignar-Trowbridge DM, Pimentel M, Parker MG, et al: Peptide growth factor cross-talk with the estrogen receptor requires the A/B domain and occurs independently of protein kinase C or estradiol. Endocrinology. 1996, 137: 1735-1744. 10.1210/en.137.5.1735.
Cho H, Aronica SM, Katzenellenbogen BS: Regulation of progesterone receptor gene expression in MCF-7 breast cancer cells: a comparison of the effects of cyclic adenosine 3',5'-monophosphate, estradiol, insulin-like growth factor-I, and serum factors. Endocrinology. 1994, 134: 658-664. 10.1210/en.134.2.658.
Patrone C, Ma ZQ, Pollio G, et al: Cross-coupling between insulin and estrogen receptor in human neuroblastoma cells. Mol Endocrinol. 1996, 10: 499-507. 10.1210/me.10.5.499.
Lee AV, Weng CN, Jackson JG, et al: Activation of estrogen receptor-mediated gene transcription by IGF-I in human breast cancer cells. J Endocrinol. 1997, 152: 39-47.
Ahmad S, Singh N, Glazer RI: Role of AKT1 in 17beta-estradiol-and insulin-like growth factor I (IGF-I)-dependent proliferation and prevention of apoptosis in MCF-7 breast carcinoma cells. Biochem Pharmacol. 1999, 58: 425-430. 10.1016/S0006-2952(99)00125-2.
Gooch JL, Van Den Berg CL, Yee D: Insulin-like growth factor (IGF)-I rescues breast cancer cells from chemotherapy-induced cell death: proliferative and anti-apoptotic effects. Breast Cancer Res Treat. 1999, 56: 1-10. 10.1023/A:1006208721167.
Turner BC, Haffty BG, Narayanan L, et al: Insulin-like growth factor-I receptor overexpression mediates cellular radioresistance and local breast cancer recurrence after lumpectomy and radiation. Cancer Res. 1997, 57: 3079-3083. This clinical study showed that overexpression of IGF1R is associated with in-breast recurrence after radiation therapy. This study suggests that IGF1R could protect cells from radiation-induced apoptosis.
Vuori K, Ruoslahti E: Association of insulin receptor substrate-1 with integrins. Science. 1994, 266: 1576-1578.This study was the first to demonstrate a link between the insulin receptor signaling pathway and cell adhesion molecules
Jones JI, Prevette T, Gockerman A, et al: Ligand occupancy of the alpha-V-beta3 integrin is necessary for smooth muscle cells to migrate in response to insulin-like growth factor. Proc Natl Acad Sci USA. 1996, 93: 2482-2487. 10.1073/pnas.93.6.2482.
Zheng B, Clemmons DR: Blocking ligand occupancy of the alphaV beta3 integrin inhibits insulin-like growth factor I signaling in vascular smooth muscle cells. Proc Natl Acad Sci USA. 1998, 95: 11217-11222. 10.1073/pnas.95.19.11217.
Doerr ME, Jones JI: The roles of integrins and extracellular matrix proteins in the insulin-like growth factor I-stimulated chemotaxis of human breast cancer cells. J Biol Chem. 1996, 271: 2443-2447. 10.1074/jbc.271.5.2443.
Perks CM, Gill ZP, Newcomb PV, et al: Activation of integrin and ceramide signalling pathways can inhibit the mitogenic effect of insulin-like growth factor I (IGF-I) in human breast cancer cell lines. Br J Cancer. 1999, 79: 701-706. 10.1038/sj.bjc.6690113.
Guvakova MA, Surmacz E: The activated insulin-like growth factor I receptor induces depolarization in breast epithelial cells characterized by actin filament disassembly and tyrosine dephosphorylation of FAK, Cas, and paxillin. Exp Cell Res. 1999, 251: 244-255. 10.1006/excr.1999.4566.
Dunn SE, Ehrlich M, Sharp NJ, et al: A dominant negative mutant of the insulin-like growth factor-I receptor inhibits the adhesion, invasion, and metastasis of breast cancer. Cancer Res. 1998, 58: 3353-3361. This study demonstrates that functional inhibition of IGF1R disrupts the adhesion, invasion, and metastasis of breast cancer.
Author information
Authors and Affiliations
Corresponding author
Additional information
Articles of particular interest have been highlighted as:
• of special interest
•• of outstanding interest
Rights and permissions
About this article
Cite this article
Zhang, X., Yee, D. Tyrosine kinase signalling in breast cancer: Insulin-like growth factors and their receptors in breast cancer. Breast Cancer Res 2, 170 (2000). https://doi.org/10.1186/bcr50
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/bcr50