
Abstract
Introduction
The prognostic significance of p53 protein expression in early breast cancer remains uncertain, with some but not all studies finding an association with poorer outcomes. Estrogen receptor (ER) expression is both a positive prognostic marker and predictive of response to endocrine therapies. The relationship between these biomarkers is unknown.
Methods
We constructed tissue microarrays (TMAs) from available pathological material from 1113 patients participating in two randomized clinical trials comparing endocrine therapy alone versus chemo-endocrine therapy in node-negative breast cancer. Expression of p53 defined as >10% positive nuclei was analyzed together with prior immunohistochemical assays of ER performed at central pathological review of whole tumor sections.
Results
ER was present (i.e. >1% positive tumor cell nuclei) in 80.1% (880/1092). p53 expression was significantly more frequent when ER was absent, 125/212 (59%) than when ER was present, 171/880 (19%), p <0.0001. A significant qualitative interaction was observed such that p53 expression was associated with better disease-free survival (DFS) and overall survival (OS) among patients whose tumors did not express ER, but worse DFS and OS among patients whose tumors expressed ER. The interaction remained significant after allowance for pathologic variables, and treatment. Similar effects were seen when luminal and non-luminal intrinsic subtypes were compared.
Conclusions
Interpretation of the prognostic significance of p53 expression requires knowledge of concurrent expression of ER. The reason for the interaction between p53 and ER is unknown but may reflect qualitatively different p53 mutations underlying the p53 expression in tumors with or without ER expression.
Trial registration
Current Controlled Trials ACTRN12607000037404 (Trial VIII) and ACTRN12607000029493 (Trial IX).
Similar content being viewed by others

Introduction
The p53 tumor suppressor protein, encoded by the TP53 gene, is a transcription factor that when activated as part of the cellular stress response, regulates suites of genes involved in cellular processes including the cell cycle, apoptosis, and senescence [1]. Mutations in TP53 are amongst the commonest genetic alterations in human cancer, but unlike other tumor suppressors, TP53 is not usually inactivated through deletion or truncating mutations [2]. Instead, it commonly undergoes mono-allelic missense mutations affecting the DNA binding domain, leading to the production of a protein that lacks DNA binding activity but remains capable of binding to, and hence dominantly inactivating, wild-type p53. Some TP53 mutations also result in the acquisition of new oncogenic properties, and it is unclear whether loss of wild-type function, gain of oncogenic function, or their combined effects, account for the oncogenicity of these mutations [1, 2]. Interpretation of many experiments addressing this issue is complicated by the recent identification of multiple p53 isoforms, which arise from alternative splicing and the presence of an internal promoter [3]. The principal isoforms differ in the domain responsible for oligomerization, but are identical through the transactivation and DNA binding domains in which many TP53 mutations occur.
Wild-type p53 protein is rapidly degraded, except under conditions of cellular stress. TP53 mutations are often, although not always, associated with the production of a stable protein that is readily detectable by immunohistochemistry (IHC) of cancer cells [1, 2]. IHC detection of p53 protein is therefore loosely, but imperfectly, associated with mutations in TP53. Some studies assessing p53 status using either IHC or mutational analysis have concluded that TP53 mutation is associated with poor prognosis, but other authors have reported no impact of TP53 mutation on outcome in early breast cancer, and the evidence is not sufficiently strong for p53 status to be recommended as a marker in routine clinical practice [4]. Studies of women treated with a variety of chemotherapeutic regimens, or hormonal therapy suggest that p53 status may be predictive of response to therapy [5, 6]. However, until recently few large studies have either considered, or been able to effectively control for, treatment effects. Retrospective analyses of randomized clinical trials, using either IHC measurement of p53 expression (CALB 9344) or TP53 gene sequencing (BIG 02-98), have identified a significant association with a worse prognosis in patients treated with adjuvant doxorubicin and cyclophosphamide or doxorubin but no significant association with response to taxanes [7, 8]. Similarly, a prospective clinical trial (EORTC 10994/BIG 1-00) found that TP53 status was not predictive of response to taxane-based therapy [9].
The different methods used to assess p53 status have both advantages and disadvantages. Direct identification of mutations by sequencing has the advantage of pinpointing the specific aberration, but is not currently applicable to clinical practice because of its expense, technical difficulty, and likely reduced sensitivity in the routine setting. In addition, it may mis-classify some functionally equivalent genomic alterations, where TP53 is inactivated indirectly. Functional assessment of p53 activity in yeast offers a distinction between inactivating and other mutations [10, 11]. Several laboratories have attempted to develop gene signatures of p53 inactivation, which can potentially be measured using a PCR-based test [12–14]. However, these assays are not immediately practical for routine diagnosis, and some of these signatures are potentially measures of subtype rather than p53 status, since mutations in TP53 are more frequent in tumors lacking estrogen receptor (ER) expression [15–17], in tumors of basal-like, HER2 and luminal B subtypes [18, 19], tumors showing stem-cell like transcriptional patterns [20] and in those with high proliferative fraction [21]. IHC detection of p53 protein mis-classifies some TP53 mutations, and instances where TP53 is inactivated through deletion or truncation, or where persistent cellular stress leads to sustained expression of p53 protein. However, it does have the advantage of ready transfer into clinical practice, if studies in large, well-characterized cohorts provide good evidence of potential utility as a biomarker. Since the material available for the present study was limited to tissue micro-arrays, IHC was the most suitable available method.
Evidence for cross-talk between ER and p53 pathways at several levels suggests that the impact of TP53 mutation may be affected by the presence of ER. First, ER is a p53 target and conversely estrogen treatment induces p53 expression, although it has also been reported to increase the cytoplasmic localization of p53, thereby functionally inactivating it [22–24]. Second, p53 is required for hormonal protection against carcinogen-induced mammary cancer in rodents [25, 26]. Finally, the ER and p53 proteins physically interact, leading to repression of their transcriptional activity and protection of p53 from degradation [27–29]. However, other authors have concluded that the dominant interaction is via ER and p53 binding their cognate response elements in cis to cooperatively regulate p53-responsive genes [30]. Simultaneous allowance for p53 and ER in survival analysis of patients with early breast cancer, where the presence of ER is a favorable prognostic factor [31–34], and predicts the efficacy of endocrine therapies [31], has yielded conflicting results. Some authors report independent adverse prognostic significance of ER-negativity and p53 expression [15, 35] while more recently, different p53 gene signatures have been associated with disparate prognosis and response to cytotoxic therapy in ER-positive and ER-negative disease [12].
As a step towards addressing some of these issues, we have explored the relationship between ER expression and p53 expression detected by immunohistochemistry on prognosis in the context of large prospective randomized clinical trials. In the present study we have examined available pathological material from International Breast Cancer Study Group (IBCSG) Trials VIII and IX, comparing endocrine adjuvant therapy alone versus combined modality chemo-endocrine therapy in patients with node-negative early breast cancer. This revealed a qualitative interaction between ER and p53 expression, such that p53 expression was prognostically adverse in patients whose tumors expressed ER, but favourable in those whose tumors lacked ER expression.
Materials and methods
Trials and patients
The designs of IBCSG Trials VIII and IX have been described in detail elsewhere [36, 37]. Briefly, from 1990 to 1999 in Trial VIII, 1,063 pre- and peri-menopausal women with node-negative early breast cancer were randomly assigned to endocrine therapy with 24 months of goserelin alone, six cycles of chemotherapy with classical cyclophosphamide, methotrexate and 5-fluorouracil (CMF) or a sequence of 6 cycles of CMF followed by 18 months of goserelin. Similarly, from 1988 to 1999 in Trial IX, 1,669 eligible postmenopausal women were randomly assigned endocrine therapy with 5 years of tamoxifen 20 mg daily or three cycles of CMF followed by tamoxifen to complete 5 years therapy. In each trial, randomization was stratified according to locally-determined ER status. Institutional review boards reviewed and approved the protocols, and informed consent was required according to the criteria established within the individual countries. Patient follow-up, vital status and date of any relapse or recurrence are recorded in the IBCSG database. Median follow-up from randomization in Trial VIII is 12 years [36] and in Trial IX is 13 years [37]. IBCSG Trials VIII and IX were conducted from 1988 to 1999. Ethical approval was obtained in participating countries according to national regulations. The IBCSG Independent Data Monitoring Committee reviewed the trials periodically. All patients included in the analysis provided consent to participate in the trials. The study was reviewed and approved by the IBCSG Biological Protocols Working Group.
Pre-existing pathological data
Central review of immunohistochemical expression of ER in whole sections has been documented [38, 39]. The data presented in these published reports is incorporated in a cumulative IBCSG database, which has been used to classify ER status in the present study. Similarly, the central review assessment of tumor size, Bloom and Richardson grade [39], and peri-tumoral vascular invasion, as previously described [40], were used for the present study. HER2 was considered as positive if 3+ by IHC at central assessment or amplified by fluorescent in situ hybridisation (FISH) performed on the tissue microarrays (TMAs) (HER2: C17 ratio > 2.0). There is debate about the threshold at which tumors should be considered positive for ER [41, 42]. Following IBCSG practice [39] and the recommendations of the American Society of Clinical Oncology and the College of American Pathologists [43], and before any data analysis, we defined a cut-point to identify ER as present if at least 1% of cells showed ER staining on immunohistochemistry.
Tissue microarrays
Available tissue blocks from 1,493 patients randomized to IBCSG Trials VIII (593 patients) and IX (900 patients) were sent from the IBCSG Pathology Office to the Cancer Research Program, Garvan Institute of Medical Research, Sydney, Australia for construction of tissue TMAs. The TMAs were produced using the MTA-1 Manual Tissue Arrayer and a 1.0 mm needle to biopsy tumor tissue identified by examination of hematoxylin and eosin (H&E)-stained slides from a standard histological block. Three representative cores were taken from each donor block and deposited in the recipient array block. Each array in Trial VIII comprised 108 (9 ± 12) cores representing about 32 patients. Arrays from Trial IX comprised 96 cores (8 ± 12) representing about 28 patients. An asymmetric template was employed for core orientation. Cores of renal tissue (± 6) were randomly placed within each array to act as orientation markers when scoring. Normal breast cores taken from reduction mammoplasties (± 6) were also placed on each array to allow comparison between immunohistochemical staining in morphological normal breast and invasive breast carcinoma. Details of numbers of patients randomized and those analyzed in the present study are given in the REMARK Diagram (Figure 1) and in Table 1. Briefly, after exclusion of ineligible patients and those from non-compliant institutions, TMAs were prepared from 1,220 patients (Trial VIII, 450; Trial IX, 770). Staining for p53 as described below was successfully performed on TMAs from 1,113 patients (Trial VIII, 417; Trial IX, 696). Comparison of patients assessed for p53 and those not so assessed is presented in Table 1. Centrally reviewed ER was unavailable for 21 patients (Trial VIII, 11; Trial IX, 10) leaving a study cohort for the present analysis of 1,092 patients (Trial VIII, 406; Trial IX, 686).

Remark diagram showing patients randomized to the parent clinical trials and those included in the present cohort for analysis of p53 expression. CMF, cyclophosphamide, methotrexate, 5-fluorouracil; FISH, fluorescent in situ hybridisation; HER, human epidermal growth factor receptor 2.
TMA staining
Four-μm sections were baked in an oven at 63°C for 2 hours followed by rehydration in graded ethanols, followed by water. Antigen retrieval was performed by boiling in a water bath for 30 minutes at pH 9.0 using Dako antigen retrieval solution (S2367, Dako, Denmark). All additional steps were performed on a Dako autostainer: p53 monoclonal antibody (clone DO-7 Dako, Denmark) was incubated at 1:200 dilution for 30 minutes at room temperature, following standard blocking procedures with 3% hydrogen peroxide for 5 minutes. Detection involved Envision labeled polymer-horseradish peroxidise (HRP) anti-mouse antibody (Dako, Denmark) which was added for 30 minutes at room temperature, followed by diaminobenzidine (DAB)+ chromogen (Dako, Denmark) for 10 minutes. Slides were then counterstained with haematoxylin, dehydrated and mounted. Isotype-matched non-specific immunoglobulin was substituted for the primary antibody as the negative control. A p53 positive breast carcinoma was used as the positive control.
P53 scoring
All scoring was performed by an experienced breast pathologist blinded to all clinical and outcome information. Maximum nuclear staining > 10% (any intensity) was considered positive, as employed in most studies using this antigen. Data from the scoring assessment of p53 on the TMAs were entered into, the data handling program (Cansto) at the Garvan Institute of Medical Research, Sydney.
Statistical analysis
Data on baseline characteristics and follow-up were extracted from the IBCSG clinical and pathological database and merged with the p53 data obtained from TMAs. The trial endpoint was disease-free survival (DFS), defined as the length of time from the date of randomization to any invasive breast cancer relapse (including ipsilateral or contralateral breast recurrence), the appearance of a second non-breast malignancy, or death, whichever occurred first, or was censored at the date of last follow-up. Overall survival (OS) was defined as the length of time from randomization to death from any cause, or was censored when last known alive. OS and DFS were estimated using the Kaplan-Meier method and compared using the logrank test. The association of covariates and of interactions between variables with OS and DFS was assessed using proportional hazard models. Since similar effects and interactions were observed separately for each trial (data not shown) the definitive analyses included all available patients. Multivariate DFS and OS analyses were stratified by trial, using SAS software version 9.2. Proportional hazards assumptions were checked using martingale residuals. All P-values are two-sided.
Results
Characteristics of analyzed patients compared to other trial participants
Blocks available for this study were those with residual tumor after routine pathology, central review and the conducting of previous translational research studies using whole tumor sections [39, 44, 45]. Thus the 1,113 patients with p53 results available for this project were more likely to be those with more advanced tumors than those not included. This is reflected in the significantly higher proportion in larger primary tumor size categories and with higher Bloom and Richardson grade in patients available from both trials. There was no significant difference in age distribution or the presence of ER and the trends to worse DFS and OS were not statistically significant (Table 1).
Impact of p53 status and ER, and their interaction on outcome
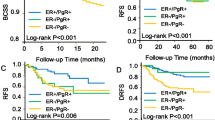
Data on p53 status were available in 1,113 patients and for centrally reviewed ER status in 1,092 of these. P53 positivity was more common among patients with absent ER expression (125/212, 59%) than among those expressing ER (171/880, 19%, P < 0.0001). In univariate analyses the overall slight negative impact of p53 expression on DFS was not significant (Figure 2A). In the presence of ER expression, the adverse effect of p53 staining was more marked while in the absence of ER, p53 expression was associated with better DFS (Figure 2B and 3).

Disease-free survival by p53 expression (≥ 10%) at 12-year median follow-up. (A) All analysed patients. (B) Patients whose tumors expressed estrogen receptor (ER). (C) Patients whose tumors did not express ER.

Forest plots of hazard ratios for disease-free and overall survival by p53, estrogen receptor (ER) expression and intrinsic subtype.
A statistically significant interaction was present between p53 status and ER (P = 0.004), as shown in Table 2. The statistical significance of the interaction persisted for both DFS and OS in further models allowing for significant pathological variables, and for treatment allocation and its interaction with ER status (Table 2). We found no evidence of interaction between p53 expression and the relative efficacy of the various endocrine or chemo-endocrine therapies used in the trials (data not shown), indicating that in our studies p53 expression was not a predictive marker for the efficacy of adding CMF chemotherapy to endocrine adjuvant therapy. Martingale residuals reflected no violations of proportionality apart from the ER variable. Tumors lacking ER are known to exhibit higher initial failure rates [46].
The classification of ER presence (if at least 1% of tumor cells showed ER staining on IHC) was defined prior to any data analysis. Exploration of an alternative ER-positive cut-point based on staining of at least 10% of cells [41], resulted in reclassification of 45 patients with ER staining levels of 1 to 9%, and this materially reduced the clarity of the interaction with p53 status (DFS interaction hazard ratio (HR) 1.76, P = 0.04, OS interaction HR 1.68, P = 0.11).
Reflecting the effect of ER expression, a similar dichotomy of impact of p53 expression was seen when patients were divided into luminal and non-luminal subtypes based on IHC expression of ER, progesterone receptor and Ki67, as well as the detection of HER2 by IHC (3+) or FISH [47] (Figure 3).
Discussion
The present study is unusual in demonstrating a significant qualitative interaction between two biological markers. Detectable p53 expression was associated with better prognosis in patients whose tumors did not express ER but with worse prognosis if ER was expressed. This interaction appeared robust in the two trials examined and was independent of other pathologic features and of treatment. However, this and the similar interaction between p53 status and intrinsic subtype (as assessed by IHC) were unexpected and therefore require confirmation in large independent data sets for which p53 status is known to be available, such as that recently described by Lara et al. for the Cancer and Leukemia Group B [7], the EORTC 10994/BIG 1-00 trial described by Bonnefoi et al. [9], or the BIG 2-98 trial described by Francis et al. [48].
If confirmed, such an interaction might at least partially explain the earlier disparate reports of the prognostic significance of p53 staining viewed in isolation. Based on our exploratory analysis of a higher ER cut-point we suggest that any confirmatory study should look primarily at ER-present (IHC > 1% staining) versus ER-absent staining. Our observation that the interaction is most clearly seen with an ER cut-point reflecting ER expression versus absence of expression may explain the failure to observe such an interaction in earlier small studies which typically used a higher ER cut-point [15, 16, 35].
If the interaction between ER and p53 as markers of prognosis is biologically real, its basis is currently unclear, although there is evidence for functional relationships between p53 and ER that affect mammary oncogenesis and/or response to tamoxifen. In genetically engineered mice, p53 heterozygosity leads to increased mammary epithelial proliferation, decreased apoptosis and eventual development of pre-neoplastic mammary lesions [49]. These responses are all enhanced in the presence of deregulated ER expression [49]. A direct interaction between p53 and ER has been described to inhibit p53-responsive transcriptional activation in mammospheres, but this response is antagonised by tamoxifen, suggesting a mechanism that could contribute to a better response to tamoxifen in women with wild-type p53 [50]. Our observation that p53 positivity is associated with a worse prognosis in women treated with tamoxifen in the context of a randomised clinical trial is consistent with this idea.
TP53 mutations are not only more common among breast cancers not expressing ER, and among the basal-like and HER2 molecular subtypes, which typically lack ER expression [18] but also tend to be different in type [12]. Mutations are non-randomly distributed along the p53 domains [5, 51]. Different p53 mutations have been described as carrying differing adverse prognostic significance [5, 8, 17]. It is possible that p53 staining by IHC seen in tumors not expressing ER reflects mutations that are not disabling, or are less disabling, to cell homeostasis than those responsible for p53 IHC staining in tumors expressing ER.
As well as the well-studied canonical p53 protein, normal breast tissue expresses the p53β and p53γ isoforms, which arise from alternative splicing and differ at the carboxy-terminus [3]. Most TP53 mutations will result in changes in all three isoforms, and there are as yet no reagents for IHC that distinguish between them. However, different patterns of isoform expression are apparent in breast cancer: p53β mRNA expression is associated with ER expression, while p53γ mRNA expression is associated with TP53 mutation [52]. Each has been detected by PCR in 36 to 37% of breast cancers, but only 19% were found to express both [52]. Since the antibody used in this study recognises all three isoforms and the p53β protein isoform is more stable than the other isoforms [53], it is possible that p53 IHC positivity represents a different spectrum of isoforms in cancers expressing, or not expressing ER. The isoforms are differently regulated and may have different functions [3]. In particular, the physical interaction between ER and p53 occurs via the carboxy-terminal domain, which differs in sequence between the different p53 isoforms [29]. Although patients expressing mutant p53 but not p53γ have been shown to have a particularly poor prognosis, those expressing mutant p53 and p53γ have a good prognosis, indistinguishable from patients expressing wild-type p53 [52]. This complexity in the prognostic impact of TP53 mutations may contribute to the interaction with ER observed here.
Future studies should attempt to clarify this relationship by use of material from patients with p53 staining to ascertain the nature of the p53 mutations involved and examine their prognostic significance. Meanwhile it seems prudent to encourage independent validation of the current data in other large randomized clinical trials and to interpret the prognostic significance of IHC detection of p53 in the context of ER expression.
Conclusions
IHC detection of p53 protein in early breast cancer loosely reflects the presence of p53 mutations but its prognostic significance is inconsistently reported. In patients with node-negative breast cancer who participated in International Breast Cancer Study Group Trials VIII and IX we show that the relationship between p53 expression and DFS and OS was dependent on ER status. Among patients whose tumors expressed ER, p53 expression was associated with inferior DFS and OS, whereas p53 expression was associated with better DFS and OS in patients whose tumors did not express ER. The interaction was statistically significant, and remained so in models including other pathological variables. Similarly, p53 was associated with worse prognosis among patients with luminal tumor subtypes but better prognosis among those with triple-negative or HER2-positive subtypes. Interpretation of the prognostic significance of p53 staining requires consideration of ER status.
Abbreviations
- CALGB:
-
Cancer and Leukemia Group B
- CMF:
-
cyclophosphamide, methotrexate, 5-fluorouracil
- DAB:
-
diaminobenzidine
- DFS:
-
disease-free survival
- EORTC:
-
European Organisation for the Research and Treatment of Cancer
- ER:
-
estrogen receptor
- FISH:
-
fluorescent in situ hybridisation
- H&E:
-
haematoxylin and eosin
- HER2:
-
human epidermal growth factor receptor 2
- HRP:
-
horseradish peroxidise
- IBCSG:
-
International Breast Cancer Study Group
- IHC:
-
immunohistochemistry
- OS:
-
overall survival
- PCR:
-
polymerase chain reaction
- TMA:
-
tissue microarray.
References
Brosh R, Rotter V: When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009, 9: 701-713.
Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M: TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007, 26: 2157-2165. 10.1038/sj.onc.1210302.
Bourdon JC, Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, Saville MK, Lane DP: p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19: 2122-2137. 10.1101/gad.1339905.
Harris L, Fritsche H, Mennel R, Norton L, Ravdin P, Taube S, Somerfield MR, Hayes DF, Bast RC: American Society of Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J Clin Oncol. 2007, 25: 5287-5312. 10.1200/JCO.2007.14.2364.
Borresen-Dale AL: TP53 and breast cancer. Hum Mutat. 2003, 21: 292-300. 10.1002/humu.10174.
Varna M, Bousquet G, Plassa LF, Bertheau P, Janin A: TP53 status and response to treatment in breast cancers. J Biomed Biotechnol. 2011, 2011: 284584-
Lara JF, Thor AD, Dressler LG, Broadwater G, Bleiweiss IJ, Edgerton S, Cowan D, Goldstein LJ, Martino S, Ingle JN, Henderson IC, Norton L, Winer EP, Hudis CA, Ellis MJ, Berry DA, Hayes DF: p53 Expression in node-positive breast cancer patients: results from the Cancer and Leukemia Group B 9344 Trial (159905). Clin Cancer Res. 2011, 17: 5170-5178. 10.1158/1078-0432.CCR-11-0484.
Fernandez-Cuesta L, Oakman C, Falagan-Lotsch P, Smoth KS, Quinaux E, Buyse M, Dolci MS, De Azambuja E, Hainaut P, Dell'orto P, Larsimont D, Francis PA, Crown J, Piccart-Gebhart M, Viale G, Di Leo A, Olivier M: Prognostic and predictive value of TP53 mutations in node-positive breast cancer patients treated with anthracycline- or anthracycline/taxane based adjuvant therapy: results from the BIG 02-98 phase III trial. Breast Cancer Res. 2012, 14: R70-10.1186/bcr3179.
Bonnefoi H, Piccart M, Bogaerts J, Mauriac L, Fumoleau P, Brain E, Petit T, Rouanet P, Jassem J, Blot E, Zaman K, Cufer T, Lortholary A, Lidbrink E, Andre S, Litiere S, Lago LD, Becette V, Cameron DA, Bergh J, Iggo R: TP53 status for prediction of sensitivity to taxane versus non-taxane neoadjuvant chemotherapy in breast cancer (EORTC 10994/BIG 1-00): a randomised phase 3 trial. Lancet Oncol. 2011, 12: 527-539. 10.1016/S1470-2045(11)70094-8.
Flaman JM, Frebourg T, Moreau V, Charbonnier F, Martin C, Chappuis P, Sappino AP, Limacher IM, Bron L, Benhattar J: A simple p53 functional assay for screening cell lines, blood, and tumors. Proc Natl Acad Sci USA. 1995, 92: 3963-3967. 10.1073/pnas.92.9.3963.
Waridel F, Estreicher A, Bron L, Flaman JM, Fontolliet C, Monnier P, Frebourg T, Iggo R: Field cancerisation and polyclonal p53 mutation in the upper aero-digestive tract. Oncogene. 1997, 14: 163-169. 10.1038/sj.onc.1200812.
Coutant C, Rouzier R, Qi Y, Lehmann-Che J, Bianchini G, Iwamoto T, Hortobagyi GN, Symmans F, Uzan S, Andre F, de TH, Pusztai L: Distinct p53 gene signatures are needed to predict prognosis and response to chemotherapy in ER-positive and ER-negative breast cancers. Clin Cancer Res. 2011, 17: 2591-1601. 10.1158/1078-0432.CCR-10-1045.
Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, Pawitan Y, Hall P, Klaar S, Liu ET, Bergh J: An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci USA. 2005, 102: 13550-13555. 10.1073/pnas.0506230102.
Troester MA, Herschkowitz JI, Oh DS, He X, Hoadley KA, Barbier CS, Perou CM: Gene expression patterns associated with p53 status in breast cancer. BMC Cancer. 2006, 6: 276-10.1186/1471-2407-6-276.
Tsutsui S, Ohno S, Murakam S, Hachitanda Y, Oda S: Prognostic value of p53 protein expression in breast cancer; an immunohistochemical analysis of frozen sections in 514 Japanese women. Breast Cancer. 2001, 8: 194-201. 10.1007/BF02967508.
Caleffi M, Teague MW, Jensen RA, Vnencak-Jones CL, Dupont WD, Parl FF: p53 gene mutations and steroid receptor status in breast cancer. Clinicopathologic correlations and prognostic assessment. Cancer. 1994, 73: 2147-2156. 10.1002/1097-0142(19940415)73:8<2147::AID-CNCR2820730820>3.0.CO;2-5.
Olivier M, Langerod A, Carrieri P, Bergh J, Klaar S, Eyfjord J, Theillet C, Rodriguez C, Lidereau R, Bieche I, Varley J, Bignon Y, Uhrhammer N, Winqvist R, Jukkola-Vuorinen A, Niederacher D, Kato S, Ishioka C, Hainaut P, Borresen-Dale AL: The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clin Cancer Res. 2006, 12: 1157-1167. 10.1158/1078-0432.CCR-05-1029.
Langerod A, Zhao H, Borgan O, Nesland JM, Bukholm IR, Ikdahl T, Karesen R, Borresen-Dale AL, Jeffrey SS: TP53 mutation status and gene expression profiles are powerful prognostic markers of breast cancer. Breast Cancer Res. 2007, 9: R30-10.1186/bcr1675.
Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Eystein Lonning P, Borresen-Dale AL: Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001, 98: 10869-10874. 10.1073/pnas.191367098.
Mizuno H, Spike BT, Wahl GM, Levine AJ: Inactivation of p53 in breast cancers correlates with stem cell transcriptional signatures. Proc Natl Acad Sci USA. 2010, 107: 22745-22750. 10.1073/pnas.1017001108.
Allred DC, Clark GM, Elledge R, Fuqua SA, Brown RW, Chamness GC, Osborne CK, McGuire WL: Association of p53 protein expression with tumor cell proliferation rate and clinical outcome in node-negative breast cancer. J Natl Cancer Inst. 1993, 85: 200-206. 10.1093/jnci/85.3.200.
Hurd C, Khattree N, Alban P, Nag K, Jhanwar SC, Dinda S, Moudgil VK: Hormonal regulation of the p53 tumor suppressor protein in T47D human breast carcinoma cell line. J Biol Chem. 1995, 270: 28507-28510. 10.1074/jbc.270.48.28507.
Molinari AM, Bontempo P, Schiavone EM, Tortora V, Verdicchio MA, Napolitano M, Nola E, Moncharmont B, Medici N, Nigro V, Armetta I, Abbondanza C, Puca GA: Estradiol induces functional inactivation of p53 by intracellular redistribution. Cancer Res. 2000, 60: 2594-2597.
Shirley SH, Rundhaug JE, Tian J, Cullinan-Ammann N, Lambertz I, Conti CJ, Fuchs-Young R: Transcriptional regulation of estrogen receptor-alpha by p53 in human breast cancer cells. Cancer Res. 2009, 69: 3405-3414. 10.1158/0008-5472.CAN-08-3628.
Medina D, Kittrell FS: p53 function is required for hormone-mediated protection of mouse mammary tumorigenesis. Cancer Res. 2003, 63: 6140-6143.
Sivaraman L, Conneely OM, Medina D, O'Malley BW: p53 is a potential mediator of pregnancy and hormone-induced resistance to mammary carcinogenesis. Proc Natl Acad Sci USA. 2001, 98: 12379-12384. 10.1073/pnas.221459098.
Yu CL, Driggers P, Barrera-Hernandez G, Nunez SB, Segars JH, Cheng S: The tumor suppressor p53 is a negative regulator of estrogen receptor signaling pathways. Biochem Biophys Res Commun. 1997, 239: 617-620. 10.1006/bbrc.1997.7522.
Liu G, Schwartz JA, Brooks SC: Estrogen receptor protects p53 from deactivation by human double minute-2. Cancer Res. 2000, 60: 1810-1814.
Liu W, Konduri SD, Bansal S, Nayak BK, Rajasekaran SA, Karuppayil SM, Rajasekaran AK, Das GM: Estrogen receptor-alpha binds p53 tumor suppressor protein directly and represses its function. J Biol Chem. 2006, 281: 9837-9840. 10.1074/jbc.C600001200.
Menendez D, Inga A, Resnick MA: Estrogen receptor acting in cis enhances WT and mutant p53 transactivation at canonical and noncanonical p53 target sequences. Proc Natl Acad Sci USA. 107: 1500-1505.
McGuire WL: Estrogen receptors in human breast cancer. J Clin Invest. 1973, 52: 73-77. 10.1172/JCI107175.
Reiner A, Neumeister B, Spona J, Reiner G, Schemper M, Jakesz R: Immunocytochemical localization of estrogen and progesterone receptor and prognosis in human primary breast cancer. Cancer Res. 1990, 50: 7057-7061.
Harvey JM, Clark GM, Osborne CK, Allred DC: Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J Clin Oncol. 1999, 17: 1474-1481.
Biganzoli E, Boracchi P, Coradini D, Grazia Daidone M, Marubini E: Prognosis in node-negative primary breast cancer: a neural network analysis of risk profiles using routinely assessed factors. Ann Oncol. 2003, 14: 1484-1493. 10.1093/annonc/mdg422.
Takahashi M, Tonoki H, Tada M, Kashiwazaki H, Furuuchi K, Hamada J, Fujioka Y, Sato Y, Takahashi H, Todo S, Sakuragi N, Moriuchi T: Distinct prognostic values of p53 mutations and loss of estrogen receptor and their cumulative effect in primary breast cancers. Int J Cancer. 2000, 89: 92-99. 10.1002/(SICI)1097-0215(20000120)89:1<92::AID-IJC15>3.0.CO;2-8.
Karlsson P, Sun Z, Braun D, Price KN, Castiglione-Gertsch M, Rabaglio M, Gelber RD, Crivellari D, Collins J, Murray E, Zaman K, Colleoni M, Gusterson BA, Viale G, Regan MM, Coates AS, Goldhirsch A: Long-term results of International Breast Cancer Study Group Trial VIII: adjuvant chemotherapy plus goserelin compared with either therapy alone for premenopausal patients with node-negative breast cancer. Ann Oncol. 2011, 22: 2216-2226. 10.1093/annonc/mdq735.
Aebi S, Sun Z, Braun D, Price KN, Castiglione-Gertsch M, Rabaglio M, Gelber RD, Crivellari D, Lindtner J, Snyder R, Karlsson P, Simoncini E, Gusterson BA, Viale G, Regan MM, Coates AS, Goldhirsch A: Differential efficacy of three cycles of CMF followed by tamoxifen in patients with ER-positive and ER-negative tumors: long-term follow up on IBCSG Trial IX. Ann Oncol. 2011, 22: 1981-1987. 10.1093/annonc/mdq754.
Regan MM, Viale G, Mastropasqua MG, Maiorano E, Golouh R, Carbone A, Brown B, Suurkula M, Langman G, Mazzucchelli L, Braye S, Grigolato P, Gelber RD, Castiglione-Gertsch M, Price KN, Coates AS, Goldhirsch A, Gusterson B, For the International Breast Cancer Study G: Re-evaluating Adjuvant Breast Cancer Trials: Assessing Hormone Receptor Status by Immunohistochemical Versus Extraction Assays. J Natl Cancer Inst Cancer Spectrum. 2006, 98: 1571-1581.
Viale G, Regan MM, Maiorano E, Mastropasqua MG, Golouh R, Perin T, Brown RW, Kovacs A, Pillay K, Ohlschlegel C, Braye S, Grigolato P, Rusca T, Gelber RD, Castiglione-Gertsch M, Price KN, Goldhirsch A, Gusterson BA, Coates AS: Chemoendocrine compared with endocrine adjuvant therapies for node-negative breast cancer: predictive value of centrally reviewed expression of estrogen and progesterone receptors-International Breast Cancer Study Group. J Clin Oncol. 2008, 26: 1404-1410. 10.1200/JCO.2007.10.6393.
Viale G, Giobbie-Hurder A, Gusterson B, Maiorano E, Mastropasqua MG, Sonzogni A, Colleoni M, Castiglione-Gertsch M, Price KN, Gelber RD, Goldhirsch A, Coates AS, International Breast Cancer Study G: Adverse prognostic value of vessel invasion: is it abrogated by adequate endocrine adjuvant therapy? Results from two International Breast Cancer Study Group randomized trials of chemoendocrine adjuvant therapy for early breast cancer. Ann Oncol. 2009, 21: 245-254.
Iwamoto T, Booser D, Valero V, Murray JL, Koenig K, Esteva FJ, Ueno NT, Zhang J, Shi W, Qi Y, Matsuoka J, Yang EJ, Hortobagyi GN, Hatzis C, Symmans WF, Pusztai L: Estrogen receptor (ER) mRNA and ER-related gene expression in breast cancers that are 1% to 10% ER-positive by immunohistochemistry. J Clin Oncol. 2012, 30: 729-734. 10.1200/JCO.2011.36.2574.
Harbeck N, Rody A: Lost in translation? Estrogen receptor status and endocrine responsiveness in breast cancer. J ClinOncol. 2012, 30: 686-689. 10.1200/JCO.2011.38.9619.
Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, Fitzgibbons PL, Francis G, Goldstein NS, Hayes M, Hicks DG, Lester S, Love R, Mangu PB, McShane L, Miller K, Osborne CK, Paik S, Perlmutter J, Rhodes A, Sasano H, Schwartz JN, Sweep FCG, Taube S, Torlakovic EE, Valenstein P, Viale G, Visscher D, Wheeler T, Williams RB, et al: American Society of Clinical Oncology/College of American Pathologists Guideline Recommendations for Immunohistochemical Testing of Estrogen and Progesterone Receptors in Breast Cancer. J Clin Oncol. 2010, 28: 2784-2795. 10.1200/JCO.2009.25.6529.
Ravaioli A, Monti F, Regan MM, Maffini F, Mastropasqua MG, Spataro V, Castiglione-Gertsch M, Panzini I, Gianni L, Goldhirsch A, Coates A, Price KN, Gusterson BA, Viale G: p27 and Skp2 immunoreactivity and its clinical significance with endocrine and chemo-endocrine treatments in node-negative early breast cancer. Ann Oncol. 2008, 19: 660-668.
Viale G, Regan MM, Mastropasqua MG, Maffini F, Maiorano E, Colleoni M, Price KN, Golouh R, Perin T, Brown RW, Kovacs A, Pillay K, Ohlschlegel C, Gusterson BA, Castiglione-Gertsch M, Gelber RD, Goldhirsch A, Coates AS, On the behalf of the International Breast Cancer Study G: Predictive Value of Tumor Ki-67 Expression in Two Randomized Trials of Adjuvant Chemoendocrine Therapy for Node-Negative Breast Cancer. J Natl Cancer Inst. 2008, 100: 207-212. 10.1093/jnci/djm289.
Saphner T, Tormey DC, Gray R: Annual hazard rates of recurrence for breast cancer after primary therapy. J Clin Oncol. 1996, 14: 2738-2746.
Cheang MCU, Chia SK, Voduc D, Gao D, Leung S, Snider J, Watson M, Davies S, Bernard PS, Parker JS, Perou CM, Ellis MJ, Nielsen TO: Ki67 Index, HER2 Status, and Prognosis of Patients With Luminal B Breast Cancer. J Natl Cancer Inst. 2009, 101: 736-750. 10.1093/jnci/djp082.
Francis P, Crown J, Di Leo A, Buyse M, Balil A, Andersson M, Nordenskjold B, Lang I, Jakesz R, Vorobiof D, Gutierrez J, Van Hazel G, Dolci S, Jamin S, Bendahmane B, Gelber RD, Goldhirsch A, Castiglione-Gertsch M, Piccart-Gebhart M, On behalf of the BIG: Adjuvant Chemotherapy With Sequential or Concurrent Anthracycline and Docetaxel: Breast International Group 02 98 Randomized Trial. J Natl Cancer Inst. 2008, 100: 121-133. 10.1093/jnci/djm287.
Diaz-Cruz ES, Furth PA: Deregulated estrogen receptor alpha and p53 heterozygosity collaborate in the development of mammary hyperplasia. Cancer Res. 2010, 70: 3965-3974. 10.1158/0008-5472.CAN-09-3450.
Konduri SD, Medisetty R, Liu W, Kaipparettu BA, Srivastava P, Brauch H, Fritz P, Swetzig WM, Gardner AE, Khan SA, Das GM: Mechanisms of estrogen receptor antagonism toward p53 and its implications in breast cancer therapeutic response and stem cell regulation. Proc Natl Acad Sci USA. 2010, 107: 15081-15086. 10.1073/pnas.1009575107.
Bai L, Wei-Guo Z: p53: structure, function and therapeutic applications. Journal of Cancer Molecules. 2006, 2: 141-153.
Bourdon JC, Khoury MP, Diot A, Baker L, Fernandes K, Aoubala M, Quinlan P, Purdie CA, Jordan LB, Prats AC, Lane DP, Thompson AM: p53 mutant breast cancer patients expressing p53gamma have as good a prognosis as wild-type p53 breast cancer patients. Breast Cancer Res. 2011, 13: R7-10.1186/bcr2811.
Camus S, Menendez S, Fernandes K, Kua N, Liu G, Xirodimas DP, Lane DP, Bourdon JC: The p53 isoforms are differentially modified by Mdm2. Cell Cycle. 2012, 11: 1646-1655.
Acknowledgements
We record with sorrow the recent death of Professor Rob Sutherland who, as Inaugural Director of The Kinghorn Cancer Centre and Director of the Cancer Research Program, Garvan Institute of Medical Research, was the driving force behind this collaboration with the International Breast Cancer Study Group.
The authors would like to thank Ms Joanne Scorer for assistance in preparing the manuscript. We thank the patients who participated in the clinical trials, and the contributions of the physicians, nurses, data managers, and pathologists of the International Breast Cancer Study Group.
This work was supported by the National Health and Medical Research Council (Program Grant 535903 EA Musgrove, RL Sutherland; Fellowship 427601 RL Sutherland), the Cancer Institute New South Wales (Translational Program Grant 10/TPG/1-04 RL Sutherland, EA Musgrove, EKA Millar, SA O'Toole; Sydney Catalyst Translational Research Centre 11/TRC/1-02 RL Sutherland; Fellowships 10/CRF/1-07 SA O'Toole, 11/CDF/3-26 EA Musgrove, 11/CDF/3-25 TJ Molloy), the Australia and New Zealand Breast Cancer Trials Group (RL Sutherland, SA O'Toole), the Australian Cancer Research Foundation (RL Sutherland, EA Musgrove), the Sydney Breast Cancer Foundation (SA O'Toole), the RT Hall Trust (RL Sutherland) and the Petre Foundation (RL Sutherland). Additional support for IBCSG Trials VIII and IX was provided in part by the Swedish Society for Cancer Research; the Frontier Science and Technology Research Foundation; the Swiss Group for Clinical Cancer Research (SAKK); the Swiss Cancer League; and the United States National Institutes of Health (CA-75362).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
ASC, EKAM, SO'T, EAM and RLS participated in the concept and design of the study. ASC, GV, AG, MC, RDG and BG provided materials for the study. ASC, EKAM, SO'T, TJM, EAM, RLS, MMR and ZS performed data analysis and interpretation for the study. ASC, EKAM, SO'T, EAM and RLS drafted, revised and edited the manuscript. All authors read and approved the final draft of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Coates, A.S., Millar, E.K., O'Toole, S.A. et al. Prognostic interaction between expression of p53 and estrogen receptor in patients with node-negative breast cancer: results from IBCSG Trials VIII and IX. Breast Cancer Res 14, R143 (2012). https://doi.org/10.1186/bcr3348
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/bcr3348