
Abstract
The epithelial-to-mesenchymal transition (EMT) is a critical developmental process that has recently come to the forefront of cancer biology. In breast carcinomas, acquisition of a mesenchymal-like phenotype that is reminiscent of an EMT, termed oncogenic EMT, is associated with pro-metastatic properties, including increased motility, invasion, anoikis resistance, immunosuppression and cancer stem cell characteristics. This oncogenic EMT is a consequence of cellular plasticity, which allows for interconversion between epithelial and mesenchymal-like states, and is thought to enable tumor cells not only to escape from the primary tumor, but also to colonize a secondary site. Indeed, the plasticity of cancer cells may explain the range of pro-metastatic traits conferred by oncogenic EMT, such as the recently described link between EMT and cancer stem cells and/or therapeutic resistance. Continued research into this relationship will be critical in developing drugs that block mechanisms of breast cancer progression, ultimately improving patient outcomes.
Similar content being viewed by others

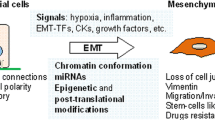
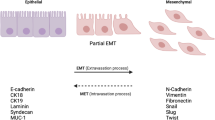
Introduction
Medical advances in the breast cancer field have dramatically altered the overall 5-year survival rate for women in the United States from 63% in the 1960s to 90% as of 2010 [1]. Despite these advances, the 5-year survival rate is a mere 23% for women diagnosed with distant metastatic disease [1]. Accordingly, basic researchers and clinicians have been working to combat breast cancer mortality by unraveling the molecular mechanisms that underlie metastasis, in an effort to improve treatment regimens and ultimately prognostic outcomes.
A recent focus in breast cancer metastasis research is the epithelial-to-mesenchymal transition (EMT). Classical EMT is a critical developmental program that entails the transdifferentiation of epithelial cells to mesenchymal cells, giving rise to different cell types, often in new locales [2]. As tumors progress, a subset of epithelial cancer cells may attain attributes of mesenchymal cells, a process that is broadly referred to as an oncogenic EMT. Amongst other things, an oncogenic EMT can result in increased migratory and invasive capabilities that may in turn contribute to metastatic dissemination. Oncogenic EMTs are not equivalent to developmental EMTs, as mesenchyme, by definition, is embryonic in origin. Instead, oncogenic EMTs should be viewed more as a partial EMT, in which carcinoma cells gain characteristics of mesenchymal cells, but may not fully lose epithelial characteristics (see 'Type III epithelial-to-mesenchymal transition' section for further discussion). This intermediate phenotype represents a plastic state, and it is speculated that plastic cells that have undergone an EMT to escape from a primary tumor must subsequently undergo the reverse mesenchymal-to-epithelial transition (MET) prior to colonizing a secondary site [3]. Such plasticity may also allow for cellular alterations that facilitate newly uncovered and important functional characteristics that have been linked to EMT, such as increased tumor-initiation and self-renewal capacity [4, 5] and increased resistance to conventional therapies [6, 7]. Thus, the role of epithelial plasticity will be an underlying theme throughout this review.
While the debate regarding the exact role of EMT in human breast cancers continues [8], such debate should not distract from the fact that the study of oncogenic EMT has led to significant findings that have widespread implications in the treatment of breast cancer patients. This review highlights such important findings.
Epithelial-to-mesenchymal transition
EMT occurs in a number of contexts with characteristic differences, and while three different subtypes have been classified (types I, II and III), there are large areas of overlap [9, 10]. In general, EMT programming allows epithelial cells to become invasive and motile mesenchymal or mesenchymal-like cells that are no longer spatially restricted by extracellular matrix [9]. This programming occurs in part through loss of apical-basal polarity and tight cell-cell contacts, with a concomitant gain in front-back end polarity and focal cell-cell contacts. In addition, the process of EMT leads to the formation of filopodia, accompanied by a switch from integrin receptors that mediate cell-cell adhesion to cell-extracellular matrix adhesion-specific integrins that are critical for cell motility [11, 12]. The epithelial cytokeratin-based intermediate filament network is replaced with vimentin (VIM) along with actin (ACTA1) stress fiber formation, yielding a more spindle-like shape in vitro [11]. An increase in the mesenchymal N-cadherin (CDH2) also facilitates focal cell-cell contacts and mobility, while the epithelial E-cadherin (CDH1) functionally dissipates through either down-regulation or relocalization away from the adherens junctions in the membrane [13].
Type I and II epithelial-to-mesenchymal transition
Type I EMT occurs during development and is responsible for the complex tissue types and organization present in metazoans [9]. A classic example of this EMT in humans is the formation of the primitive streak that defines the first embryonic axis and designates where cells will ingress to form new tissue layers during gastrulation [2]. In some instances of type I EMT, mesenchymal cells revert back to an epithelial phenotype in a MET, such as during nephrogenesis, when the metanephric mesenchyme transitions into epithelial nephric tubules, re-establishing epithelial structures at novel sites [11]. Thus, although some type I EMTs are permanent, interconversion between epithelial and mesenchymal phenotypes (that is, epithelial plasticity) is observed during development.
Type II EMTs are those that occur in wound healing and fibrosis [9, 10]. In some instances, fibrosis can arise as a result of inappropriate presence of myofibroblasts at an injured/inflamed site due to an EMT response to persistent injury or inflammation [9]. During wound healing, an EMT causes integrin changes and lamellipodia formation that allow keratinocytes at injured edges to migrate to close a wound [14]. An important aspect of the wound healing response is that only cells of the leading edge appear to undergo an EMT [14]. As the leading cells migrate, they pull a sheet of keratinocytes behind them. The cell-cell contacts required during this co-migration indicate that these cells only undergo a partial EMT, in that an individual cell exhibits spatially restricted epithelial and mesenchymal-like properties simultaneously, demonstrating another instance of epithelial plasticity. Type I and II EMTs are more thoroughly reviewed elsewhere [9, 14, 15].
Type III epithelial-to-mesenchymal transition
Type III EMT, or oncogenic EMT, is the name given to an EMT-like process that is observed in carcinoma cells, and is associated with tumor progression and metastasis [9, 10]. EMT-associated gains in migration and invasion are thought to allow tumor cells to better navigate elements of the metastatic cascade, such as invasion through the basement membrane and intravasation into the circulatory system. Additionally, oncogenic EMT is linked to other pro-metastatic phenotypes, including resistance to chemo- and radiation therapy, self-renewal, evasion of the immune system and anoikis resistance (Figure 1). It should be noted that the scope or completeness of a type III EMT is often less than that of a type I EMT; depending on the cellular and microenvironmental context, different EMT-associated traits may or may not be acquired. Thus, oncogenic EMT could be defined as an EMT-like process in which carcinoma cells gain mesenchymal-like characteristics and/or lose epithelial characteristics; morphological alterations may or may not accompany such changes (see 'Breast cancer EMT mediators' section for further discussion).

Functional consequences of a type III epithelial-to-mesenchymal transition. An oncogenic epithelial-to-mesenchymal transition (EMT) in a breast carcinoma cell gives rise to a tumor cell with mesenchymal-like features. This transition can lead to the acquisition of a number of pro-metastatic features, including increased motility, invasiveness, anoikis resistance and evasion of the immune system. Recently, the acquisition of cancer stem cell-like properties has been added to this list (that is, self-renewal, multipotency, therapeutic resistance), resulting in significant cross-talk between the EMT and cancer stem cell fields.
If carcinoma cells that have undergone an oncogenic EMT retain some epithelial features, while gaining mesenchymal characteristics, does that mean that a complete conversion never occurs? Unfortunately, complete loss of epithelial characteristics from a carcinoma cell would be difficult to detect in human cancers, as these cells would no longer morphologically or molecularly appear epithelial and may be confused with stromal cells. Nonetheless, evidence for an oncogenic EMT does exist in mouse models and in human tumors [16–18]. A recent study used fate mapping to examine MYC-initiated breast tumors in mice, specifically focusing on histologically identified tumor-adjacent stroma and breast tumor epithelium. Using a Cre and Rosa26LoxP system to mark tumor cells, an epithelial promoter-driven Cre marked tumor-adjacent stroma, which also stained positive for epithelial cytokeratins, indicative of late type III EMT where carcinoma cells have transitioned into mesenchymal-like cells [19]. Conversely, tumor epithelia were marked with a fibroblast promoter-driven Cre, suggestive of early stage type III EMT where carcinoma cells are beginning to acquire mesenchymal characteristics [19]. Additional studies further demonstrate the presence of an oncogenic EMT, where a gain of mesenchymal characteristics occurs while epithelial characteristics are in part maintained. For example, Sine oculis homeobox homolog 1 (SIX1) overexpression in mammary epithelial cells of transgenic mice leads to tumors, 21% of which are sarcomatoid in morphology and are negative for CDH1 and positive for α-Smooth muscle actin (ACTA2) [20]. Importantly, these tumors are also cytokeratin18 (KRT18) positive, supporting an epithelial origin [20]. Of the non-sarcomatoid SIX1 tumors, almost 80% appeared morphologically epithelial, but contained regions in which membranous CDH1 is decreased and nuclear β-catenin (CTNNB1) is upregulated, indicative of a cell in the earlier stages of EMT [20]. Indeed, there are now several additional studies demonstrating such an oncogenic EMT within mouse and human breast cancer cell lines and tumors [16–18].
Breast cancer EMT mediators
Many groups have dedicated significant effort towards elucidating causes and effects of EMT in breast cancer, yielding a better, though still incomplete, understanding of the process. Numerous mediators of EMT have been discovered, including transcription factors, signaling molecules and microRNAs (miRNAs). Many downstream markers are used to distinguish between epithelial and mesenchymal-like phenotypes, including loss of epithelial proteins that exist in junctional complexes. A variety of proteins that are down-regulated in response to an EMT include CDH1, plakoglobin (JUP), occludin (OCLN), zonula occludens1 (TJP1), α-catenin (CTNNA3) and claudins 3/4/7 (CLDN-3/4/7) [10]. On the other end of the spectrum, the promotion of a mesenchymal-like phenotype is indicated by the up-regulation of proteins such as fibronectin (FN1), CDH2, VIM, ACTA2 and nuclear CTNNB1 [10]. As noted above, carcinoma cells may not completely lose their epithelial phenotype during an oncogenic EMT and may express epithelial and mesenchymal markers simultaneously [3].
A common theme among oncogenic EMT inducers is their crucial role in type I EMT. It has become increasingly evident that improper activation of developmental EMT inducers in adults gives rise to an out of context EMT-like program that contributes to the progression of breast cancer, as well as other cancers. A few examples of transcription factors and signaling pathways known to play a role in both type I and type III EMT include Twist1 (TWIST1), SIX1, Snail1 (SNAI1) and Ladybird homeobox (LBX1) and the Wnt and transforming growth factor-β (TGF-β) signaling pathways [2]. The relationship between developmental regulators and type III EMT is more thoroughly reviewed in other bodies of work [2, 15, 21].
Transcription factors
The dissolution of adherens junctions is a critical step of EMT, with loss/decrease or relocalization of CDH1 as the most commonly used determinant of the EMT phenotype. Not surprisingly, a number of EMT inducers are direct transcriptional repressors of CDH1. The zinc-finger proteins SNAI1 [22, 23], Snail2 (SNAI2) [24], Zeb1 (ZEB1) [25] and Zeb2 (ZEB2) [26] each directly repress transcription of CDH1 in mammary cells by binding the E-boxes (CANNTG) located in the CDH1 proximal promoter, as do the basic helix-loop-helix factors E12/E47 (TCF3) [27] and TWIST1 [28]. A number of other transcription factors cause relocalization of junctional CDH1, including SIX1 [29], Goosecoid (GSC) [30] and Forkhead box C2 (FOXC2) [31]. Interestingly, knockdown of CDH1 alone is sufficient to induce an EMT [32], highlighting the significance of repressors of CDH1 in the induction of an EMT.
Much research has focused on direct repressors of CDH1, and in addition to laboratory based studies, clinical associations with breast cancer have been demonstrated for many of these repressors. Analysis of breast cancer patients has associated SNAI1 with tumor recurrence, while SNAI2 is associated with tumor recurrence and metastasis [33, 34]. High levels of ZEB1/2 have similarly been found to correlate with poor survival, outcome and grade in numerous cancers, including breast [35, 36]. Surprisingly, however, a recent study by Montserrat and colleagues demonstrates that lower ZEB1 transcript levels correlate with worse overall survival and disease-free survival in breast cancer patients [37]. TWIST1 analyses are also inconsistent between studies: nuclear TWIST1 staining in the epithelial compartment of breast carcinomas is associated with poor survival [35], while TWIST1-negative breast tumors have also been associated with worse overall survival [37]. Unfortunately, because these studies employ different cohorts of women, who may be at different stages of the disease and have been subjected to different therapeutic regimens, it is difficult to draw firm conclusions from these opposing data. Well-controlled studies are needed, including microdissection of the leading edge of tumors where oncogenic EMT is thought to occur, to truly determine the prognostic value of EMT inducers.
Indirect repression of CDH1 is also accomplished by EMT inducers, including SIX1, GSC and FOXC2 [29–31]. SIX1 drives an oncogenic EMT that is dependent on its ability to activate TGF-β signaling and relocalize CDH1 away from the membrane in MCF7 breast cancer cells [29]. GSC can induce a type III EMT likely through activation of SNAI1/2 and TWIST1 [30], while FOXC2 has been shown to relocalize CDH1 away from adherens junctions [31]. Recently, p53 (TP53), Twist2 (TWIST2) and Forkhead box Q1 (FOXQ1) have been added to this list of oncogenic EMT inducers. Both TWIST2 and FOXQ1 decrease activity of the CDH1 promoter, but it is not clear if this interaction is direct [18, 38, 39]. The mechanism of action for TP53 is indirect control over ZEB1/2 protein levels, through direct regulation of the miR-200 and miR-192 families in breast [40] and hepatocellular cancer models [41].
One puzzling aspect of CDH1 expression in breast cancer patients occurs in those diagnosed with lobular cancers, which are largely (55 to 85%) CDH1 negative [42]. These tumors do not appear morphologically mesenchymal-like [8], though detection of VIM occurs on occasion [43]. Interestingly, it has been demonstrated that inactivating CDH1 mutations occur in 56% of all lobular tumors [42], and breast cancer cell lines with CDH1 truncating mutations have a distinct epithelial expression profile when compared to cell lines where the CDH1 promoter is silenced, which exhibit a fibroblastic profile [44]. This suggests CDH1 down-regulation by mutation is largely not associated with EMT. Clearly, our understanding of the role of CDH1 in lobular cancers is still incomplete and requires further analysis.
Major signaling pathways
In addition to transcription factors, several signaling pathways are known to induce an EMT, such as the TGF-β [45], epithelial growth factor (EGF) [46], Wnt [47], Notch [48] and Hedgehog pathways [49]. Not surprisingly, these pathways often activate the aforementioned transcription factors. Examples include TGF-β and EGF signaling, which both lead to activation of SNAI1/2, TWIST1 and ZEB1/2, while TGF-β also up-regulates FOXC2 [45, 46, 50]. In addition, Notch, Hedgehog and Wnt signaling mediate an EMT through activation of SNAI1/2 [47–49]. Some of these transcription factors can in turn activate signaling pathways to promote an EMT [50], such as SIX1, which activates both TGF-β and Wnt signaling [20, 29], demonstrating significant cross-talk between EMT regulators.
EMT signaling pathways can also be enhanced via activation of a ligand released from the tumor microenvironment. For example, matrix metalloproteases and a disintegrin and metalloproteases (ADAMs) can be up-regulated as a result of transformation [21, 51]. Up-regulation of MMPs and ADAMs can then lead to an increase in processing of pro-ligands such as TGF-β1 (TGFB1) and TNF-α [52, 53], ultimately enhancing EMT. Because signaling pathways in EMT have been extensively reviewed, we refer readers to the following reviews for more detailed descriptions of this topic [45, 46, 49].
MicroRNAs
Relatively recently, a class of small non-coding RNAs, termed miRNAs, was discovered. These post-transcriptional inhibitors target mRNAs through sequence specificity, directing cleavage of the mRNA or translational inhibition [54]. As miRNAs play a role in development [55], it is not surprising that they have also been implicated in the induction of EMT. The most frequently cited EMT-related miRNAs are those belonging to the miR-200 family, which consists of miR-200a/b/c, miR-141 and miR-429. Repression of these miRNAs leads to an EMT, at least in part by relieving down-regulation of ZEB1/2 [56, 57]. Interestingly, ZEB1/2 can directly repress transcription of miR-200 family members, completing a double-negative feedback loop [57, 58]. While miR-200c maintains the epithelial phenotype by keeping CDH1 levels high, it also represses FN1 [59], thus repressing the mesenchymal phenotype. On the other hand, miR-9 and miR-495 repress the epithelial arm of EMT by directly targeting CDH1 for degradation, thus promoting a more mesenchymal-like state [60, 61].
EMT-associated signaling pathways can also be influenced by miRNAs. Suppression of miR-448 gives rise to an EMT, both in vitro and in vivo, through indirect up-regulation of amphiregulin (AREG), resulting in increased EGF signaling [62]. In the TGF-β pathway, the downstream co-activator SMAD family member 4 (SMAD4) up-regulates miR-155, which is required for EMT in a non-tumorigenic mammary model in vitro [63]. In contrast, miR-155 prevents EMT in vivo in a breast cancer model [64]. As TGF-β signaling is known to switch from tumor-suppressive to tumor-promotional during cancer progression [45], perhaps the difference in the transformed state of the cells is responsible for the opposite effects observed with miR-155 expression. A similar up-regulation of miR-29a leads to an EMT in murine mammary cells, but only in conjunction with RAS expression [65]. Thus, it appears transformation itself may play a role in mediating the effects of miR-155 and miR-29a on EMT, emphasizing the importance of cellular context.
Epithelial-to-mesenchymal transition networks
The complex process of EMT is historically thought to be controlled by master regulators [11]. While some of the above examples appear deserving of this label, there is a level of complexity in the EMT process that is not fully understood and suggests that multiple molecules act together to mediate EMT, rather than master regulators acting on their own. For example, SNAI2 is necessary for TWIST1-mediated down-regulation of CDH1 and up-regulation of various mesenchymal genes [66], while cooperation between SNAI1 and TWIST1 is needed to achieve maximal up-regulation of ZEB1 [67]. Interestingly, however, ZEB1 levels can eventually increase without the SNAI1-TWIST1 partnership [67]. Investigations into this 'EMT interactome' have revealed that many individual EMT inducers are able to up-regulate other EMT activators, though they are not necessarily dependent on this cross-talk to maintain activity [50, 68]. As multiple feedback loops exist between EMT mediators, these intricate relationships are just beginning to be understood.
Impact of EMT on breast cancer: metastasis, cancer stem cells and therapeutics
EMT and metastasis
Since oncogenic EMT is observed in many breast cancer models, what then, is the relevance of this process to the human disease? It has been argued that EMT is critical for metastasis and, indeed, many EMT regulators are capable of inducing metastasis. Examples include TWIST1 [69], FOXC2 [31], FOXQ1 [38, 39] and SIX1 [29], all of which generate an oncogenic EMT in breast cancer models, induce metastasis to distant organs in these same models and are associated with poor outcomes in breast cancer. Importantly though, patient metastases typically reflect the primary carcinoma histologically, implying that if a carcinoma cell that underwent an oncogenic EMT escaped the primary tumor and was responsible for the colonization of a distant site, a MET must have occurred at some point.
But, has an MET ever been observed in breast cancer models? Indeed, multiple recent studies suggest that MET can occur in breast cancer models. For example, Chao and colleagues [70] demonstrated, using the mesenchymal-like, CDH1-negative MDA-MB-231 breast cancer line and primary human explants, that co-culture with hepatocytes could restore a more epithelial morphology to MDA-MB-231 cells, in part by decreasing CDH1 promoter methylation, resulting in increased levels of CDH1. As micrometastases from MDA-MB-231-initiated primary tumors were found to contain membranous human CDH1 in vivo [70], the possibility of MET or mesenchymal-to-epithelial reverting transition is certainly feasible within cancer [70]. In addition, a study by Asiedu and colleagues [16] used a NEU-driven mouse breast cancer model to induce an EMT with TGFB1 and TNF in vitro, which up-regulated CDH2 and silenced CDH1. After these mesenchymal-like cells were subcutaneously injected into mice to form tumors, CDH1 became re-expressed, but this reversion disappeared after in vitro culture of the cells retrieved from the tumor [16]. These data suggest that MET does occur, and that it is highly dependent on signals from the host microenvironment.
It is inherent to a metastasis model in which an oncogenic EMT is followed by a MET that a high degree of plasticity must be exhibited by the cancer cells. Interestingly, when Dykxhoorn and colleagues examined an isogenic mouse breast cancer cell line series (4T1 series) with varying metastatic potential [71], they found that the highly metastatic 4T1 line displays predominantly epithelial characteristics, though it also expresses the mesenchymal marker VIM [72]. While this seems counter to the argument in support of the role of EMT in metastasis, one interpretation of the data is that the 4T1 cell line exists in a highly plastic state, retaining epithelial characteristics while also expressing mesenchymal ones, which ultimately allows for increased metastatic potential. The plasticity may indicate that given the right contextual signals, this cell line is primed to interchange between states, such as an in vivo oncogenic EMT and MET, thereby affecting early and late stage metastasis, respectively. In an additional cell line in the 4T1 series, exogenous expression of the miR-141-200c cluster in the non-metastatic, mesenchymal-like 4TO7 cell line induced a MET as expected, but also increased tumor-initiation and metastases [72]. To better understand these findings, Korpal and colleagues [73] compared orthotopic against intravenous injections, in the same system, and found that the miR-200 family prevented 4TO7 invasion and intravasation during early state metastasis, while promoting efficient colonization of a secondary site in late stage metastasis. Akin to the 4T1 line, the 4TO7 line may already be primed for EMT, with the miR-200 family enabling 4TO7 cells to undergo the MET portion of the EMT-MET axis to establish metastases. Whether 4T1 and/or 4T07 cells actually interconvert between epithelial and mesenchymal-like states in vivo remains to be determined, and will be important in understanding the generality of EMT as a mediator of the metastatic process.
Recent work by Tsuji and colleagues [74] provides an explanation other than interconversion between oncogenic EMT and MET for how an oncogenic EMT may contribute to metastasis: cooperativity between epithelial and more mesenchymal-like cancer cells. In this study, p12 (CDK2AP1) induction of EMT in hamster HCPC-1 cheek carcinoma cells led to increased in vivo invasion and survival in the circulatory system; however, these cells were not able to colonize the lung. Alternatively, epithelial HCPC-1 cells formed lung metastases when injected intravenously, yet could not invade or access the vasculature when injected subcutaneously [74]. Subcutaneous injection of a mixture of differentially tagged mesenchymal-like and epithelial HPCP-1 cells allowed both cell types to be found in the circulation, with the epithelial HPCP-1 cells forming lung metastases [74]. This led the authors to postulate that, at least in the HPCP-1 model, EMT is necessary but not sufficient for metastatic colonization [74] (Figure 2a). Such results are not observed in all studies, however. For example, MCF7 breast cancer cells that undergo a SIX1-induced EMT form more distant metastases than control cells, in both orthotopic and intracardiac injection models, without co-inclusion of the parental epithelial MCF7 cell line [29] (Figure 2b). These studies can be reconciled if SIX1 imparts a more plastic phenotype on epithelial cells compared to CDK2AP1, allowing the SIX1-expressing MCF7 cells to convert back to an epithelial state at the secondary site. Alternatively, it is possible that MCF7 cells are more amenable to interconverting than HPCP-1 cells, thus not requiring the cooperation of mesenchymal-like and epithelial cells.

Epithelial-to-mesenchymal transition may contribute to metastasis through multiple mechanisms. (a) Carcinoma cells that undergo an oncogenic epithelial-to-mesenchymal transition (EMT) may cooperate with epithelial tumor cells to stimulate metastasis. In this example, mesenchymal-like tumor cells, arising from an exogenously induced oncogenic EMT, are required to enable the parental epithelial tumor cell access to the vasculature; however, once both cell types have accessed the vasculature, only the epithelial cell is able to colonize the secondary site. In this model, the tumor cells are not plastic, and exist as two distinct populations. (b) Tumor cells that are plastic can carry out both early and late stages of the metastatic cascade by utilizing the mesenchymal-like state to leave the primary tumor and enter the vasculature, while the epithelial state is needed to colonize a secondary site; a combination of strictly epithelial and mesenchymal-like cancer cells is not needed.
In addition to EMT contributing to metastasis via increased cellular migration and invasion, an oncogenic EMT is known to impart anoikis resistance, which would be expected to aid the survival of tumor cells in the vasculature [3]. A SNAI1-induced EMT also increases immunosuppression, providing yet another mechanism by which EMT may promote metastatic dissemination [2]. Taken together, the majority of oncogenic EMT-derived gains in function appear to be pro-metastatic (Figure 1).
EMT and cancer stem cells
Normal mammary stem cells (MaSCs) possess properties such as multipotency and self-renewal. This is demonstrated in vivo by the formation of a functional mouse mammary gland from a single MaSC [75], whereas mammosphere assays are used to determine properties of MaSCs in vitro. Sub-populations of primary human mammary epithelial cells (HMECs) grown as single cells in mammosphere culture can produce both differentiated luminal and myoepithelial cells, while the remaining un-differentiated progeny are able to recapitulate the same multipotent phenotype in successive mammosphere passages [76]. If a tumor cell were endowed with these attributes, it would possess assets beneficial for the establishment of a secondary site. For example, colonizing a distant site involves the expansion of cancer cells that, at least initially, would be expected to contain a stem/progenitor-like cell to spawn the new tumor. Not surprisingly then, cells isolated from breast cancers can also form mammospheres, or tumorspheres. These cells have an enriched CD44+/CD24- cell surface profile [77], which marks the same population of cells that have increased tumor-initiation capability in vivo [78]. Thus, a subset of breast cancer cells possess self-renewing and multipotent characteristics similar to MaSCs, as well as demonstrate a heightened ability to initiate tumors, and are denoted as cancer stem cells (CSCs). It should be said, however, that the CSC naming convention does not imply totipotency, such as is observed with true stem cells. Accordingly, the terms tumor-initiating cell and cancer stem-like cell are used interchangeably with CSC in the field.
Because both EMT and CSC phenotypes are implicated in metastasis, a connection between EMT and CSCs was proposed and recently demonstrated. Indeed, two independent groups showed that the CD44high/CD24low population of normal and transformed HMECs displayed EMT-associated phenotypes when compared to CD44low/CD24high cells [4, 5]. Importantly, the reverse experiment of inducing an oncogenic EMT with TWIST1, SNAI1 or TGFB1 led to an increase in CD44high/CD24low cells and tumor-initiation frequency [4, 5], demonstrating a causal role for EMT inducers in CSC formation. SIX1 [20, 79], SNAI2 [80], TWIST2 [18], FOXQ1 [39], TNF [16] and TP53 [40] have all since been shown to induce an EMT and also increase breast CSC features, further establishing an EMT-CSC relationship. Analysis of breast cancer tissue additionally strengthened the EMT-CSC association by identifying a significant correlation between the claudin-low subtype of breast cancers with gene expression signatures for both EMT and CSCs [81]. Interestingly, the miR-200 family is down-regulated in these tumors [82], and these miR family members have been shown to target BMI1 polycomb ring finger oncogene (BMI1) [83] and Suppressor of zeste 12 homolog (SUZ12) [84], polycomb repressive complex members with positive roles in self-renewal [84, 85]. BMI1 is more highly expressed in breast cancer metastases when compared to matched primary tumors [86], again connecting EMT and CSC phenotypes with metastasis.
EMT, cancer stem cells and therapeutic resistance
Conventional breast cancer treatment includes chemotherapy and radiotherapy, and while these treatment options are commonly used, many patients will ultimately relapse due to the presence of residual cancer cells that are presumably treatment-resistant. Recent research has begun to look at EMT and CSCs as one mechanism by which tumors are treatment-resistant. An in vitro study using radiation therapy reported increased resistance in cells grown as mammospheres, which contain a relatively high CSC population, versus monolayer cultures [6]. This result was extended by irradiating mice with mammary tumors and examining CSC abundance, noting an increased percentage of CSCs in residual cells from irradiated mice compared to untreated mice [87]. Regarding chemoresistance, examination of breast tumors after neo-adjuvant chemotherapy revealed an increase in the CSC-enriched CD44high/CD24-/low population [88]. Additionally, a 'mammosphere-CD44+/CD24-/low' gene expression signature constructed from breast tumors applied to biopsies pre- and post-endocrine therapy or chemotherapy demonstrated an increased correlation of the mammosphere-CD44+/CD24-/low gene expression signature with the post-treatment samples [7]. Importantly, samples obtained after treatment were also enriched in EMT-related mesenchymal markers [7], again highlighting a relationship between oncogenic EMT and CSCs. Indeed, oncogenic EMTs have themselves been linked to therapeutic resistance, as highlighted in a recent study by Li and colleagues [89]: doxorubicin treatment increased the fraction of EMT-like cells in vitro, and the cells that underwent an oncogenic EMT were resistant to vincristine and pacilitaxel. It remains unclear whether conventional therapy induces an EMT or CSC phenotype or whether therapies select for cells that have undergone an oncogenic EMT and/or CSC-like conversion. Whichever the case, these studies provide strong justification for increased research to understand the role of oncogenic EMT and CSCs in therapy resistance, so that knowledge gained can be applied towards improving breast cancer treatment.
Therapeutic implications
Studies on the role of EMT and CSCs in metastasis and therapeutic resistance may significantly impact how breast cancer patients are treated in the future. If mediators of oncogenic EMT and/or CSC phenotypes are known, blocking the effects of such mediators should sensitize tumor cells to treatment. In fact, recent studies demonstrate that such approaches may ultimately have efficacy in the clinic. Inhibition of TWIST1 during doxorubicin-induced/enriched oncogenic EMT significantly increased survival and decreased pulmonary and lymph node metastases in a mouse xenograft model in vivo [89]. Since TWIST1 increases were mediated in part by mitogen-activated protein kinase signaling, it was demonstrated that treatment of cells in vitro with a MEK inhibitor could prevent TWIST1 up-regulation [62]. These data suggest that combination treatment with MEK inhibitors and doxorubicin may be a potent mechanism to decrease metastasis, but this finding must first be tested in vivo.
In another study, performed by Joseph and colleagues [90], breast cancer cell lines were treated with the telomerase inhibitor Imetelstat. The authors demonstrated an overall decrease in the CD44+/CD24- cell population and in mammosphere propagation in vitro, while in vivo Imetelstat treatment led to a 50% decrease in tumor initiation. It will thus be of interest to combine Imetelstat with conventional therapy in the future, to determine the combined effects of the drugs on metastasis.
Additional studies have demonstrated that combination therapy can influence both the CSC population and metastasis. For example, mice treated with docetaxel in combination with repertaxin, a CXCR1/2 small-molecule inhibitor, exhibited a reduction in the CD44+/CD24-population in their primary mammary tumors and a decrease in systemic metastases [91]. Since the Imetelstat and repartaxin studies did not directly address the role of oncogenic EMT in the observed effects on CSCs, it would be of interest to investigate this relationship. Overall though, the established EMT-CSC link has led researchers down a worthwhile path towards discovering novel therapeutic targets.
Studies by Gupta and colleagues [92] have recently laid the groundwork for further innovative, anti-oncogenic EMT/CSC approaches to developing new therapies. By employing an induced EMT model to enrich for CD44high/CD24low cells, a high-throughput screen was used to identify drugs that target the breast CSC population, resulting in the discovery of salinomycin as a drug that preferentially kills mesenchymal-like CSCs. Salinomycin treatment was found to decrease the CD44high/CD24low cell population and to lower mammosphere-forming efficiency in vitro, as well as lead to a reduction in tumor-initiating frequency and lung metastases when compared to paclitaxel treatment in vivo [92]. Interestingly, primary tumor and metastatic cells surviving salinomycin treatment did not display the EMT phenotype observed in cells that survived paclitaxel treatment [92]. Thus, combined paclitaxel and salinomycin might be expected to kill both the bulk tumor as well as the CSCs within the tumor. Additional studies investigating therapies that reduce EMT/CSC populations can be found in alternative reviews [93–95].
While many new therapeutic efforts are focusing on targeting EMT-like cells/CSCs, one must remain mindful of tumor cell heterogeneity when developing such therapies. Geyer and colleagues [96] demonstrated that microdissected metaplastic primary breast tumors and matched metastatic samples contain intratumoral genomic differences, in addition to similarities. This indicates that either a single cancer cell did not give rise to the entire tumor, or that distinct genetic alterations occurred in the progeny of the tumor-initiating cell, giving rise to multiple intratumoral clonal populations. The clonal propagation of cells with newly arising mutations may explain intratumoral genotypic heterogeneity, such as metastases that contain both amplified and non-amplified HER2 intratumoral populations [96], but it does not necessarily explain phenotypic, or state of differentiation, heterogeneity within tumors of the same genotype.
Shedding light on intratumoral genotypic and phenotypic heterogeneity are recent studies from the Weinberg and Struhl laboratories. Both groups presented evidence for the conversion of normal and neoplastic non-stem cell populations into functional stem cell populations in vitro and in vivo, using hTert (TERT) immortalized HMECs and vSrc (SRC) transformed MCF10A human mammary cells [97, 98]. Building upon the data, a model addressing genotypic and phenotypic heterogeneity arises where the non-CSC progeny of a metastasized CSC could potentially convert back to a CSC, complete with any gained genomic differences (genomic heterogeneity). This genetically distinct CSC could then spawn a new clonal population of more and less differentiated cells (phenotypic heterogeneity), either within the same tumor or at a new site (Figure 3a). In support of such a model, next generation sequencing of 100 individual nuclei from a polygenomic, triple negative human breast tumor indicated punctuated clonal evolution [99]. Rather than observing a gradual progression of genetic changes in tumor cell populations, no intermediate genotypes were found between the different populations, and in fact the authors noted the 'rate of effective population growth markedly exceeds [the] rate of genomic evolution' [99]. Multiple mutations in a cancer cell that converts to a progenitor-like cell (that is, CSC) could explain such differences. Most importantly though, if genetically distinct CSC and non-CSC populations exist within an individual, multiple therapies may ultimately be required for increased prognosis.

Clinical implications of the plasticity of cancer stem cells. (a) The initial tumor is composed of non-cancer stem cells (CSCs; yellow) and the rare CSCs (red). Non-CSCs within the tumor can mutate, resulting in a genetically distinct non-CSC (blue). Spontaneous conversion of this new non-CSC (blue) into a new CSC (green) provides a given tumor with genetically distinct CSCs (red and green) that can foster the outgrowth of different clonal populations. (b) Conventional therapy is known to be ineffective at eliminating CSCs, which allow tumor cells to eventually repopulate (left); however, targeting solely CSCs will leave the bulk of the tumor intact (middle). A remaining tumor cell could then convert into a CSC, allowing for tumor recurrence and metastasis. Combination therapies targeting both CSCs and non-CSCs are likely needed to better prevent tumor recurrence and metastasis (right).
The take home message from these studies is that targeting the oncogenic EMT/CSC-like population alone is unlikely to be sufficient to inhibit tumor progression and metastasis. The central theme of future research, then, should be that both the CSC and the bulk tumor population must be effectively targeted to attain the best patient response (Figure 3b). It has been suggested that the ability of non-CSC populations to convert to CSCs may help attain this goal [97]. Obtaining non-CSCs from an individual patient should spontaneously yield, as observed with cell lines [97, 98], naturally arising personalized CSCs to be used for predictive testing of an individual's response to a particular therapy. Along those lines, mouse claudin-low tumors were recently shown to functionally contain more CSCs than other breast cancer subtypes, yielding a new model to both identify CSC targets and test resulting therapies [82].
Conclusions
While epithelial cancers may metastasize via various mechanisms, including, but not restricted to, their ability to induce oncogenic EMT, it is clear that epithelial plasticity is an important means by which carcinoma cells can acquire numerous pro-metastatic characteristics. Defining an oncogenic EMT by precise mesenchymal and epithelial alterations runs counter to the fact that these cells are plastic and not created equal: each possesses a different metastatic potential that is either harnessed or repressed by the host. In closing, it is indisputable that studies related to oncogenic EMT have critically contributed to, and will continue to contribute to, our understanding of the most devastating aspect of breast cancer: metastatic dissemination.
Abbreviations
- ADAM:
-
a disintegrin and metalloprotease
- CSC:
-
cancer stem cell
- EGF:
-
epidermal growth factor
- EMT:
-
epithelial-to-mesenchymal transition
- HMEC:
-
human mammary epithelial cell
- MaSC:
-
mammary stem cell
- MET:
-
mesenchymal-to-epithelial transition
- miR:
-
microRNA
- MMP:
-
matrix metalloprotease
- TGF:
-
transforming growth factor
- TNF:
-
tumor necrosis factor.
References
American Cancer Society: Cancer Facts & Figures 2010. Atlanta. 2010
Micalizzi DS, Farabaugh SM, Ford HL: Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia. 2010, 15: 117-134.
Lee JM, Dedhar S, Kalluri R, Thompson EW: The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006, 172: 973-981.
Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A: Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 2008, 3: e2888-
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA: The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008, 133: 704-715.
Phillips TM, McBride WH, Pajonk F: The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006, 98: 1777-1785.
Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI, Fan C, Zhang X, He X, Pavlick A, Gutierrez MC, Renshaw L, Larionov AA, Faratian D, Hilsenbeck SG, Perou CM, Lewis MT, Rosen JM, Chang JC: Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci USA. 2009, 106: 13820-13825.
Tarin D, Thompson EW, Newgreen DF: The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res. 2005, 65: 5996-6000. discussion 6000-5991
Kalluri R, Weinberg RA: The basics of epithelial-mesenchymal transition. J Clin Invest. 2009, 119: 1420-1428.
Zeisberg M, Neilson EG: Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009, 119: 1429-1437.
Hay ED, Zuk A: Transformations between epithelium and mesenchyme: normal, pathological, and experimentally induced. Am J Kidney Dis. 1995, 26: 678-690.
Parsons JT, Horwitz AR, Schwartz MA: Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol. 2010, 11: 633-643.
Hazan RB, Kang L, Whooley BP, Borgen PI: N-cadherin promotes adhesion between invasive breast cancer cells and the stroma. Cell Adhes Commun. 1997, 4: 399-411.
Martin P, Parkhurst SM: Parallels between tissue repair and embryo morphogenesis. Development. 2004, 131: 3021-3034.
Thiery JP, Acloque H, Huang RY, Nieto MA: Epithelial-mesenchymal transitions in development and disease. Cell. 2009, 139: 871-890.
Asiedu MK, Ingle JN, Behrens MD, Radisky DC, Knutson KL: TGF{beta}/TNF{alpha}-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res. 2011, 71: 4707-4719.
Dubois-Marshall S, Thomas JS, Faratian D, Harrison DJ, Katz E: Two possible mechanisms of epithelial to mesenchymal transition in invasive ductal breast cancer. Clin Exp Metastasis. 2011 [Epub ahead of print].
Fang X, Cai Y, Liu J, Wang Z, Wu Q, Zhang Z, Yang CJ, Yuan L, Ouyang G: Twist2 contributes to breast cancer progression by promoting an epithelial mesenchymal transition and cancer stem-like cell self-renewal. Oncogene. 2011 [Epub ahead of print]
Trimboli AJ, Fukino K, de Bruin A, Wei G, Shen L, Tanner SM, Creasap N, Rosol TJ, Robinson ML, Eng C, Ostrowski MC, Leone G: Direct evidence for epithelial-mesenchymal transitions in breast cancer. Cancer Res. 2008, 68: 937-945.
McCoy EL, Iwanaga R, Jedlicka P, Abbey NS, Chodosh LA, Heichman KA, Welm AL, Ford HL: Six1 expands the mouse mammary epithelial stem/progenitor cell pool and induces mammary tumors that undergo epithelial mesenchymal transition. J Clin Invest. 2009, 119: 2663-2677.
Micalizzi DS, Ford HL: Epithelial-mesenchymal transition in development and cancer. Future Oncol. 2009, 5: 1129-1143.
Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De Herreros A: The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000, 2: 84-89.
Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA: The transcription factor snail controls epithelial mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000, 2: 76-83.
Hajra KM, Chen DY, Fearon ER: The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res. 2002, 62: 1613-1618.
Eger A, Aigner K, Sonderegger S, Dampier B, Oehler S, Schreiber M, Berx G, Cano A, Beug H, Foisner R: DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene. 2005, 24: 2375-2385.
Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F: The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001, 7: 1267-1278.
Perez-Moreno MA, Locascio A, Rodrigo I, Dhondt G, Portillo F, Nieto MA, Cano A: A new role for E12/E47 in the repression of E-cadherin expression and epithelial-mesenchymal transitions. J Biol Chem. 2001, 276: 27424-27431.
Vesuna F, van Diest P, Chen JH, Raman V: Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer. Biochem Biophys Res Commun. 2008, 367: 235-241.
Micalizzi DS, Christensen KL, Jedlicka P, Coletta RD, Barón AE, Harrell JC, Horwitz KB, Billheimer D, Heichman KA, Welm AL, Schiemann WP, Ford HL: The Six1 homeoprotein induces human mammary carcinoma cells to undergo epithelial-mesenchymal transition and metastasis in mice through increasing TGF-beta signaling. J Clin Invest. 2009, 119: 2678-2690.
Hartwell KA, Muir B, Reinhardt F, Carpenter AE, Sgroi DC, Weinberg RA: The Spemann organizer gene, Goosecoid, promotes tumor metastasis. Proc Natl Acad Sci USA. 2006, 103: 18969-18974.
Mani SA, Yang J, Brooks M, Schwaninger G, Zhou A, Miura N, Kutok JL, Hartwell K, Richardson AL, Weinberg RA: Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc Natl Acad Sci USA. 2007, 104: 10069-10074.
Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA: Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008, 68: 3645-3654.
Moody SE, Perez D, Pan TC, Sarkisian CJ, Portocarrero CP, Sterner CJ, Notorfrancesco KL, Cardiff RD, Chodosh LA: The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell. 2005, 8: 197-209.
Martin TA, Goyal A, Watkins G, Jiang WG: Expression of the transcription factors snail, slug, and twist and their clinical significance in human breast cancer. Ann Surg Oncol. 2005, 12: 488-496.
Soini Y, Tuhkanen H, Sironen R, Virtanen I, Kataja V, Auvinen P, Mannermaa A, Kosma VM: Transcription factors zeb1, twist and snai1 in breast carcinoma. BMC Cancer. 2011, 11: 73-
Peinado H, Olmeda D, Cano A: Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype?. Nat Rev Cancer. 2007, 7: 415-428.
Montserrat N, Gallardo A, Escuin D, Catasus L, Prat J, Gutierrez-Avigno FJ, Peiro G, Barnadas A, Lerma E: Repression of E-cadherin by SNAIL, ZEB1, and TWIST in invasive ductal carcinomas of the breast: a cooperative effort?. Hum Pathol. 2011, 42: 103-110.
Zhang H, Meng F, Liu G, Zhang B, Zhu J, Wu F, Ethier SP, Miller F, Wu G: Forkhead transcription factor foxq1 promotes epithelial-mesenchymal transition and breast cancer metastasis. Cancer Res. 2011, 71: 1292-1301.
Qiao Y, Jiang X, Lee ST, Karuturi RK, Hooi SC, Yu Q: FOXQ1 regulates epithelial-mesenchymal transition in human cancers. Cancer Res. 2011, 71: 3076-3086.
Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW, Yu WH, Rehman SK, Hsu JL, Lee HH, Liu M, Chen CT, Yu D, Hung MC: p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011, 13: 317-323.
Kim T, Veronese A, Pichiorri F, Lee TJ, Jeon YJ, Volinia S, Pineau P, Marchio A, Palatini J, Suh SS, Alder H, Liu CG, Dejean A, Croce CM: p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med. 2011, 208: 875-883.
Berx G, Van Roy F: The E-cadherin/catenin complex: an important gatekeeper in breast cancer tumorigenesis and malignant progression. Breast Cancer Res. 2001, 3: 289-293.
Thomas PA, Kirschmann DA, Cerhan JR, Folberg R, Seftor EA, Sellers TA, Hendrix MJ: Association between keratin and vimentin expression, malignant phenotype, and survival in postmenopausal breast cancer patients. Clin Cancer Res. 1999, 5: 2698-2703.
Lombaerts M, van Wezel T, Philippo K, Dierssen JW, Zimmerman RM, Oosting J, van Eijk R, Eilers PH, van de Water B, Cornelisse CJ, Cleton-Jansen AM: E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-to-mesenchymal transition in breast cancer cell lines. Br J Cancer. 2006, 94: 661-671.
Taylor MA, Parvani JG, Schiemann WP: The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-beta in normal and malignant mammary epithelial cells. J Mammary Gland Biol Neoplasia. 2010, 15: 169-190.
Hardy KM, Booth BW, Hendrix MJ, Salomon DS, Strizzi L: ErbB/EGF signaling and EMT in mammary development and breast cancer. J Mammary Gland Biol Neoplasia. 2010, 15: 191-199.
Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, Cha SY, Ryu JK, Choi YJ, Kim J, Fearon ER, Weiss SJ: A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006, 8: 1398-1406.
Leong KG, Niessen K, Kulic I, Raouf A, Eaves C, Pollet I, Karsan A: Jagged1- mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J Exp Med. 2007, 204: 2935-2948.
Kasper M, Jaks V, Fiaschi M, Toftgard R: Hedgehog signalling in breast cancer. Carcinogenesis. 2009, 30: 903-911.
Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, Hollier BG, Ram PT, Lander ES, Rosen JM, Weinberg RA, Mani SA: Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci USA. 2010, 107: 15449-15454.
Westermarck J, Kahari VM: Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J. 1999, 13: 781-792.
Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, Chen WJ, Clay WC, Didsbury JR, Hassler D, Hoffman CR, Kost TA, Lambert MH, Leesnitzer MA, McCauley P, McGeehan G, Mitchell J, Moyer M, Pahel G, Rocque W, Overton LK, Schoenen F, Seaton T, Su JL, Becherer JD, et al: Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997, 385: 733-736.
Illman SA, Lehti K, Keski-Oja J, Lohi J: Epilysin (MMP-28) induces TGF-beta mediated epithelial to mesenchymal transition in lung carcinoma cells. J Cell Sci. 2006, 119: 3856-3865.
Guo H, Ingolia NT, Weissman JS, Bartel DP: Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010, 466: 835-840.
Alvarez-Garcia I, Miska EA: MicroRNA functions in animal development and human disease. Development. 2005, 132: 4653-4662.
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ: The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008, 10: 593-601.
Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T: A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9: 582-589.
Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, Goodall GJ: A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008, 68: 7846-7854.
Howe EN, Cochrane DR, Richer JK: Targets of miR-200c mediate suppression of cell motility and anoikis resistance. Breast Cancer Res. 2011, 13: R45-
Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, Teruya-Feldstein J, Reinhardt F, Onder TT, Valastyan S, Westermann F, Speleman F, Vandesompele J, Weinberg RA: miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010, 12: 247-256.
Hwang-Verslues WW, Chang PH, Wei PC, Yang CY, Huang CK, Kuo WH, Shew JY, Chang KJ, Lee EY, Lee WH: miR-495 is upregulated by E12/E47 in breast cancer stem cells, and promotes oncogenesis and hypoxia resistance via downregulation of E-cadherin and REDD1. Oncogene. 2011, 30: 2463-2474.
Li QQ, Chen ZQ, Cao XX, Xu JD, Xu JW, Chen YY, Wang WJ, Chen Q, Tang F, Liu XP, Xu ZD: Involvement of NF-kappaB/miR-448 regulatory feedback loop in chemotherapy-induced epithelial-mesenchymal transition of breast cancer cells. Cell Death Differ. 2011, 18: 16-25.
Kong W, Yang H, He L, Zhao JJ, Coppola D, Dalton WS, Cheng JQ: MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol. 2008, 28: 6773-6784.
Xiang X, Zhuang X, Ju S, Zhang S, Jiang H, Mu J, Zhang L, Miller D, Grizzle W, Zhang HG: miR-155 promotes macroscopic tumor formation yet inhibits tumor dissemination from mammary fat pads to the lung by preventing EMT. Oncogene. 2011, 30: 3440-3453.
Gebeshuber CA, Zatloukal K, Martinez J: miR-29a suppresses tristetraprolin, which is a regulator of epithelial polarity and metastasis. EMBO Rep. 2009, 10: 400-405.
Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J: Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res. 2011, 71: 245-254.
Dave N, Guaita-Esteruelas S, Gutarra S, Frias A, Beltran M, Peiro S, de Herreros AG: Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J Biol Chem. 2011, 286: 12024-12032.
Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, Bell G, Guo W, Rubin J, Richardson AL, Weinberg RA: Paracrine and autocrine signals induce and maintain mesenchymal and stem cell States in the breast. Cell. 2011, 145: 926-940.
Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA: Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004, 117: 927-939.
Chao YL, Shepard CR, Wells A: Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol Cancer. 2010, 9: 179-
Aslakson CJ, Miller FR: Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992, 52: 1399-1405.
Dykxhoorn DM, Wu Y, Xie H, Yu F, Lal A, Petrocca F, Martinvalet D, Song E, Lim B, Lieberman J: miR-200 enhances mouse breast cancer cell colonization to form distant metastases. PLoS One. 2009, 4: e7181-
Korpal M, Ell BJ, Buffa FM, Ibrahim T, Blanco MA, Celià-Terrassa T, Mercatali L, Khan Z, Goodarzi H, Hua Y, Wei Y, Hu G, Garcia BA, Ragoussis J, Amadori D, Harris AL, Kang Y: Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med. 2011, 17: 1101-1108.
Tsuji T, Ibaragi S, Shima K, Hu MG, Katsurano M, Sasaki A, Hu GF: Epithelial-mesenchymal transition induced by growth suppressor p12CDK2-AP1 promotes tumor cell local invasion but suppresses distant colony growth. Cancer Res. 2008, 68: 10377-10386.
Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML, Wu L, Lindeman GJ, Visvader JE: Generation of a functional mammary gland from a single stem cell. Nature. 2006, 439: 84-88.
Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS: In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003, 17: 1253-1270.
Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, Pilotti S, Pierotti MA, Daidone MG: Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005, 65: 5506-5511.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF: Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003, 100: 3983-3988.
Farabaugh SM, Micalizzi DS, Jedlicka P, Zhao R, Ford HL: Eya2 is required to mediate the pro-metastatic functions of Six1 via the induction of TGF-beta signaling, epithelial-mesenchymal transition, and cancer stem cell properties. Oncogene. 2011 [Epub ahead of print]
Bhat-Nakshatri P, Appaiah H, Ballas C, Pick-Franke P, Goulet R, Badve S, Srour EF, Nakshatri H: SLUG/SNAI2 and tumor necrosis factor generate breast cells with CD44+/CD24- phenotype. BMC Cancer. 2010, 10: 411-
Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM: Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12: R68-
Herschkowitz JI, Zhao W, Zhang M, Usary J, Murrow G, Edwards D, Knezevic J, Greene SB, Darr D, Troester MA, Hilsenbeck SG, Medina D, Perou CM, Rosen JM: Comparative oncogenomics identifies breast tumors enriched in functional tumor-initiating cells. Proc Natl Acad Sci USA. 2011 [Epub ahead of print]
Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, Diehn M, Liu H, Panula SP, Chiao E, Dirbas FM, Somlo G, Pera RA, Lao K, Clarke MF: Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009, 138: 592-603.
Iliopoulos D, Lindahl-Allen M, Polytarchou C, Hirsch HA, Tsichlis PN, Struhl K: Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol Cell. 2010, 39: 761-772.
Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, Suri P, Wicha MS: Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006, 66: 6063-6071.
Joensuu K, Hagstrom J, Leidenius M, Haglund C, Andersson LC, Sariola H, Heikkila P: Bmi-1, c-myc, and Snail expression in primary breast cancers and their metastases-elevated Bmi-1 expression in late breast cancer relapses. Virchows Arch. 2011, 459: 31-39.
Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, Joshua B, Kaplan MJ, Wapnir I, Dirbas FM, Somlo G, Garberoglio C, Paz B, Shen J, Lau SK, Quake SR, Brown JM, Weissman IL, Clarke MF: Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009, 458: 780-783.
Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, Wong H, Rosen J, Chang JC: Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008, 100: 672-679.
Li QQ, Xu JD, Wang WJ, Cao XX, Chen Q, Tang F, Chen ZQ, Liu XP, Xu ZD: Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin Cancer Res. 2009, 15: 2657-2665.
Joseph I, Tressler R, Bassett E, Harley C, Buseman CM, Pattamatta P, Wright WE, Shay JW, Go NF: The telomerase inhibitor imetelstat depletes cancer stem cells in breast and pancreatic cancer cell lines. Cancer Res. 2010, 70: 9494-9504.
Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, Wicinski J, Cabaud O, Charafe-Jauffret E, Birnbaum D, Guan JL, Dontu G, Wicha MS: CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest. 2010, 120: 485-497.
Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES: Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009, 138: 645-659.
Al-Ejeh F, Smart CE, Morrison BJ, Chenevix-Trench G, Lopez JA, Lakhani SR, Brown MP, Khanna KK: Breast cancer stem cells: treatment resistance and therapeutic opportunities. Carcinogenesis. 2011, 32: 650-658.
May CD, Sphyris N, Evans KW, Werden SJ, Guo W, Mani SA: Epithelial-mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res. 2011, 13: 202-
Pannuti A, Foreman K, Rizzo P, Osipo C, Golde T, Osborne B, Miele L: Targeting Notch to target cancer stem cells. Clin Cancer Res. 2010, 16: 3141-3152.
Geyer FC, Weigelt B, Natrajan R, Lambros MB, de Biase D, Vatcheva R, Savage K, Mackay A, Ashworth A, Reis-Filho JS: Molecular analysis reveals a genetic basis for the phenotypic diversity of metaplastic breast carcinomas. J Pathol. 2010, 220: 562-573.
Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, Arendt LM, Kuperwasser C, Bierie B, Weinberg RA: Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci USA. 2011, 108: 7950-7955.
Iliopoulos D, Hirsch HA, Wang G, Struhl K: Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci USA. 2011, 108: 1397-1402.
Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, Hicks J, Wigler M: Tumour evolution inferred by single-cell sequencing. Nature. 2011, 472: 90-94.
Acknowledgements
HLF is funded by grants from the National Cancer Institute (2RO1-CA095277, 1R01-CA157790, and 1R01-CA124545-01), The American Cancer Society (#RSG-07-183-01-DDC), The Department of Defense (BC084105), and The Breast Cancer Research Foundation-American Association for Cancer Research, as well as from the State of Colorado. TPR is supported by the Cancer League of Colorado.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Drasin, D.J., Robin, T.P. & Ford, H.L. Breast cancer epithelial-to-mesenchymal transition: examining the functional consequences of plasticity. Breast Cancer Res 13, 226 (2011). https://doi.org/10.1186/bcr3037
Published:
DOI: https://doi.org/10.1186/bcr3037