
Abstract
Aberrant activation of a latent embryonic program - known as the epithelial-mesenchymal transition (EMT) - can endow cancer cells with the migratory and invasive capabilities associated with metastatic competence. The induction of EMT entails the loss of epithelial characteristics and the de novo acquisition of a mesenchymal phenotype. In breast cancer, the EMT state has been associated with cancer stem cell properties including expression of the stem cell-associated CD44+/CD24-/low antigenic profile, self-renewal capabilities and resistance to conventional therapies. Intriguingly, EMT features are also associated with stem cells isolated from the normal mouse mammary gland and human breast reduction tissues as well as the highly aggressive metaplastic and claudin-low breast tumor subtypes. This has implications for the origin of these breast tumors as it remains unclear whether they derive from cells that have undergone EMT or whether they represent an expansion of a pre-existing stem cell population that expresses EMT-associated markers to begin with. In the present review, we consider the current evidence connecting EMT and stem cell attributes and discuss the ramifications of these newly recognized links for our understanding of the emergence of distinct breast cancer subtypes and breast cancer progression.
Similar content being viewed by others


Introduction
Despite recent medical advances, metastasis, tumor relapse and resistance to therapy remain the principal causes of death for breast cancer patients. The lack of effective therapies calls for an improved understanding of the molecular mechanisms driving breast cancer progression. It is increasingly acknowledged that aberrant activation of a latent embryonic program - known as the epithelial-mesenchymal transition (EMT) - can endow cancer cells with the migratory and invasive capabilities associated with metastatic competence [1–3]. Moreover, several lines of evidence have converged in recent years to support the notion that not all cancer cells within a given tumor are equal in terms of their tumor-initiating potential. The emerging paradigm posits that tumor progression is driven by a small subpopulation of cancer cells - termed cancer stem cells (CSCs) or tumor-initiating cells - that exhibit two defining characteristics: the ability to self-renew and the ability to regenerate the phenotypic heterogeneity of the parental tumor [4]. CSCs have thus been implicated both in initiating and sustaining primary tumor growth and in driving the seeding and establishment of metastases at distal sites [5–9].
Whereas the CSC hypothesis does not stipulate the cell of origin for a particular cancer, it is reasonable to hypothesize that tumors may originate from the transformation of normal adult tissue stem cells or from more differentiated progenitors that have acquired self-renewal capabilities [4] (Figure 1). Importantly, recent studies have established a crucial link between passage through EMT and the acquisition of molecular and functional properties of stem cells [10, 11]. Thus, in addition to bestowing migratory and invasive potential, induction of EMT in immortalized and transformed human mammary epithelial cells significantly enhanced their self-renewal and tumor-initiating capabilities and led to the expression of stem-cell markers, typically associated with breast CSCs [10]. As EMT can be sporadically triggered by extracellular stimuli and microenvironment factors, these findings provide a plausible explanation for the de novo generation of CSCs from differentiated tumor cells and suggest that passage through EMT is an alternative and/or additional driving force in tumorigenesis (Figure 1).

Epithelial-mesenchymal transition and stem cell traits in breast cancer progression. Breast tumors may originate from the transformation of normal adult tissue stem cells or from more differentiated progenitors that have acquired self-renewal capabilities (left panel). Moreover, a subset of resident mammary gland stem cells (MaSCs, in blue) exhibit epithelial-mesenchymal transition (EMT) features a priori. The EMT features of metaplastic and claudin-low breast tumors may thus signify either that they derive from cells that have undergone EMT or that they originate from deregulated expansion of a pre-existing stem cell pool that expresses EMT-associated markers. Additionally, the induction of sporadic EMT within a tumor bestows migratory and invasive potential coupled with self-renewal capabilities to cancer cells, generating cancer stem cells (CSCs) (right panel). Following extravasation and upon encountering an altered local microenvironment, CSCs (in red) may at least partially revert to an epithelial phenotype (mesenchymal-epithelial transition (MET)) to allow adhesion and proliferation at distal sites. As sporadic EMT and MET are triggered by extracellular stimuli and microenvironment factors, this model provides a plausible explanation for the de novo generation of CSCs from differentiated tumor cells and suggests that passage through EMT and MET is an alternative and/or additional driving force in breast tumorigenesis.
Intriguingly, the gene expression signatures of stem cells from normal mouse and human mammary tissues and of claudin-low and metaplastic breast tumors share strong similarities with the gene expression profiles of cells that have undergone EMT [10, 12–16]. This has implications for the origin of these breast tumor subtypes, as it remains unclear whether they derive from cells that have undergone EMT or whether they represent an expansion of a pre-existing stem cell population that already expresses EMT-associated markers [15, 17] (Figure 1).
In the present review, we bring together the current evidence linking EMT and stem cell attributes and discuss the ramifications of these newly recognized links for our understanding of the emergence of distinct breast cancer subtypes as well as breast cancer progression, particularly in view of the fact that both the EMT and CSC phenotypes have been independently linked with metastatic progression, drug resistance and disease recurrence [14, 18–20].
Epithelial-mesenchymal transition
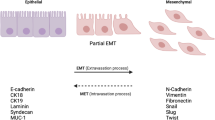
EMT is an essential developmental process that enables reprogramming of polarized epithelial cells towards a mesenchymal motile phenotype. During normal embryonic development, EMT serves to loosen cell-cell contacts and to enhance intrinsic cell motility, thus paving the way for the extensive cell movements required for gastrulation and organogenesis [1, 3]. In normal adult tissues, the typically dormant EMT program is reactivated during wound repair and tissue regeneration. Dysregulation of EMT, however, can lead to pathologic conditions such as organ fibrosis and tissue destruction. Indeed, as mentioned above, nonmetastatic cancer cells may harness the EMT program to attain the migratory and invasive potential required for metastatic progression [1, 3]. This profound phenotypic conversion from an epithelial phenotype to a spindle-shaped morphology is orchestrated by integrated networks of signal transduction pathways and EMT-related transcription factors (TFs) that direct the altered expression of genes involved in cell adhesion, differentiation and motility. A critical molecular event underpinning the dissolution of cell-cell contacts during EMT is the loss of E-cadherin, a key component of adherens junctions. While the initial stages of EMT may involve endocytosis and lysosomal degradation of E-cadherin [21], EMT and metastatic progression are most often associated with a reversible downregulation of E-cadherin (encoded by CDH1) involving either hypermethylation of the CDH1 promoter or repression by EMT-inducing TFs [1, 3].
The loss of E-cadherin releases β-catenin into the cytosol and elicits activation of the canonical Wnt signaling pathway [1, 3]. Moreover, the impairment of E-cadherin function, together with the downregulation of components of tight junctions and desmosomes (for example, claudins, occludins, desmogleins and desmocollins) and polarity genes, contributes to the dissolution of inter-cellular contacts and the loss of apico-basal polarity [1, 3]. However, EMT is not merely a shedding of epithelial characteristics but also entails the de novo expression of mesenchymal-associated genes (for example, N-cadherin, fibronectin, α-smooth muscle actin, vimentin). The ensuing reorganization of the actin cytoskeleton and the intermediate filament network and the acquisition of front-back polarity dramatically alter the cellular architecture, while the secretion of extracellular matrix components and matrix metalloproteinases remodels the extracellular matrix. Collectively, these changes disrupt the contiguity of the tissue epithelium and render the cells intrinsically able to migrate - independent of one another - and to invade the underlying stromal compartment by breaching the basement membrane [1, 3].
Whereas the migratory and invasive capabilities imparted by EMT facilitate the initial steps of the meta-static cascade, it is envisaged that, following extravasation and upon encountering an altered local microenvironment, disseminated cancer cells may at least partially revert to an epithelial phenotype to allow adhesion and proliferation at distal sites. This reversal of EMT - known as mesenchymal-epithelial transition (MET) - has been evoked to explain the findings that disseminated tumor cells in the bone marrow often exhibit an epithelial phenotype [22] and that distant metastases resemble glandular structures with the same molecular subtype as their respective primary tumors [23]. Accordingly, the induction of MET was recently shown to increase the number of macroscopic lung nodules formed by 4T1 cells, suggesting that acquisition of an epithelial-like phenotype through MET promotes colonization [24]. Cancer-associated EMT may therefore not represent a complete interconversion of epithelial and mesenchymal phenotypes nor an irreversible commitment to a full-blown mesenchymal state. Indeed, the existence of a metastable or partial EMT phenotype, exhibiting both epithelial and mesenchymal features, is a notion more easily reconciled with the concept of a highly plastic stem-like state, which is reversible and under the influence of the local microenvironment [25]. Consistent with this hypothesis, in a mouse model of EMT induced by infiltrating CD8 T cells, EMT-derived mesenchymal cells exhibited combined luminal and basal markers, providing in vivo evidence for the occurrence of a partial EMT [26]. Moreover, Damonte and colleagues documented the widespread occurrence of cells exhibiting dual staining for epithelial and mesenchymal markers in a subset of EMT-type spindle tumors in mouse models [27]. More recently, epithelial cells coexpressing cytokeratins 5/19 and vimentin were identified by dual immunofluorescence labeling in claudin-low and basal-like breast cancer subtypes [2], confirming the existence of cells with combined epithelial and mesenchymal characteristics in human tumors.
EMT can be induced by a plethora of extracellular stimuli, including hepatocyte growth factor, epidermal growth factor, platelet-derived growth factor, Wnt, Notch, Sonic hedgehog and transforming growth factor beta (TGFβ) as well as components of the extracellular matrix such as collagen and hyaluronic acid and adverse conditions such as hypoxia [1, 3]. These diverse stimuli trigger a multitude of signal transduction pathways that converge on several EMT-inducing TFs, including Snail, Slug, Zeb1, Zeb2, Twist, FoxC2 and Goosecoid, many of which are frequently overexpressed in breast cancers [1, 3].
Cellular hierarchy in the normal mammary gland
The human breast is a complex secretory organ that comprises an elaborate branching network of epithelial ducts embedded in a fat pad, rich in adipocytes, blood vessels, hematopoietic cells and stromal fibroblasts. The ducts are lined with an inner layer of luminal epithelial cells and an outer layer of contractile basal myoepithelial cells that promote the extrusion of the secreted milk from the alveoli during lactation. The ducts terminate in small grape-like clusters - termed the terminal ductal lobular units - that comprise the direct precursors of the lobuloalveolar units that produce milk during lactation. Pregnancy hormones elicit the expansion and differentiation of alveolar luminal cells into secretory cells that produce and secrete milk. After weaning, the expanded mammary epithelium is returned to the state of the mature virgin gland by a process known as involution, which involves extensive apoptosis and tissue remodeling [17].
The capacity of the mammary gland epithelium to expand and remodel during puberty and repeated cycles of pregnancy is highly suggestive of the existence of resident mammary gland stem cells (MaSCs) [17]. Further support for the existence of MaSCs has come from mammary fat pad transplantation assays, whereby cell suspensions or mammary gland explants are transplanted into a recipient epithelium-divested fat pad. The study of the resultant epithelial outgrowths has confirmed the existence of pluripotent stem cells [15, 28–32] capable of regenerating a fully functional mammary tree containing luminal and myoepithelial cells that respond to pregnancy hormones by generating alveoli, as well as duct-limited and lobule-limited multipotent progenitors [33]. Moreover, the development of mammosphere assays has provided a way to enrich for stem cells/progenitors in vitro as well as a surrogate assay of the anchorage-independent growth properties and self-renewal capabilities of mammary stem and progenitor cell types [34]. Collectively, these studies have begun to shed light on the cellular hierarchy inherent in the normal mammary gland epithelium. Whereas the precise nature of the intermediates remains to be elucidated, it is believed that undifferentiated estrogen receptor-negative MaSCs give rise to a common bipotent progenitor that is the predecessor of two distinct lineages: the basal/myoepithelial progenitors and the luminal progenitors. In turn, basal/myoepithelial progenitors generate differentiated myoepithelial cells, whereas luminal progenitors differentiate into ductal luminal cells or generate alveolar luminal cells in response to pregnancy hormones [17, 35].
Mammary gland stem cell phenotypic markers associated with EMT
The ability to evaluate the in vivo repopulating activity of distinct subsets of mammary gland cells has enabled the prospective isolation and characterization of putative stem cells and/or progenitors from both mouse [31, 32] and human [7, 28, 29] mammary tissues, and has led to the identification of stem cell-associated antigenic markers. In the mouse, a single cell with the Lin-/CD24+/CD29high antigenic profile [31, 32] can thus reconstitute a fully functional mammary gland. Interestingly, these cells - designated as mammary gland repopulating units - express cytokeratins 5 and 14, smooth muscle actin, vimentin and smooth muscle myosin - markers typically associated with basal/myoepithelial cells.
In breast tumors, the CD44+/CD24-/low antigenic phenotype defines a subpopulation of breast cancer cells enriched for cells with stem-like qualities: namely, the ability to self-renew and efficiently reconstitute differentiated tumors. In limiting dilution assays, therefore, as few as 100 injected CD44+/CD24-/low cells were capable of initiating tumors in non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice [6], whereas tens of thousands of cells with alternate phenotypes failed to form tumors. More recently, expression of the enzyme aldehyde dehydrogenase 1 (ALDH-1) has been designated as a marker of normal and malignant breast stem cells [7].
The induction of EMT has been associated with the cell surface expression of the CD44+/CD24-/low antigenic phenotype [10]. Indeed, a major proportion of circulating tumor cells - probably generated by EMT - display combined expression of one or more EMT markers and ALDH-1 [36], suggesting that combining the ALDH-1 and CD44+/CD24-/low expression profiles may be exploited for the isolation/detection of the highly tumorigenic subset of EMT-associated breast CSCs. Importantly, however, no markers have to date been identified that could distinguish between MaSCs and CSCs. Undoubtedly, future efforts will focus on this important area and its potential therapeutic implications.
EMT and normal mammary gland development
Interestingly, transient EMT-like events have been implicated in the branching morphogenesis that underlies mouse mammary gland development. Specifically, the restricted expression of Twist-1, Twist-2 and Snail in the terminal end buds of the mouse mammary gland strongly suggests that these TFs may function to transiently repress epithelial differentiation in favor of a motile phenotype during the development of the ductal networks [37]. Furthermore, in an elegant series of organotypic culture experiments, using real-time imaging of green fluorescent protein expressed under the control of the vimentin gene promoter, the position and pattern of branch formation was found to mirror the sites of transient green fluorescent protein expression, suggesting dynamic EMT-like events at the branch points [38].
Indeed, cells at the leading edge of the mammary gland ductal tree exhibit mesenchymal-like features and secrete matrix metalloproteinase 3 [39].
Many authors have drawn parallels between the branching morphogenesis processes underlying normal embryonic mammary gland development and the initiation of invasive events in breast cancer progression, based on the fact that, in the developing gland, epithelial cells proliferate, migrate and invade from a pre-existing epithelium into the adjacent fat pad [40]. Indeed, it is envisaged that the signaling pathways orchestrating normal mammary gland development and remodeling during pregnancy and involution are also the ones that go awry during tumor initiation and progression. Consistent with this, aberrations in numerous signaling pathways and transcription factors implicated in normal mammary gland development and EMT have been documented in breast cancer progression [1, 3].
Breast cancer: a hierarchy in flux
There is increasing evidence of similar cellular hierarchies in the normal and the malignant mammary gland [17, 35]. Consistent with this, the gene expression profile of CD44+/CD24-/low breast cancer cells, enriched for cells with tumor-initiating capabilities, more closely resembles that of CD44+/CD24-/low cells from the normal breast than CD44-/CD24+ cells isolated from the same tumor [41].
Breast cancers have long been recognized as a remarkably diverse and heterogeneous set of malignancies, owing mostly to their classification into numerous histological subtypes on the basis of several histopathological criteria. In recent years, cytogenetic and mutational analyses - in conjunction with molecular profiling technologies - have revealed that this histologic heterogeneity is underpinned by diverse gene expression signatures, thought to represent the different cell lineages of the mammary gland and stages of mammary epithelial cell differentiation [4, 17]. Using genomic profiling, at least six breast cancer intrinsic subtypes have been identified on the basis of their distinct molecular signatures rather than their clinical or histopathologic behavior: luminal A, luminal B, HER2+, basal-like, normal breast-like and the most recently recognized claudin-low [2, 12, 42, 43].
Previously, several studies had alluded to basal-like tumors being composed of primitive undifferentiated cells [44–46]. Honeth and colleagues reported that the CD44+/CD24-/low phenotype, associated with populations enriched for breast CSCs, was most prominent in basal-like and, in particular, BRCA1 hereditary breast tumors [45]. Moreover, basal-like breast cancers, which are enriched for CD44+/CD24-/low cells, were reported to exhibit EMT features that might account for their aggressive clinical behavior and metastatic propensities [47]. At the molecular level, hypoxia-induced SLUG expression had been associated with the acquisition of a basal-like breast cancer phenotype characterized by the stem cell-regulatory genes CD133 and BMI1 [48]. Recent in vivo studies, however, have demonstrated that, despite their stem cell-like characteristics, basal-like breast tumors may not be derived from normal mammary gland stem cells [49] and instead point to the failure of luminal progenitors to undergo terminal differentiation as a more likely contributor to the emergence of sporadic and BRCA1-mutated basal-like breast cancers. Indeed, Lim and colleagues found an aberrant expansion of this luminal progenitor subpopulation in the pre-neoplastic tissues from BRCA1 carriers [29], who are predisposed to basal-like breast tumors. Accordingly, interrogation of the breast cancer intrinsic subtype gene sets with the gene signatures of human MaSC-enriched, luminal progenitor, mature luminal and stromal populations uncovered hitherto unrecognized similarities between the basal-like and luminal progenitor gene signatures [15].
On the other hand, the MaSC (human, CD49fhighEpCAM-; mouse, CD29highCD24+CD61+) expression profile most closely resembles the claudin-low subtype [2, 15]. Together, these findings seem consistent with the notion that distinct subsets of breast cancer may derive from the different cell lineages of the mammary gland and/or stages of mammary epithelial cell differentiation; that is, MaSCs may be the cell of origin for claudin-low tumors, and committed luminal progenitors may beget basal-like tumors. It is also possible, however, that MaSCs comprise the cell of origin for both subtypes but claudin-low tumors are locked in a stem-like state portraying MaSC features whereas the luminal progenitors are impeded from undergoing terminal differentiation [15, 29, 35].
The newly recognized claudin-low subtype is characterized by reduced expression of genes involved in tight junctions and intercellular adhesions (for example, claudin 3, claudin 4, claudin 7, occludins, E-cadherin), diminished expression of luminal differentiation markers and enrichment for EMT markers, immune response genes and cancer stem cell features (CD44+/CD24-/low; CD49f+/EpCAM-/low; high ALDH-1) [2, 15]. Although the claudin-low and basal-like subtypes share some similarities (for example, low HER-2, luminal cytokeratins, estrogen receptor and GATA-3), it is clear that they are distinct subtypes. Indeed, claudin-low tumors lack expression of proliferation genes and are hence likely to comprise slow-cycling tumors quite unlike the basal-like subtype [2]. Yet many of the studies on basal-like tumors were conducted prior to the recognition of the claudin-low subtype as a distinct entity [12]. Previously classified basal-like tumors may thus need to be reassessed with tumor classifiers that take into account the recently identified claudin-low subtype. Indeed, recent profiling would suggest that the basal-like subtype does not show an appreciable mesenchymal or stem-cell like signature [16].
Furthermore, genome-wide transcriptional profiling has shown that metaplastic breast cancers, a rare and aggressive histological subtype, frequently exhibit EMT and stem cell-like gene expression [13], probably contributing to their poor outcomes. Indeed, metaplastic and claudin-low tumors share many gene expression features, although the former are often associated with phospho-inositide-3-kinase pathway mutations [13].
Most notably, many of the conserved genes in the MaSC signature are typically considered EMT-associated genes [15]. The expression of these EMT-associated genes in tumor cells may therefore reflect a basal differentiation program and not necessarily indicate cells that have undergone EMT [15]. This is quite distinct from the idea that EMT might facilitate the generation of migrating cancer stem cells due to an altered microenvironment at the invasive front [10, 50, 51].
Similar to breast cancer subtypes, genomic profiling has also been applied to classify breast cancer cell lines into luminal, basal A (mixed basal/luminal features) and basal B/mesenchymal types [52–55]. These cell line genomic profiles broadly mirror the heterogeneity and diversity of primary breast tumors, and have helped establish a strong correlation between invasiveness, metastatic potential, EMT gene expression and CSC properties. The basal B/mesenchymal cell lines thus tend to be highly invasive, exhibit the CD44+/CD24-/low antigenic profile and display EMT features, including reduced E-cadherin levels and selective upregulation of ZEB1 [52, 53, 55]. Indeed, a subset of nine previously classified basal B cell lines (BT549, HBL100, Hs578T, MDA-MB-157, MDA-MB-231, MDA-MB-435, MDA-MB-436, SUM1315, SUM159PT) was more recently reported to represent the claudin-low subtype [2], strengthening the case for considering basal-like and claudin-low tumors and cell lines separately.
Whereas CD44 expression is elevated in both basal A and basal B cell lines, CD24 expression levels are significantly lower in the basal B subgroup, suggesting that these cell lines are enriched for CSCs [52, 53]. Indeed, within the basal B cell line subgroup, the levels of CD24 correlate positively with E-cadherin and negatively with vimentin. Moreover, CD44+/CD24-/low cells exhibit activation of the TGFβ signaling pathway [41], consistent with the proposed association between this antigenic profile and EMT features.
Molecular circuitries linking EMT and stemness
EMT-inducing TFs function either directly or indirectly to repress CDH1 transcription, reduce epithelial differentiation and/or promote the mesenchymal gene expression program. Recently, several TFs and miRNAs have been implicated in the molecular pathways linking EMT to the acquisition of stem cell properties, although the precise molecular circuitries remain largely undefined.
Recent studies demonstrated that the induction of EMT by ectopic expression of SNAIL, TWIST or TGFβ treatment in immortalized and transformed human mammary epithelial cells results in the acquisition of stem cell properties: the ability to self-renew and initiate tumors [10, 11]. In addition, chronic overexpression of the homeobox protein Six1 in the mouse mammary gland generated highly aggressive tumors with an EMT phenotype, stem cell features and activated Wnt signaling [56], providing vital in vivo evidence for the emergence of cells with combined EMT/CSC phenotypes.
At the molecular level, TWIST1 directly stimulates the expression of BMI1, which encodes a polycomb-group protein that maintains self-renewal through repression of the p16INK4A-ARF locus. Indeed, TWIST1 and BMI1 cooperate to repress expression of both E-cadherin and p16INK4a, thus simultaneously promoting EMT and conferring tumor-initiating capabilities [57]. Moreover, TWIST1 modulates the CSC phenotype by downregulating the expression of CD24 [58].
Recently, several novel EMT-inducers that appear to act either upstream or in concert with the known EMT TFs have been implicated in the transcriptional hierarchy of EMT and the establishment of the combined EMT/CSC phenotype. The mammalian Y-box binding protein 1 (YB1) has been shown to promote cap-independent translation of SNAIL, TWIST and ZEB2/SIP1 together with the upregulation of the stem cell markers p63, CD44 and CD10, while at the same time repressing cap-dependent translation of growth-promoting genes and CD24. YB1 thus appears to link the acquisition of a mesenchymal/migratory phenotype and stem cell-associated gene expression with entrance into a quiescent state [59]. Further molecular targets of YB1 in the MDA-MB-231 and SUM149 breast cancer cell lines have been shown to include the stem-cell-associated markers CD44 and CD49f as well as c-KIT, BMI1 and members of the WNT and NOTCH signaling pathways [60].
Another newly identified player in EMT - the ladybird homeobox 1 (LBX1) protein - transcriptionally targets ZEB1, ZEB2, SNAIL1 and TGFβ2. Accordingly, ectopic LBX1 expression in mammary epithelial cells induced EMT and mesenchymal markers with concordant increases in mammosphere formation and the proportion of CD44+/CD24-/low cells [61]. Both YB1 and LBX1 may thus function as master regulators of the EMT/stemness program.
Two recent studies have emphasized the role of the microenvironment in promoting tumor progression by influencing EMT-dependent manifestation of CSC properties. First, the activating transcription factor 3 gene is an adaptive-response gene that may serve to integrate stromal signals from the tumor microenvironment with the acquisition of combined EMT/CSC properties in mammary epithelial cells [62]. Second, signaling by the urokinase-type plasminogen activator receptor can also instigate EMT and promote CSC properties in MDA-MB-468 cells exposed to hypoxia [63].
The discovery of miRNAs has added an additional level of complexity to the molecular networks regulating EMT, metastasis and stemness [51, 64]. Indeed, members of the miR-200 family (miR-141, miR-200a, miR-200b, miR-200c and miR-429) are rapidly emerging as master regulators of differentiation by directly targeting the transcripts encoding ZEB1 and ZEB2/SIP1, thereby leading to de-repression of CDH1 and eliciting MET. In turn, ZEB1 can also suppress members of the miR-200 family, establishing a reciprocal feedback loop [65, 66] that enables reversible phenotypic modulation of the epithelial and mesenchymal states. Consistent with the induction of EMT as an important early step in tumor metastasis, downregulation of the miR-200 family has been extensively documented to occur during EMT and in invasive breast cancers [65, 66]. Indeed, recently, ZEB1 has been shown to induce EMT by suppressing miR-200 family members at the invasive front of pancreatic tumors, thus leading to the generation of migrating CSCs [51].
Importantly, downregulation of the miR-200 family has also been described in normal murine mammary stem cells and human normal and breast cancer stem cells (CD44+/CD24-/low), reinforcing the molecular links between normal stem cells and CSCs [64] as well as the roles of the miR-200 family in regulating stemness and EMT. Indeed, the relevant molecular targets of the miR-200 family include the stem cell-associated factors BMI1, SOX2 and KLF4 [51, 64, 65].
Other miRNAs have also been implicated in the regulation of EMT and CSC traits. Enforced expression of let-7, which targets HMGA2 and HRAS, thus sup-pressed mammosphere formation and tumor initiation in serial transplantation assays in NOD/SCID mice, whereas suppression of let-7 function in differentiated cells increased mammosphere formation. Accordingly, let-7 expression levels are reduced in breast CSCs but increase during differentiation [67].
EMT, stemness and resistance to chemotherapy
Several lines of evidence suggest that CSCs, including those generated through EMT, exhibit intrinsic resistance to conventional chemotherapies [14, 19, 20, 68]. Indeed, EMT TFs have been directly implicated in subverting key tumor suppressor mechanisms. For example, TWIST can stimulate AKT2 expression, leading to enhanced cell survival and increased resistance to paclitaxel in vitro [69], can directly elicit multidrug resistance by inducing expression of MDR1 in adriamycin-treated cells [70] and can override oncogene-induced senescence in vivo by inhibiting key components of the p53-dependent and Rb-dependent pathways [71].
Many currently administered chemotherapies target the rapidly proliferating cells of the tumor bulk but fail to eradicate the intrinsically resistant CSCs, thus inadvertently leading to expansion of the CSC pool and/or selection of resistant CSCs. Accordingly, Creighton and colleagues demonstrated that the residual breast cancer cell populations, persisting in patients after conventional treatments, exhibited gene expression profiles indicative of cells with combined tumor-initiating and mesenchymal/claudin-low features [14]. Significantly, the corresponding gene expression signatures may harbor novel molecular targets for overcoming the intrinsic therapeutic resistance of breast cancers.
Towards therapeutics that target the EMT/CSC phenotype
Taken together, the findings that link EMT, CSC traits, drug resistance and enhanced metastatic competence suggest that targeting the EMT/CSC phenotype may hold considerable therapeutic promise. In a seminal study, Gupta and colleagues performed a high-throughput chemical screen to identify agents with selective toxicity against CSCs generated by EMT. They thus identified salinomycin, a potassium ionophore that reduced the CD44highCD24-/low fraction and impeded mammosphere formation. Significantly, pretreatment of EMT-derived CSCs with salinomycin in vitro reduced their ability to initiate tumors and lung metastases in NOD/SCID mice [72].
The anti-diabetic drug metformin, which has long been recognized for its beneficial effects in breast cancer, was recently shown to selectively kill breast CSCs in vitro and in vivo. Moreover, the combination of metformin and the DNA-damaging agent doxorubicin reduced tumor mass and prevented relapse more effectively than either drug alone in a xenograft mouse model [73]. Residual cell populations recovered from these tumors after the combined treatment were devoid of CSCs demonstrating that the therapeutic prowess of metformin - in the context of doxorubicin treatment - is linked to its ability to kill CSCs. Interestingly, an independent study demonstrated that metformin treatment reduced the expression of the EMT regulators ZEB1, TWIST1, SNAI2 (Slug) and TGFβ, decreased the proportion of CD44+CD24-/low cells and impeded mammosphere formation in claudin-low MDA-MB-231 cells [74].
Recent findings further confirm that monotherapy may exacerbate tumor relapse, as doxorubicin treatment actually promoted metastasis of murine 4T1 and human MDA-MB-231 cells, concurrent with activation of TGFβ signaling [75]. Importantly, however, the combination of doxorubicin with a TGFβ type I receptor kinase inhibitor prevented mammary tumor growth and lung metastases in these xenograft models [75]. Together with the important clinical findings of Creighton and colleagues [14], these studies highlight the need to combine conventional chemotherapies, radiotherapies or endocrine therapies with drugs targeting the self-renewal, survival and drug resistance of cells with an EMT/CSC phenotype. Moreover, given that CSCs are thought to represent only a small fraction of the tumor mass and that the effectiveness of current therapies is often measured by a reduction in tumor volume, CSC- targeted therapies may require new methods of gauging therapeutic success in the short term in addition to decreased tumor recurrence or increased tumor-free survival.
Concluding remarks
The induction of EMT and its ability to confer stem cell properties to cancer cells remains an important factor in disease progression irrespective of breast tumor subtype. Given that at least a subset of resident MaSCs are endowed with EMT features a priori, however, claudin-low and metaplastic tumors may arise through deregulated expansion of this MaSC pool. Alternatively, if EMT is sporadically triggered by the tumor microenvironment (cytokines produced by infiltrating immune cells, growth factor signaling, hypoxia), it may endow subsets of cancer cells with the migratory, invasive and self-renewal properties required to further sustain and expand primary tumor growth and/or generate migrating CSCs that can seed new tumors at distant sites. Nevertheless, the molecular circuitries underlying EMT and stemness appear closely intertwined, and it will be vital to further delineate key molecular players that link these two cellular states. Strategies to target the combined EMT/CSC phenotype, however, may also impact the normal MaSC pool or stem cells residing in other tissues. Efforts to develop CSC-targeted therapeutics must therefore not only be focused on the EMT/CSC phenotype but must, in parallel, strive to identify novel molecular targets that could serve to eradicate CSCs without harming normal stem cells. Overall, the close links between EMT, CSC traits, drug resistance and enhanced metastatic competence suggest that an improved understanding of the EMT/CSC connection may uncover new therapeutic targets for breast cancers, irrespective of their intrinsic subtype, since most subtypes harbor a pool of CSCs.
Note
This article is part of the series on EMT and the connection with stem cells, edited by Jenny Chang.
Abbreviations
- ALDH-1:
-
aldehyde dehydrogenase 1
- BRCA1:
-
breast cancer susceptibility gene 1
- CD:
-
cluster of differentiation
- CSC:
-
cancer stem cell
- EMT:
-
epithelial-mesenchymal transition
- EpCAM:
-
epithelial cell adhesion molecule
- LBX1:
-
ladybird homeobox 1
- MaSC:
-
mammary gland stem cell
- MET:
-
mesenchymal-epithelial transition
- NOD/SCID:
-
non-obese diabetic/severe combined immunodeficient
- TF:
-
transcription factor
- TGFβ:
-
transforming growth factor beta
- YB1:
-
Y-box binding protein 1.
References
Thiery JP, Acloque H, Huang RY, Nieto MA: Epithelial-mesenchymal transitions in development and disease. Cell. 2009, 139: 871-890. 10.1016/j.cell.2009.11.007.
Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM: Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12: R68-10.1186/bcr2635.
Polyak K, Weinberg RA: Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009, 9: 265-273. 10.1038/nrc2620.
McDermott SP, Wicha MS: Targeting breast cancer stem cells. Mol Oncol. 2010, 4: 404-419. 10.1016/j.molonc.2010.06.005.
Abraham BK, Fritz P, McClellan M, Hauptvogel P, Athelogou M, Brauch H: Prevalence of CD44+/CD24-/low cells in breast cancer may not be associated with clinical outcome but may favor distant metastasis. Clin Cancer Res. 2005, 11: 1154-1159.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF: Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003, 100: 3983-3988. 10.1073/pnas.0530291100.
Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G: ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007, 1: 555-567. 10.1016/j.stem.2007.08.014.
Liu R, Wang X, Chen GY, Dalerba P, Gurney A, Hoey T, Sherlock G, Lewicki J, Shedden K, Clarke MF: The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med. 2007, 356: 217-226. 10.1056/NEJMoa063994.
Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH, Goulet R, Badve S, Nakshatri H: CD44+/CD24- breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res. 2006, 8: R59-10.1186/bcr1610.
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA: The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008, 133: 704-715. 10.1016/j.cell.2008.03.027.
Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A: Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 2008, 3: e2888-10.1371/journal.pone.0002888.
Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, Rasmussen KE, Jones LP, Assefnia S, Chandrasekharan S, Backlund MG, Yin Y, Khramtsov AI, Bastein R, Quackenbush J, Glazer RI, Brown PH, Green JE, Kopelovich L, Furth PA, Palazzo JP, Olopade OI, Bernard PS, Churchill GA, Van Dyke T, Perou CM: Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007, 8: R76-10.1186/gb-2007-8-5-r76.
Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, Fridlyand J, Sahin A, Agarwal R, Joy C, Liu W, Stivers D, Baggerly K, Carey M, Lluch A, Monteagudo C, He X, Weigman V, Fan C, Palazzo J, Hortobagyi GN, Nolden LK, Wang NJ, Valero V, Gray JW, Perou CM, Mills GB: Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009, 69: 4116-4124. 10.1158/0008-5472.CAN-08-3441.
Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI, Fan C, Zhang X, He X, Pavlick A, Gutierrez MC, Renshaw L, Larionov AA, Faratian D, Hilsenbeck SG, Perou CM, Lewis MT, Rosen JM, Chang JC: Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci USA. 2009, 106: 13820-13825. 10.1073/pnas.0905718106.
Lim E, Wu D, Pal B, Bouras T, Asselin-Labat ML, Vaillant F, Yagita H, Lindeman GJ, Smyth GK, Visvader JE: Transcriptome analyses of mouse and human mammary cell subpopulations reveal multiple conserved genes and pathways. Breast Cancer Res. 2010, 12: R21-10.1186/bcr2560.
Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, Hollier BG, Ram PT, Lander ES, Rosen JM, Weinberg RA, Mani SA: Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci USA. 2010, 107: 15449-15454. 10.1073/pnas.1004900107.
Visvader JE: Keeping abreast of the mammary epithelial hierarchy and breast tumorigenesis. Genes Dev. 2009, 23: 2563-2577. 10.1101/gad.1849509.
Fillmore CM, Kuperwasser C: Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008, 10: R25-10.1186/bcr1982.
Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, Wong H, Rosen J, Chang JC: Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008, 100: 672-679. 10.1093/jnci/djn123.
Phillips TM, McBride WH, Pajonk F: The response of CD24-/low/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006, 98: 1777-1785. 10.1093/jnci/djj495.
Janda E, Nevolo M, Lehmann K, Downward J, Beug H, Grieco M: Raf plus TGFβ-dependent EMT is initiated by endocytosis and lysosomal degradation of E-cadherin. Oncogene. 2006, 25: 7117-7130. 10.1038/sj.onc.1209701.
van der Pluijm G: Epithelial plasticity, cancer stem cells and bone metastasis formation. Bone. 2011, 48: 37-43. 10.1016/j.bone.2010.07.023.
Weigelt B, Peterse JL, van 't Veer LJ: Breast cancer metastasis: markers and models. Nat Rev Cancer. 2005, 5: 591-602. 10.1038/nrc1670.
Dykxhoorn DM, Wu Y, Xie H, Yu F, Lal A, Petrocca F, Martinvalet D, Song E, Lim B, Lieberman J: miR-200 enhances mouse breast cancer cell colonization to form distant metastases. PLoS One. 2009, 4: e7181-10.1371/journal.pone.0007181.
Klymkowsky MW, Savagner P: Epithelial-mesenchymal transition: a cancer researcher's conceptual friend and foe. Am J Pathol. 2009, 174: 1588-1593. 10.2353/ajpath.2009.080545.
Santisteban M, Reiman JM, Asiedu MK, Behrens MD, Nassar A, Kalli KR, Haluska P, Ingle JN, Hartmann LC, Manjili MH, Radisky DC, Ferrone S, Knutson KL: Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res. 2009, 69: 2887-2895. 10.1158/0008-5472.CAN-08-3343.
Damonte P, Gregg JP, Borowsky AD, Keister BA, Cardiff RD: EMT tumorigenesis in the mouse mammary gland. Lab Invest. 2007, 87: 1218-1226. 10.1038/labinvest.3700683.
Eirew P, Stingl J, Raouf A, Turashvili G, Aparicio S, Emerman JT, Eaves CJ: A method for quantifying normal human mammary epithelial stem cells with in vivo regenerative ability. Nat Med. 2008, 14: 1384-1389. 10.1038/nm.1791.
Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, Asselin-Labat ML, Gyorki DE, Ward T, Partanen A, Feleppa F, Huschtscha LI, Thorne HJ; kConFab, Fox SB, Yan M, French JD, Brown MA, Smyth GK, Visvader JE, Lindeman GJ: Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009, 15: 907-913. 10.1038/nm.2000.
Raouf A, Zhao Y, To K, Stingl J, Delaney A, Barbara M, Iscove N, Jones S, McKinney S, Emerman J, Aparicio S, Marra M, Eaves C: Transcriptome analysis of the normal human mammary cell commitment and differentiation process. Cell Stem Cell. 2008, 3: 109-118. 10.1016/j.stem.2008.05.018.
Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML, Wu L, Lindeman GJ, Visvader JE: Generation of a functional mammary gland from a single stem cell. Nature. 2006, 439: 84-88. 10.1038/nature04372.
Stingl J, Raouf A, Eirew P, Eaves CJ: Deciphering the mammary epithelial cell hierarchy. Cell Cycle. 2006, 5: 1519-1522. 10.4161/cc.5.14.2983.
Jeselsohn R, Brown NE, Arendt L, Klebba I, Hu MG, Kuperwasser C, Hinds PW: Cyclin D1 kinase activity is required for the self-renewal of mammary stem and progenitor cells that are targets of MMTV-ErbB2 tumorigenesis. Cancer Cell. 2010, 17: 65-76. 10.1016/j.ccr.2009.11.024.
Dontu G, Al-Hajj M, Abdallah WM, Clarke MF, Wicha MS: Stem cells in normal breast development and breast cancer. Cell Prolif. 2003, 36 (Suppl 1): 59-72. 10.1046/j.1365-2184.36.s.1.6.x.
Prat A, Perou CM: Mammary development meets cancer genomics. Nat Med. 2009, 15: 842-844. 10.1038/nm0809-842.
Aktas B, Tewes M, Fehm T, Hauch S, Kimmig R, Kasimir-Bauer S: Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res. 2009, 11: R46-10.1186/bcr2333.
Kouros-Mehr H, Werb Z: Candidate regulators of mammary branching morphogenesis identified by genome-wide transcript analysis. Dev Dyn. 2006, 235: 3404-3412. 10.1002/dvdy.20978.
Nelson CM, Vanduijn MM, Inman JL, Fletcher DA, Bissell MJ: Tissue geometry determines sites of mammary branching morphogenesis in organotypic cultures. Science. 2006, 314: 298-300. 10.1126/science.1131000.
Fata JE, Werb Z, Bissell MJ: Regulation of mammary gland branching morphogenesis by the extracellular matrix and its remodeling enzymes. Breast Cancer Res. 2004, 6: 1-11.
Micalizzi DS, Farabaugh SM, Ford HL: Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia. 2010, 15: 117-134. 10.1007/s10911-010-9178-9.
Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, Nikolskaya T, Serebryiskaya T, Beroukhim R, Hu M, Halushka MK, Sukumar S, Parker LM, Anderson KS, Harris LN, Garber JE, Richardson AL, Schnitt SJ, Nikolsky Y, Gelman RS, Polyak K: Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007, 11: 259-273. 10.1016/j.ccr.2007.01.013.
Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lønning PE, Børresen-Dale AL, Brown PO, Botstein D: Molecular portraits of human breast tumours. Nature. 2000, 406: 747-752. 10.1038/35021093.
Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Eystein Lønning P, Børresen-Dale AL: Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001, 98: 10869-10874. 10.1073/pnas.191367098.
Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA: An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008, 40: 499-507. 10.1038/ng.127.
Honeth G, Bendahl PO, Ringner M, Saal LH, Gruvberger-Saal SK, Lovgren K, Grabau D, Ferno M, Borg A, Hegardt C: The CD44+/CD24- phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008, 10: R53-10.1186/bcr2108.
Park SY, Lee HE, Li H, Shipitsin M, Gelman R, Polyak K: Heterogeneity for stem cell-related markers according to tumor subtype and histologic stage in breast cancer. Clin Cancer Res. 2010, 16: 876-887. 10.1158/1078-0432.CCR-09-1532.
Sarrio D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, Palacios J: Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008, 68: 989-997. 10.1158/0008-5472.CAN-07-2017.
Storci G, Sansone P, Trere D, Tavolari S, Taffurelli M, Ceccarelli C, Guarnieri T, Paterini P, Pariali M, Montanaro L, Santini D, Chieco P, Bonafé M: The basal-like breast carcinoma phenotype is regulated by SLUG gene expression. J Pathol. 2008, 214: 25-37. 10.1002/path.2254.
Molyneux G, Geyer FC, Magnay FA, McCarthy A, Kendrick H, Natrajan R, Mackay A, Grigoriadis A, Tutt A, Ashworth A, Reis-Filho JS, Smalley MJ: BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell. 2010, 7: 403-417. 10.1016/j.stem.2010.07.010.
Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T: Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer. 2005, 5: 744-749. 10.1038/nrc1694.
Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, zur Hausen A, Brunton VG, Morton J, Sansom O, Schüler J, Stemmler MP, Herzberger C, Hopt U, Keck T, Brabletz S, Brabletz T: The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009, 11: 1487-1495. 10.1038/ncb1998.
Blick T, Hugo H, Widodo E, Waltham M, Pinto C, Mani SA, Weinberg RA, Neve RM, Lenburg ME, Thompson EW: Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44hi/CD24lo/- stem cell phenotype in human breast cancer. J Mammary Gland Biol Neoplasia. 2010, 15: 235-252. 10.1007/s10911-010-9175-z.
Blick T, Widodo E, Hugo H, Waltham M, Lenburg ME, Neve RM, Thompson EW: Epithelial mesenchymal transition traits in human breast cancer cell lines. Clin Exp Metastasis. 2008, 25: 629-642. 10.1007/s10585-008-9170-6.
Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, Hur MH, Diebel ME, Monville F, Dutcher J, Brown M, Viens P, Xerri L, Bertucci F, Stassi G, Dontu G, Birnbaum D, Wicha MS: Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009, 69: 1302-1313. 10.1158/0008-5472.CAN-08-2741.
Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, Speed T, Spellman PT, DeVries S, Lapuk A, Wang NJ, Kuo WL, Stilwell JL, Pinkel D, Albertson DG, Waldman FM, McCormick F, Dickson RB, Johnson MD, Lippman M, Ethier S, Gazdar A, Gray JW: A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006, 10: 515-527. 10.1016/j.ccr.2006.10.008.
McCoy EL, Iwanaga R, Jedlicka P, Abbey NS, Chodosh LA, Heichman KA, Welm AL, Ford HL: Six1 expands the mouse mammary epithelial stem/progenitor cell pool and induces mammary tumors that undergo epithelial- mesenchymal transition. J Clin Invest. 2009, 119: 2663-2677. 10.1172/JCI37691.
Yang MH, Hsu DS, Wang HW, Wang HJ, Lan HY, Yang WH, Huang CH, Kao SY, Tzeng CH, Tai SK, Chang SY, Lee OK, Wu KJ: Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat Cell Biol. 2010, 12: 982-992. 10.1038/ncb2099.
Vesuna F, Lisok A, Kimble B, Raman V: Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia. 2009, 11: 1318-1328.
Evdokimova V, Tognon C, Ng T, Ruzanov P, Melnyk N, Fink D, Sorokin A, Ovchinnikov LP, Davicioni E, Triche TJ, Sorensen PH: Translational activation of snail1 and other developmentally regulated transcription factors by YB-1 promotes an epithelial-mesenchymal transition. Cancer Cell. 2009, 15: 402-415. 10.1016/j.ccr.2009.03.017.
To K, Fotovati A, Reipas KM, Law JH, Hu K, Wang J, Astanehe A, Davies AH, Lee L, Stratford AL, Raouf A, Johnson P, Berquin IM, Royer HD, Eaves CJ, Dunn SE: Y-box binding protein-1 induces the expression of CD44 and CD49f leading to enhanced self-renewal, mammosphere growth, and drug resistance. Cancer Res. 2010, 70: 2840-2851. 10.1158/0008-5472.CAN-09-3155.
Yu M, Smolen GA, Zhang J, Wittner B, Schott BJ, Brachtel E, Ramaswamy S, Maheswaran S, Haber DA: A developmentally regulated inducer of EMT, LBX1, contributes to breast cancer progression. Genes Dev. 2009, 23: 1737-1742. 10.1101/gad.1809309.
Yin X, Wolford CC, Chang YS, McConoughey SJ, Ramsey SA, Aderem A, Hai T: ATF3, an adaptive-response gene, enhances TGFβ signaling and cancer-initiating cell features in breast cancer cells. J Cell Sci. 2010, 123 (Pt 20): 3558-3565. 10.1242/jcs.064915.
Jo M, Eastman BM, Webb DL, Stoletov K, Klemke R, Gonias SL: Cell signaling by urokinase-type plasminogen activator receptor induces stem cell-like properties in breast cancer cells. Cancer Res. 2010, 70: 8948-8958. 10.1158/0008-5472.CAN-10-1936.
Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, Diehn M, Liu H, Panula SP, Chiao E, Dirbas FM, Somlo G, Pera RA, Lao K, Clarke MF: Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009, 138: 592-603. 10.1016/j.cell.2009.07.011.
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ: The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008, 10: 593-601. 10.1038/ncb1722.
Park SM, Gaur AB, Lengyel E, Peter ME: The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22: 894-907. 10.1101/gad.1640608.
Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, Huang Y, Hu X, Su F, Lieberman J, Song E: let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007, 131: 1109-1123. 10.1016/j.cell.2007.10.054.
Zhang M, Atkinson RL, Rosen JM: Selective targeting of radiation-resistant tumor-initiating cells. Proc Natl Acad Sci USA. 2010, 107: 3522-3527. 10.1073/pnas.0910179107.
Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, Wang LH: Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 2007, 67: 1979-1987. 10.1158/0008-5472.CAN-06-1479.
Li QQ, Xu JD, Wang WJ, Cao XX, Chen Q, Tang F, Chen ZQ, Liu XP, Xu ZD: Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin Cancer Res. 2009, 15: 2657-2665. 10.1158/1078-0432.CCR-08-2372.
Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin S, Maestro R, Voeltzel T, Selmi A, Valsesia-Wittmann S, Caron de Fromentel C, Puisieux A: Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell. 2008, 14: 79-89. 10.1016/j.ccr.2008.06.005.
Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES: Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009, 138: 645-659. 10.1016/j.cell.2009.06.034.
Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K: Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69: 7507-7511. 10.1158/0008-5472.CAN-09-2994.
Vazquez-Martin A, Oliveras-Ferraros C, Barco SD, Martin-Castillo B, Menendez JA: The anti-diabetic drug metformin suppresses self-renewal and proliferation of trastuzumab-resistant tumor-initiating breast cancer stem cells. Breast Cancer Res Treat. 2010, [Epub ahead of print],
Bandyopadhyay A, Wang L, Agyin J, Tang Y, Lin S, Yeh IT, De K, Sun LZ: Doxorubicin in combination with a small TGFβ inhibitor: a potential novel therapy for metastatic breast cancer in mouse models. PLoS One. 2010, 5: e10365-10.1371/journal.pone.0010365.
Acknowledgements
The authors apologize to the many researchers whose work they were unable to cite in the present review due to space limitations. They are grateful to Joe Taube for helpful discussions and to the anonymous reviewers for their constructive criticisms. The present work was supported in part by The University of Texas MD Anderson Cancer Center Research Trust Fund, the V Foundation's V scholar award, an R01 supplemental grant (3R01CA138239-02S1) and the MD Anderson Cancer Center Support Grant CA016672.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Caitlin D May, Nathalie Sphyris contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
May, C.D., Sphyris, N., Evans, K.W. et al. Epithelial-mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res 13, 202 (2011). https://doi.org/10.1186/bcr2789
Published:
DOI: https://doi.org/10.1186/bcr2789