
Abstract
Inhibitors of poly(ADP-ribose) polymerase (PARP)-mediated DNA repair have shown promise in early clinical studies in the treatment of specific subgroups of breast cancer. Notably, phase II trials indicate that olaparib, an oral PARP inhibitor, has activity as a single agent in BRCA-related tumours, and that a combination of iniparib, an intravenous PARP inhibitor, and chemotherapy offers a survival advantage, compared with chemotherapy alone, in triple-negative breast cancer. Phase III data on the latter indication are expected in 2011. Intriguingly, iniparib does not increase toxicity when used as a chemo-potentiating agent, suggesting that it differs in its mechanism of action from other agents in this class. Overall, PARP inhibitors represent a potentially important new class of anti-cancer agents with two potential modes of action, as single agents causing synthetic lethality and as chemo-potentiating agents.
Similar content being viewed by others
Introduction
Breast cancer is the most commonly diagnosed cancer in the UK, with more than 45,000 new cases per year, and a lifetime risk of 1 in 9 [1]. Incidence rates have risen over the past 20 years in industrialised countries, but the same period has seen the development of many new treatments, and eight out of ten women diagnosed with breast cancer are now expected to survive 5 years or more [1, 2]. The improvements in treatment include novel cytotoxic drugs and many targeted agents, such as trastuzumab, lapatinib and the aromatase inhibitors.
Within the past 5 years, it has become apparent that another powerful class of agents - the poly(ADP-ribose) polymerase (PARP) inhibitors - has activity in defined groups of patients with breast cancer [3–11]. These agents target a DNA repair pathway via a novel mechanism of action that can be exploited to the benefit of patients with breast cancer. This review will briefly discuss the development of PARP inhibitors, and the data supporting their potential clinical use in breast cancer, as single agents and in combination with chemotherapy.
PARP activity and inhibition
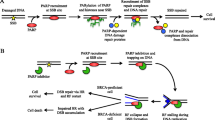
The PARP family of enzymes was first described over 40 years ago [12]. PARP1 and PARP2 occur in the cell nucleus, and are activated by DNA damage [13]. PARP1, the most abundant form of the enzyme [14], acts as a 'molecular nick sensor' to signal DNA single-strand breaks (SSBs) and assist in their repair [15]. It is inactive until bound to a DNA strand break via its zinc finger DNA-binding domain. After binding, PARP1 uses NAD+ to form long, branched polymers of poly(ADP-ribose) on acceptor proteins, including PARP itself. This autopoly(ADP-ribosyl)ation creates a negatively charged target at the SSB, which recruits the enzymes required to form the base excision repair multi-protein complex [16, 17]. Following ADP-ribosylation, PARP1 has reduced affinity for DNA; it is released, opening up the chromatin and allowing access to the damaged site for the other repair complex proteins. Subsequently, the enzyme poly(ADP-ribose) glycohydrolase (PARG) removes the poly(ADP-ribose) polymer from PARP, allowing the enzyme to be recycled and reactivated at other areas of DNA damage (Figure 1).

Mechanism of PARP1-mediated DNA repair. ARH3, ADP-ribosyl acceptor hydrolase 3; LigIII, DNA ligase III; PARG, poly(ADP-ribose) glycohydrolase; PARP, poly(ADP-ribose) polymerase; polβ, DNA polymerase; XRCC1, X-ray repair cross-complementing 1.
Most of the PARP inhibitors currently undergoing clinical investigation have been designed to compete with NAD+ for its substrate binding site [18]. It is likely that these drugs inhibit both PARP1 and PARP2 [18]. Inhibition of PARP1 compromises a cell's ability to over-come damage to the genome by repairing DNA SSBs. Many commonly used anticancer treatments, such as radiotherapy, alkylating agents and camptothecins, damage DNA by causing SSBs, and prevention of SSB repair has been shown in preclinical studies to improve cell kill [19]. For this reason, PARP inhibitors were first developed as radio- or chemo-potentiating agents [20, 21], the aim being to overcome cancer cell resistance to a DNA-damaging agent by preventing repair of the potentially lethal damage to the cancer cell caused by the treatment.
Potent PARP inhibitors first entered early clinical trials in 2003 [22], and there has been rapid expansion of the field in the past 5 years. Several PARP inhibitors are being investigated for a range of indications [23, 24], mainly cancers (Table 1). Early clinical studies suggest that PARP inhibitors will have a role in the treatment of breast cancer, with promising data emerging from trials in two specific areas: hereditary BRCA-deficient breast cancer (single-agent PARP inhibition) and triple-negative breast cancer (TNBC; PARP inhibition in combination with chemotherapy) [3–11]. However, with improved understanding of the mechanisms of PARP1 inhibition in these two clinical scenarios, it is likely that a wider group of patients with breast cancer may also benefit from this approach to treatment.
Single-agent PARP inhibitors in hereditary BRCA-deficient cancer
The publication in 2005 of paired pre-clinical papers in Nature, demonstrating hypersensitivity of BRCA-deficient cancer cells to single-agent PARP inhibitors, opened the door to clinical research into these agents as monotherapy [25, 26], and provided the clearest demonstration to date of a class effect. Before this ground-breaking research was published, it had been argued that PARP inhibitors would have no activity when used alone, and drug developers were chasing the elusive goal of chemopotentiation in tumours without an increase in toxicity in normal tissue.
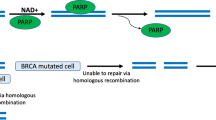
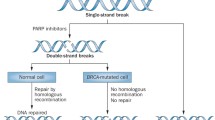
Initial preclinical experiments in cell lines and xenografts demonstrated that cells with loss of the homologous recombination repair pathway for DNA double-strand breaks (DSBs) are hypersensitive to blockade of SSB repair with a PARP inhibitor. The key to this mechanism of action is the role that BRCA1 and BRCA2 play in signalling DSBs (BRCA1) and the repair of such breaks via the homologous recombination pathway (BRCA2) [27].
The proposed mechanism for the cytotoxicity of PARP inhibition is that blockade of SSB repair leads to the formation of unrepaired DSBs at the replication fork. In normal or BRCA-heterozygous cells, the lesion can be repaired by DSB mechanisms, and DNA replication and cell division continue. However, in cells that lack functional DSB repair, such as those with a homozygous mutation in BRCA1 or BRCA2, loss of two DNA repair pathways causes synthetic lethality and cell death [25, 26].
This proposition was first tested clinically in a phase I study of AZD2281 (olaparib), looking at its potential activity as a single agent [28]. This study used an oral formulation of olaparib, and assessed the pharmacokinetics and pharmacodynamics of doses ranging from 10 mg daily for 2 out of 3 weeks to 600 mg twice daily on a continuous schedule. Nine of the 19 patients with confirmed BRCA mutations had a partial response to olaparib, representing a 47% response rate in this population. No responses were seen in 37 patients who did not have a BRCA mutation. The dose-limiting toxicities were myelosuppression and central nervous system side effects. Toxicities to normal tissue were similar in the BRCA-deficient and normal populations. An increase in γH2AX foci was found in plucked eyebrow hair follicles 6 hours after olaparib treatment, compared with baseline. These foci indicate accumulation of DNA DSBs, so the finding demonstrated a proof of mechanism of the process of synthetic lethality, whereby preservation of SSBs by PARP inhibition leads to the formation of DNA DSBs in the absence of an external DNA-damaging agent. It must be noted, however, that the mechanistic proof was demonstrated in normal tissue, not in the tumour. It is not clear from the publication whether this pharmaco-dynamic effect was seen in BRCA carriers with a heterozygote defect in all cells, or in all patient groups. These data, which suggest a risk of accumulation of DNA damage within normal tissue, raise concern over the potential dangers of prolonged, continuous dosing. In theory, there could also be an accumulation of tumourigenic mutations and second malignancies, so caution in the use of these agents in the adjuvant setting over prolonged periods may be advisable.
The phase I findings have been confirmed by two phase II studies of olaparib in BRCA1 or BRCA2 mutation carriers in patients with breast [3, 4] or ovarian [5, 6] cancer previously treated with a median of three chemotherapy regimens. Both studies assessed response and toxicity in sequential cohorts of patients treated with 400 mg twice daily or 100 mg twice daily. The activity of olaparib as a single agent was confirmed in the 400 mg cohorts, but there was less activity at the lower dose, suggesting that the extent of PARP inhibition is important for response. In the breast cancer study, 27 patients with metastatic disease were treated at each dose, and the confirmed response rate was 41% in those receiving the 400 mg schedule and 22% in the 100 mg cohort [3, 4]. Toxicity was reduced with the lower dose, compared with the higher dose, but mild overall. Fatigue, nausea and vomiting were the commonest adverse events with this well tolerated agent.
A similar dose response was found in the phase II study of ovarian cancer, in which 33 patients were treated with olaparib 400 mg twice daily and 24 with 100 mg twice daily. There was a 33% confirmed partial response rate at the higher dose and 13% at the lower dose [5, 6]. Responses were seen in carriers of BRCA1 or BRCA2 mutations, and in patients with either platinum-sensitive or platinum-resistant disease. However, these results are not consistent with findings from previous studies that acquired resistance to platinum-based treatment in ovarian cancer is associated with regain of BRCA function [29, 30].
The findings of these small, non-randomised phase I and phase II studies require confirmation at phase III. Meanwhile, they do provide the exciting suggestion that single-agent therapy with PARP inhibitors will be of benefit in BRCA-positive patients, and offer low toxicity. Phase II studies in this indication are ongoing with the PARP inhibitors PF01367338 (AG014699) and ABT-888 (veliparib). A phase I single-agent study with the novel PARP inhibitor MK-4827 has also confirmed the potential for activity with low toxicity in this group of patients [31].
PARP inhibitors as chemo-potentiating agents
As discussed above, PARP inhibitors were initially envisaged as chemo-potentiating agents. In previous studies, using inhibitors of methyl guanine methyl transferase to prevent DNA repair during cytotoxic therapy, the dose of chemotherapy needed to be reduced because of enhanced toxicity with the treatment combination [32–34]. Similarly, in some of the first studies of PARP inhibitors in combination with chemotherapy, a higher level of myelosuppression was observed than would have been expected with the chemotherapeutic agent alone [7, 22, 35–37]. Two further studies required protocol amendments in light of dose-limiting toxicities associated with olaparib in combination with chemotherapy - in particular, myelosuppression [8, 38]. These findings present two challenges to physicians and scientists designing trials with combinations of chemotherapy with PARP inhibitors. First, should clinical studies be designed with the maximum dose of chemotherapy possible, or a maximal dose of PARP inhibitor accepting a chemotherapy dose reduction? The answer to this will lie in the clinical outcome data and also how the PARP inhibitor is being used - for its single agent activity or as a true chemo-potentiating agent. Second, there is a need to investigate the mechanism by which PARP inhibitors enhance damage to normal tissue when used with some of the agents reported (platinums, bifunctional alkylating agents such as cyclophosphamide or the antimetabolite, gemcitabine), where repair of cytotoxic damage is not thought to be achieved via a PARP-dependent mechanism. There is one molecule in the class - BSI-201 (SAR 240550, iniparib) - for which normal tissue toxicity appears to be less of an issue, and so far this is the PARP inhibitor that has progressed furthest in the clinic.
PARP inhibitors in triple-negative breast cancer
Following encouraging results with iniparib in combination with chemotherapy with various solid tumours [39], clinical investigators have gone on to study this agent with chemotherapy in TNBC, a disease with a biological phenotype similar to BRCA1-defective cancers [40]. It must be borne in mind that iniparib may act via a different mechanism to the PARP inhibitors discussed above, and does not appear to have the limitation of enhanced toxicity in normal tissue [39, 41, 42].
Evidence of an improvement in anti-tumour activity has been reported in patients previously treated with two or more cytotoxic regimens who received iniparib in combination with carboplatin and gemcitabine, compared with those who received the chemotherapy regimen alone [9]. In this phase II study, 123 patients with TNBC were randomised to receive carboplatin (AUC2) and gemcitabine (1,000 mg/m2) on days 1 and 8 of a 21-day cycle, with or without iniparib 5.6 mg/kg on days 1, 4, 8 and 11. The objective response rate was 52% in the iniparib group, compared with 32% in those who received chemotherapy alone (P = 0.02), median progression-free survival was 5.9 versus 3.6 months, respectively (P = 0.01), and overall survival was 12.3 versus 7.7 months, respectively (P = 0.01) [9, 10]. No difference was observed in the rate of adverse events between the two treatment groups. A phase III study of iniparib in combination with carboplatin and gemcitabine for the same indication was initiated in 2009, and rapidly completed recruitment. It is hoped that preliminary results will be reported at the American Society of Clinical Oncology (ASCO) 2011 meeting. However, initial indications from the manufacturer, Sanofi-Aventis, are that the phase III trial did not meet the primary endpoints of overall and progression-free survival [43].
It is possible that the intermittent dosing regimen used in the studies cited above helps to protect against enhanced myelosuppression and hence toxicity. However, if iniparib is acting as a PARP inhibitor, it is intriguing that this intermittent schedule also provides an increase in overall treatment efficacy, compared with chemotherapy alone. It is possible that iniparib is acting as a single agent on the BRCA-like phenotype of TNBC, or that it is somehow able to prevent repair to treatment-induced DNA damage within the tumour without increased damage to normal tissue. If they confirm the phase II data, the results of the phase III study of iniparib and chemotherapy in TNBC may lead to an advance in the treatment of patients, but they will also present an intriguing puzzle to scientists working with PARP inhibitors.
It is also interesting to note that single-agent olaparib was described as having activity in TNBC at the ASCO 2010 meeting [11]. This was the first trial to evaluate the efficacy of a PARP inhibitor as a single agent for the treatment of sporadic TNBC, and responses were seen only in tumours that occurred on a background of BRCA1 germline mutation [11].
Conclusion
Research into PARP inhibition is beginning to show the potential of targeting DNA repair as a strategy in the development of new anti-cancer agents. This is a class of drugs with activity as monotherapy and in combination with treatments that cause DNA damage [4, 8, 38]. It is very unusual in cancer therapy for a new drug to be used solely as monotherapy, and research to date suggests that PARP inhibitors are likely to be used also as components of combination regimens. Indeed, the single-agent and DNA-damaging-potentiating facets of PARP inhibition present an interesting challenge to drug developers as we try to improve treatments for our patients. If we wish to exploit the single-agent activity of a PARP inhibitor as part of a combination regimen, we may need to consider scheduling PARP-based treatment separately to any DNA-damaging component of the combination. However, when using a PARP inhibitor as a chemo-potentiator, concomitant treatment would be appropriate.
The phase III trial of iniparib in TNBC is nearing completion. If the data confirm the earlier findings, it will provide a promising new treatment for this patient group. However, there remain many scientific questions about the underlying mechanism of action of iniparib in this setting, and the differences between the individual agents in the PARP inhibitor class.
Whatever the explanation, PARP inhibition may eventually offer an effective treatment approach for patients with BRCA-related breast cancer and TNBC, and the array of compounds in development will allow clinical investigators to exploit the differences to extend our scientific knowledge and thus benefit a wider range of patients.
Abbreviations
- ASCO:
-
American Society of Clinical Oncology
- DSB:
-
double-strand break
- PARP:
-
poly(ADP-ribose) polymerase
- SSB:
-
single-strand break
- TNBC:
-
triple-negative breast cancer.
References
Breast cancer - UK incidence statistics. [http://info.cancerresearchuk.org/cancerstats/types/breast/incidence/]
Hery C, Ferlay J, Boniol M, Autier P: Changes in breast cancer incidence and mortality in middle-aged and elderly women in 28 countries with Caucasian majority populations. Ann Oncol. 2008, 19: 1009-1018. 10.1093/annonc/mdm593.
Tutt A, Robson M, Garber J, Domchek S, Audeh M, Weitzel J, Friedlander M, Carmichael J: Phase II trial of the oral PARP inhibitor olaparib in BRCA-deficient advanced breast cancer. J Clin Oncol. 2009, 27 (18s): CRA501-
Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, Wardley A, Mitchell G, Earl H, Wickens M, Carmichael J: Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010, 376: 235-244. 10.1016/S0140-6736(10)60892-6.
Audeh M, Penson R, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel J, Carmichael J, Tutt A: Phase II trial of the oral PARP inhibitor olaparib (AZD2281) in BRCA-deficient advanced ovarian cancer. J Clin Oncol. 2009, 27 (15s): 5500-
Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Oaknin A, Loman N, Lu K, Schmutzler RK, Matulonis U, Wickens M, Tutt A: Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010, 376: 245-251. 10.1016/S0140-6736(10)60893-8.
Isakoff SJ, Overmoyer B, Tung NM, Gelman RS, Giranda VL, Bernhard KM, Habin KR, Ellisen LW, Winer EP, Goss PE: A phase II trial of the PARP inhibitor veliparib (ABT888) and temozolomide for metastatic breast cancer [abstract]. J Clin Oncol. 2010, 28 (Suppl): 1019-
Dent RA, Lindeman GJ, Clemons M, Wildiers H, Chan A, McCarthy NJ, Singer CF, Lowe ES, Kemsley K, Carmichael J: Safety and efficacy of the oral PARP inhibitor olaparib (AZD2281) in combination with paclitaxel for the first-or second-line treatment of patients with metastatic triple-negative breast cancer: Results from the safety cohort of a phase I/II multicenter trial [abstract]. J Clin Oncol. 2010, 28 (Suppl): 1018-
O'Shaughnessy J, Osborne C, Pippen J, Yoffe M, Patt D, Monaghan G, Rocha C, Ossovskaya V, Sherman B, Sammons B: Efficacy of BSI-201, a poly (ADP-ribose) polymerase-1 (PARP1) inhibitor, in combination with gemcitabine/carboplatin (G/C) in patients with metastatic triple-negative breast cancer (TNBC): results of a randomized phase II trial. J Clin Oncol. 2009, 27 (18s): 3-
O'Shaughnessy J, Osborne C, Pippen JE, Yoffe M, Patt D, Rocha C, Koo IC, Sherman BM, Bradley CR: Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N Engl J Med. 2011, 364: 205-214. 10.1056/NEJMoa1011418.
Gelmon KA, Hirte HW, Robidoux A, Tonkin KS, Tischkowitz M, Swenerton K, Huntsman D, Carmichael J, MacPherson E, Oza AM: Can we define tumors that will respond to PARP inhibitors? A phase II correlative study of olaparib in advanced serous ovarian cancer and triple-negative breast cancer [abstract]. J Clin Oncol. 2010, 28 (Suppl): 3002-
Chambon P, Weil JD, Mandel P: Nicotinamide mononucleotide activation of a new DNA-dependent polyadenylic acid sythesizing nuclear enzyme. Biochem Biophys Res Commun. 1963, 11: 39-43. 10.1016/0006-291X(63)90024-X.
Ame J, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, Muller S, Hoger T, Murcia J, de Murcia G: PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem. 1999, 274: 17860-17868. 10.1074/jbc.274.25.17860.
Sodhi RK, Singh N, Jaggi AS: Poly(ADP-ribose) polymerase-1 (PARP-1) and its therapeutic implications. Vascul Pharmacol. 2010, 53: 77-87. 10.1016/j.vph.2010.06.003.
Heale JT, Ball AR, Schmiesing JA, Kim J, Kong X, Zhou S, Hudson DF, Earnshaw WC, Yokomori K: Condensin I interacts with the PARP-1-XRCC1 complex and functions in DNA single-strand break repair. Mol Cell. 2006, 21: 837-848. 10.1016/j.molcel.2006.01.036.
Fortini P, Pascucci B, Parlatini E, Errico MD, Dogliotti E: The base excision repair: mechanisms and its relevance for cancer susceptibility. Biochimie. 2003, 85: 1053-1071. 10.1016/j.biochi.2003.11.003.
Dantzer F, Schreiber V, Niedergang C, Trucco C, Flatter E, De La Rubia G, Oliver J, Rolli V, Menissier-de Murcia J, de Murcia G: Involvement of poly(ADP-ribose) polymerase in base excision repair. Biochimie. 1999, 81: 69-75. 10.1016/S0300-9084(99)80040-6.
Rouleau M, Hendzel MJ, Kaufmann SH, Poirier GG: PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010, 10: 293-301. 10.1038/nrc2812.
Bowman KJ, Newell DR, Calvert AH, Curtin NJ: Differential effects of the poly(ADP-ribose) polymerase (PARP) inhibitor NU1025 on topoisomerase I and II inhibitor cytotoxicity in L1210 cells in vitro. Br J Cancer. 2001, 84: 106-112. 10.1054/bjoc.2000.1555.
Canan Koch SS, Thoresen LH, Tikhe JG, Maegley KA, Almassy RJ, Li J, Yu XH, Zook SE, Kumpf RA, Zhang C, Boritzki TJ, Mansour RN, Zhang KE, Ekker A, Calabrese CR, Curtin NJ, Kyle S, Thomas HD, Wang LZ, Calvert AH, Golding BT, Griffin RJ, Newell DR, Webber SE, Hostomsky Z: Novel tricyclic poly(ADP-ribose) polymerase-1 inhibitors with potent anticancer chemopotentiating activity: design, synthesis, and X-ray cocrystal structure. J Med Chem. 2002, 45: 4961-4974. 10.1021/jm020259n.
Jones C, Plummer ER: PARP inhibitors and cancer therapy - early results and potential applications. Br J Radiol. 2008, 81: S2-S5. 10.1259/bjr/30872348.
Plummer R, Jones C, Middleton M, Wilson R, Evans J, Olsen A, Curtin N, Boddy A, McHugh P, Newell D, Harris A, Johnson P, Steinfeldt H, Dewji R, Wang D, Robson L, Calvert H: Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. 2008, 14: 7917-7923. 10.1158/1078-0432.CCR-08-1223.
Tentori L, Portarena I, Graziani G: Potential clinical applications of poly(ADP-ribose) polymerase (PARP) inhibitors. Pharmacol Res. 2002, 45: 73-85. 10.1006/phrs.2001.0935.
Graziani G, Szabo C: Clinical perspectives of PARP inhibitors. Pharmacol Res. 2005, 52: 109-118. 10.1016/j.phrs.2005.02.013.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T: Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005, 434: 913-917. 10.1038/nature03443.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A: Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005, 434: 917-921. 10.1038/nature03445.
Tutt A, Ashworth A: The relationship between the roles of BRCA genes in DNA repair and cancer predisposition. Trends Mol Med. 2002, 8: 571-576. 10.1016/S1471-4914(02)02434-6.
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JH, de Bono JS: Inhibition of Poly(ADP-Ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009, 361: 123-134. 10.1056/NEJMoa0900212.
Edwards S, Brough R, Lord C, Natrajan R, Vatcheva R, Levine D, Boyd J, Reis-Filho J, Ashworth A: Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008, 451: 1111-1115. 10.1038/nature06548.
Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, Urban N, Taniguchi T: Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008, 451: 1116-1120. 10.1038/nature06633.
Sandhu SK, Wenham RM, Wilding G, McFadden M, Sun L, Toniatti C, Stroh M, Carpenter CL, de Bono JS, Schelman WR: First-in-human trial of a poly(ADP-ribose) polymerase (PARP) inhibitor MK-4827 in advanced cancer patients (pts) with antitumor activity in BRCA-deficient and sporadic ovarian cancers [abstract]. J Clin Oncol. 2010, 28 (Suppl): 3001-
Quinn JA, Desjardins A, Weingart J, Brem H, Dolan ME, Delaney SM, Vredenburgh J, Rich J, Friedman AH, Reardon DA, Sampson JH, Pegg AE, Moschel RC, Birch R, McLendon RE, Provenzale JM, Gururangan S, Dancey JE, Maxwell J, Tourt-Uhlig S, Herndon JE, Bigner DD, Friedman HS: Phase I trial of temozolomide plus O6-benzylguanine for patients with recurrent or progressive malignant glioma. J Clin Oncol. 2005, 23: 7178-7187. 10.1200/JCO.2005.06.502.
Ranson M, Middleton MR, Bridgewater J, Lee SM, Dawson M, Jowle D, Halbert G, Waller S, McGrath H, Gumbrell L, McElhinney RS, Donnelly D, McMurry TB, Margison GP: Lomeguatrib, a potent inhibitor of O6-alkylguanine-DNA-alkyltransferase: phase I safety, pharmacodynamic, and pharmacokinetic trial and evaluation in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. 2006, 12: 1577-1584. 10.1158/1078-0432.CCR-05-2198.
Schilsky RL, Dolan ME, Bertucci D, Ewesuedo RB, Vogelzang NJ, Mani S, Wilson LR, Ratain MJ: Phase I clinical and pharmacological study of O6-benzylguanine followed by carmustine in patients with advanced cancer. Clin Cancer Res. 2000, 6: 3025-3031.
Kummar S, Chen AP, Ji JJ, Allen D, Egorin MJ, Gandara DR, Lenz H, Morgan R, Newman EM, Doroshow JH: A phase I study of ABT-888 (A) in combination with metronomic cyclophosphamide (C) in adults with refractory solid tumors and lymphomas [abstract]. J Clin Oncol. 2010, 28 (Suppl): 2605-
Plummer R, Lorigan P, Evans J, Steven N, Middleton M, Wilson R, Snow K, Dewji R, Calvert H: First and final report of a phase II study of the poly(ADP-ribose) polymerase (PARP) inhibitor, AGO14699, in combination with temozolomide (TMZ) in patients with metastatic malignant melanoma (MM). J Clin Oncol. 2006, 24: 456S-456S.
Kummar S, Ji J, Zhang Y, Simmons D, Kinders R, Gutierrez ME, Allen D, Homeffer Y, Juwara L, Rubinstein L, Parchment RE, Murgo AJ, Chen A, Tomaszewski JE, Doroshow JH: A phase I combination study of ABT-888 and topotecan hydrochloride in adults with refractory solid tumors and lymphomas. Ann Oncol. 2009, 20: 42-44.
Giaccone G, Rajan A, Kelly RJ, Gutierrez M, Kummar S, Yancey M, Ji JJ, Zhang L, Parchment RE, Doroshow JH: A phase I combination study of olaparib (AZD2281; KU-0059436) and cisplatin (C) plus gemcitabine (G) in adults with solid tumors [abstract]. J Clin Oncol. 2010, 28 (Suppl): 3027-
Mahany J, Lewis N, Heath E, LoRusso P, Mita M, Rodon J, Tolcher A, Sherman B, Bradley C, Papadopoulos K: A phase IB study evaluating BSI-201 in combination with chemotherapy in subjects with advanced solid tumors. J Clin Oncol. 2008, 26 (Suppl): 3579-
Bartsch R, Ziebermayr R, Zielinski CC, Steger GG: Triple-negative breast cancer. Wien Med Wochenschr. 2010, 160: 174-181. 10.1007/s10354-010-0773-6.
Kopetz S, Mita M, Mok I, Sankhala K, Moseley J, Sherman B, Bradley C, Tolcher A: First in human phase I study of BSI-201, a small molecule inhibitor of poly ADP-ribose polymerase (PARP) in subjects with advanced solid tumors. J Clin Oncol. 2008, 26 (Suppl): 3577-
Blakeley JO, Ye X, Grossman SA, Mikkelsen T, Rosenfeld MR, Bradley CR, Eichler AF, Nabors LB, Desideri S, Supko JG: Poly (ADP-ribose) polymerase-1 (PARP1) inhibitor BSI-201 in combination with temozolomide (TMZ) in malignant glioma [abstract]. J Clin Oncol. 2010, 28 (Suppl): 2012-
Sanofi-aventis Reports Top-line Results from Phase III Study with Iniparib (BSI-201) in Metastatic Triple-Negative Breast Cancer. [http://sanofi-aventis.mediaroom.com/index.php?s=43&item=310]
Acknowledgements
During the preparation of this review article, the author received editorial assistance from Succinct Healthcare Communications, Amersham, UK. This assistance, which comprised copy editing and formatting, was supported through an educational grant from Sanofi-Aventis, which had no input into the content. The opinions expressed are the author's alone.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author is an inventor on a patent of use of the PARP inhibitor PF0367338 and has received research funding from Pfizer GRD for the development of this agent. Additionally, she has received funding to cover the costs of clinical trials activity relating to AZD2281, ABT888 and BSI-201 (iniparib) and laboratory research funding for pharmacodynamic research with BSI-201.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Plummer, R. Poly(ADP-ribose) polymerase inhibition: a new direction for BRCAand triple-negative breast cancer?. Breast Cancer Res 13, 218 (2011). https://doi.org/10.1186/bcr2877
Published:
DOI: https://doi.org/10.1186/bcr2877