
Abstract
Breast cancer frequently metastasizes to the skeleton, interrupting the normal bone remodeling process and causing bone degradation. Osteolytic lesions are the end result of osteoclast activity; however, osteoclast differentiation and activation are mediated by osteoblast production of RANKL (receptor activator for NFκB ligand) and several osteoclastogenic cytokines. Osteoblasts themselves are negatively affected by cancer cells as evidenced by an increase in apoptosis and a decrease in proteins required for new bone formation. Thus, bone loss is due to both increased activation of osteoclasts and suppression of osteoblasts. This review summarizes the current understanding of the osteolytic mechanisms of bone metastases, including a discussion of current therapies.
Similar content being viewed by others


Introduction
Breast cancer frequently metastasizes to the skeleton. It is estimated that 85% of individuals with advanced disease harbor bone metastases [1]. While ductal carcinoma in situ detected early is 98% curable, bone metastases are basically incurable [2]. Metastatic cancer cells tend to colonize the heavily vascularized areas of the skeleton, such as the red marrow of the long bones, sternum, pelvis, ribs and vertebrae, where they disrupt not only bone physiology but also hematopoiesis and the immune system [3].
Metastases leading to overall bone loss are classified as osteolytic. Those leading to excess bone deposition are considered osteoblastic. However, both bone degradation and deposition likely occur early in the metastatic process. The majority of breast cancer metastases ultimately cause bone loss. The clinical outcomes of bone pain, pathologic fractures, nerve compression syndrome, and metabolic disturbances leading to hypercalcemia and acid/base imbalance severely reduce the quality of life [3].
In the 1960s and 70s it was proposed that bone degradation might result from the physical pressure of the tumor on the bone and/or direct resorption of the bone by tumor cells. It was also noted that tumor cells caused other cells in the bone (for example, lymphocytes) to produce molecules such as prostaglandins (PGs) that can affect bone [4]. While there is evidence that the breast cancer cell matrix metalloproteinases (MMPs) can resorb bone in vitro and contribute to bone degradation in vivo [5], it is now well accepted that osteoclasts are largely responsible for osteolytic metastatic lesions [6].
Bone remodeling
Bone provides support and protects vital organs but also is a metabolically active tissue. It is a reservoir of numerous growth factors as well as calcium and phosphorous, which are released from the matrix during bone remodeling. Cortical bone provides strength and protection while trabecular bone is the most metabolically active. Trabecular bone is the major site of bone turnover under normal conditions and in diseases of bone loss or formation.
The skeleton is constantly undergoing remodeling. Even in adults it is estimated that about 10% of the bone is renewed each year [7]. The normal processes of bone resorption and formation are remarkably well balanced. In the young adult, bone mass reaches its peak, but with increasing age there is a slow loss of mass. This loss is more precipitous in women, due to the decrease in estrogen at menopause [3]. However, the presence of metastatic breast cancer cells or other bone metastatic cancers, such as prostate, lung, renal, and myeloma, accelerates the remodeling process and disturbs the balance between bone depositing cells, osteoblasts, and bone degrading cells, osteoclasts. It is impossible to understand the growth and progression of cancer cells in the bone marrow without consideration of the interaction between osteoblasts and osteoclasts. A thorough review of bone remodeling is beyond the scope of this article, and there are several excellent, recent reviews [8, 9]. However, the process is described in brief in order to further consider the mechanisms of osteolytic metastasis.
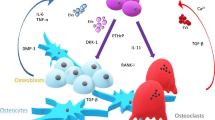
Bone remodeling is often described as a cycle beginning with bone degradation and ending with bone deposition (Figure 1A). This process is effected by osteoblasts and osteoclasts within a functional and anatomic unit known as the basic multicellular unit (BMU). Cells of the osteoblast lineage are derived from mesenchymal stem cells, and are represented in this unit by osteoblasts, bone lining cells and osteocytes. Bone lining cells appear microscopically as relatively undifferentiated cells that line the bone. Their function is not clear except that their retraction is necessary for bone resorption to begin [10]. Osteocytes are terminally differentiated osteoblasts that become embedded in the bone matrix at the end of the deposition phase of remodeling. Once osteoblasts finish bone deposition, they undergo apoptosis, remain in the matrix as osteocytes or revert to thin bone-lining cells.

The bone microenvironment. (A) The bone microenvironment under conditions of normal bone remodeling; (B) and in the presence of osteolytic bone metastases. (A) The bone remodeling unit consists of osteoblasts, which produce osteoid, bone matrix, and osteoclasts, which degrade mineralized bone. Osteoblasts derive from mesenchymal stem cells in the marrow under control of Runx2, a key osteoblastic transcription factor. Osteoclasts derive from mononuclear myeloid precursors that fuse to form pre-osteoclasts. Under the influence of macrophage colony-stimulating factor (M-CSF) and RANKL (receptor activator for NFκB ligand) produced by osteoblasts and other cells in the microenvironment, pre-osteoclasts differentiate into multinuclear, activated osteoclasts that adhere to the bone and begin matrix degradation. Osteoblasts also produce osteoprotegerin (OPG), a decoy receptor to RANKL. The ratio of RANKL to OPG determines the extent of the osteoclast activity and bone degradation. Other cells of the osteoblastic lineage include bone lining cells and osteocytes. (B) Metastatic breast cancer cells in the bone microenvironment secrete parathyroid hormone-related protein (PTHrP), cytokines and growth factors that negatively impact osteoblast function. RANKL and other pro-osteoclastogenic cytokines are increased with a concomitant reduction in OPG, resulting in more osteoclast formation and bone degradation. Osteoblast differentiation is suppressed; new osteoid production is no longer able to keep pace with bone resorption. Current therapeutic targets are indicated in green. Bisphosphonates binding to hydroxyapatite are ingested by osteoclasts and cause their apoptosis. These drugs may also cause cancer cell death; however, they may also negatively affect osteoblasts. Denosumab is an antibody directed to RANKL that prevents osteoclast differentiation. Teriparatide is a recombinant peptide of parathyroid hormone that stimulates osteoblast activity and bone formation. In addition, pre-clinical trials with agents that target cathepsin K, certain matrix metalloproteinases (MMPs), and transforming growth factor (TGF)-β are underway. IGF, insulin-like growth factor; MCP-1, monocyte chemotactic protein-1; PDGF, platelet-derived growth factor; VEGF, vascular endothelial growth factor.
Osteoclasts derive from hematopoietic stem cells. Cells of the monocyte-macrophage lineage are stimulated to form osteoclast progenitor cells. These cells fuse to form multinucleated, but non-functional pre-osteoclasts. Further stimulation results in large multinuclear cells capable of bone resorption.
What initiates remodeling in the non-tumor-containing bone? There are many suspected factors, such as microfractures, loss of mechanical loading, hormones, cytokines, calcium levels and inflammation. Osteocytes may act as mechanosensing cells and initiate the process when microfractures and loading are involved. In the context of the current discussion, cancer cells may initiate the process. The resorption phase of the process begins with recruitment of pre-osteoclasts that differentiate into activated osteoclasts under the direction of osteoblasts (Figure 1A). Osteoblasts produce macrophage colony stimulating factor (M-CSF) and receptor activator of NFκB ligand (RANKL), which bind to their respective receptors, c-fms and RANK, on pre-osteoclasts to bring about osteoclast differentiation and activation. Osteo-blasts also produce osteoprotegerin (OPG), a decoy receptor to RANKL that curtails osteoclast activation. Thus, the ratio of RANKL to OPG is critical for osteoclast activation. Once activated the large multinucleated osteoclasts attach to the bone surface creating a resorption lacuna, a sealed zone in which acid and proteolytic enzymes, such as cathepsin K, are released and degrade the bone matrix. This area has been likened to an extracellular lysosome [11]. The osteoclasts work as part of the bone remodeling compartment, underneath a canopy of bone lining cells. In the next step, preosteoblasts are recruited from the mesenchymal stem cell population and differentiate into osteoblasts. They follow the osteoclasts, reforming the bone matrix. Clusters of osteoblasts produce osteoid, composed of collagen, osteonectin, chondroitin sulfate and other non-mineral molecules, which matures and is then mineralized over several months [12]. This remarkable process of bone degradation and formation is synchronized by direct cell contact and a variety of secreted factors (Table 1). The presence of tumor cells in the bone microenvironment perturbs the balance between osteoblasts and osteoclasts, leading to excess bone loss or formation. Here we discuss some of the proposed mechanisms that contribute to metastatic breast cancer-induced bone loss.
Osteoclasts and the vicious cycle model of bone loss
The entry of breast cancer cells into the bone micro-environment synergistically increases the complexity of cell-cell interactions. A working model to describe the bone remodeling compartment in the presence of metastatic cancer cells has been referred to as the 'vicious cycle of bone metastasis' [13] (Figure 1B). There are many excellent reviews describing this paradigm [14–17] from its inception in the 1990 s. The minimal essential components are osteoblasts, osteoclasts, tumor cells and the mineralized bone matrix. According to this paradigm, the tumor cells produce a variety of growth factors, most notably parathyroid hormone-related protein (PTHrP) [18]. The role of PTHrP in bone metabolism is not fully understood, but it is known to cause upregulation of RANKL and downregulation of OPG [19], thus enhancing osteoclast function leading to bone degradation. In the process, growth factors stored in the matrix, such as transforming growth factor (TGF)-β, vascular endothelial growth factor (VEGF), insulin-like growth factors (IGFs), bone morphogenic proteins and fibroblast-derived factors, as well as calcium, are released into the bone microenvironment. These factors can stimulate the tumor cells to proliferate and produce more growth factors and more PTHrP, further perpetuating the vicious cycle of bone metastasis.
In reality the system is much more complex (Table 1). Cytokines such as IL-6, IL-8 and IL-11 secreted by breast cancer cells also promote osteoclast differentiation and bone resorption. IL-11, normally produced by bone marrow stromal cells and osteoblasts, is an important regulator of hematopoiesis and a potent promoter of osteoclast formation. In addition, its expression is enhanced in the presence of TGF-β [20]. IL-8, a proinflammatory CXC chemokine, is secreted by monocytes, endothelial cells and osteoblasts. It can activate osteoclasts independent of RANKL [21]. Cancer cells also can elicit an increase in osteoblast production of several other osteoclastogenic cytokines, such as monocyte chemotactic protein-1 (MCP-1) and IL-6, IL-8 and TNF [22].
TGF-β is well-known for its role in osteolytic bone metastasis. It can activate both Smad-dependent and Smad-independent signal pathways to induce preosteolytic factors such as PTHrP [23]. Because of its significant role, TGF-β has been a tempting therapeutic target. Ganapathy and colleagues [24] found that TGF-β antagonists are able to reduce bone metastasis and the number and activity of differentiated osteoclasts [24]. However, because TGF-β plays a more global role in cell proliferation and differentiation, its utility as a therapeutic may be limited.
The importance of osteoblasts in osteolytic breast cancer metastasis
Just as osteoblasts are a critical partner in normal bone remodeling, they are vital to the metastatic osteolytic process. Because osteoblasts secrete both RANKL and OPG, they are major mediators of osteoclastogenesis [25]. Current therapies consist of blocking osteoclast activity as a means of disrupting the vicious cycle. Bisphosphonates such as zoledronic acid (Zoledronate™) bind to hydroxyapatite of the bone matrix and are ingested by osteoclasts, which then undergo apoptosis. However, this approach has not entirely solved the problem. Administration of bisphosphonates may slow osteolytic lesion progression and stabilize or increase overall bone density, but does not bring about healing [1, 16, 26]. There is evidence in both humans and animals that bone loss in osteolytic metastasis is partly due to the failure of the osteoblasts to produce new osteoid for the bone matrix.
The hypoactivity of osteoblasts has been known for some time in multiple myeloma. This is a disease of clonal malignancy of terminally differentiated plasma cells that accumulate in the bone marrow. It is estimated that osteolytic lesions occur in 60 to 95% of myeloma patients [1, 27]. In advanced disease, bone formation is essentially absent, and the processes of bone resorption and formation become uncoupled. Myeloma cells produce factors that upregulate osteoblast production of M-CSF and RANKL and downregulate production of OPG. Myeloma cells may also produce RANKL and directly affect osteoclasts [28]. The mechanisms for suppressed osteoblast activity are not clear but Dickkopf-1 (DKK1), an inhibitor of Wnt signaling, is believed to inhibit osteoblast differentiation [29]. Other molecules made by multiple myeloma cells, such as IL-3, IL-7 and soluble frizzle-related protein-2, also inhibit osteoblast differentiation [27]. Furthermore, Pozzi and colleagues [30] have recently reported that high doses of zoledronic acid, the current standard therapeutic for most osteolytic diseases, may also negatively affect osteoblast differentiation.
Recently, we have found that metastatic breast cancer cells have profound effects on osteoblasts in culture [22] and in animals [31, 32]. Metastatic breast cancer cells or their conditioned media increase osteoblast apoptosis, and suppress osteoblast differentiation and expression of proteins required for new bone matrix formation. Neutralization of TGF-β in conditioned medium from human metastatic MDA-MB-231 breast cancer cells permitted the differentiation of osteoblasts in culture, suggesting that TGF-β negatively affects osteoblasts while promoting growth of the metastatic cells [33]. In the presence of cancer cells, osteoblasts increase expression of pro-inflammatory cytokines such as IL-6, monocyte chemotactic protein-1 (MCP-1), macrophage inflammatory protein-2 (MIP-2; GRO alpha human), keratinocyte chemoattractant (KC; IL-8 human) and VEGF. These molecules not only help support tumor cells, but also are osteoclastogenic. When the bone loss is extensive, the osteoblasts are absent from the lesion [32]. Thus, in the course of the osteolytic process, the osteoblasts are unable to fulfill their role as bone building cells.
Breast cancer is often compared with prostate cancer, which metastasizes to the skeleton with a similar frequency. In contrast to breast cancer, prostate bone metastasis often results in osteoblastic lesions. While the outcome is predominantly osteoblastic, it is known that prostate cancer lesions display both blastic and lytic characteristics early in the process. There is evidence that osteoblastic metastases form at sites of osteolytic lesions, suggesting an overall increase of bone remodeling Accelerated osteoblastogenesis can be stimulated by factors secreted by prostate cancer cells, such as endothelin-1, TGF-β, and fibroblast growth factor (FGF) [1]. These molecules cause osteoblasts not only to form new bone but also to release RANKL and other osteoclastic mediators. Although the mechanisms of osteoteoblastic and osteolytic responses are not fully understood, it is clear that many factors involved in osteolytic breast cancer bone metastasis also regulate the osteolytic aspects of prostate cancer. Akech and colleagues [34] recently reported that Runx2 (Runt-related transcription factor 2) is produced by the highly metastatic prostate cancer cell PC-3, and positively correlates to the severity of osteolytic disease. There is also evidence that molecules in conditioned medium from PC-3 cells alone [34], or from both PC-3 cells and MC3T3-E1 osteoblasts [35], promote osteoclastogenesis. While some of the growth factors produced by breast and prostate cancers may be different, ultimately they engage the bone re-modeling process.
The bone remodeling microenvironment is a complex system in which the cell functions are controlled by multifunctional transcription factors, cytokines and growth factors. The dynamics of this system are interrupted when metastatic breast cancer cells are introduced, adding another layer of active molecules to the bone environment. In the section that follows, we will discuss in greater detail the key factors involved in metastatic breast cancer osteolysis. While they are categorized into functional groups, it should be noted that many of these factors are multifunctional and must be considered within the context of the bone remodeling system as a whole.
Cancer cell survival in the bone microenvironment
Osteomimicry
It has been suggested that cancer cells preferentially metastasize to bone due to their ability to express genes that are normally considered bone or bone-related [36]. In doing so, cancer cells are equipped to home, adhere, survive and proliferate in the bone microenvironment. Osteomimetic factors include osteopontin (OPN), osteocalcin, osteonectin, bone sialoprotein, RANKL and PTHrP. Several of these molecules are related to the recruitment and differentiation of osteoclasts; some are prominent players in the vicious cycle. For example, OPN is produced by many breast cancer cells and has a strong clinical correlation with poor prognosis and decreased survival [37]. It can contribute to tumor cell survival, proliferation, adhesion, and migration. In the bone, OPN is involved in the differentiation and activity of osteoclasts, and inhibition of mineral deposition in the osteoid [37]. The results of an in vivo study showed that OPN-deficient mice showed significantly reduced bone metastasis [38].
Runx2 expression
Interestingly, many osteomimetic factors are regulated by the same transcription factor, Runx2, considered to be the major regulator of osteoblast commitment and differentiation [39]. It is required to drive mesenchymal cells to become osteoblasts. Dysfunctional Runx2 results in the developmental arrest of osteoblasts and inhibition of osteogenesis. Runx2 downregulates proliferation and induces p21, RANKL, MMP2, MMP9, MMP13, VEGF, OPN, bone sialoprotein and PTHrP protein expression to promote osteoblast differentiation, bone development and turnover [39].
It has also been suggested that Runx2 is ectopically expressed in bone-destined metastatic breast cancer cells. Evidence from an intratibial bone metastasis model indicates that when highly aggressive metastatic MDA-MB-231 cells express dysfunctional Runx2 or small hair-pin RNA for Runx2, both osteoclastogenesis and osteolytic lesions decrease [40]. These results signify an important role for cancer cell-derived Runx2 in the osteolytic process. Recent research has revealed how cancer cell Runx2 affects other cells in the bone microenvironment and promotes osteolysis. Pratap and colleagues [40] found that Runx2 responds to TGF-β stimulation by activating the expression of Indian hedgehog (IHH), which further increases the level of PTHrP. Thus, Runx2 plays a significant role in the vicious cycle via TGF-β-induced IHH-PTHrP pathways in breast cancer cells, resulting in increased osteoclastogenesis and osteolysis.
Regulatory factors of the RANKL pathway
RANKL clearly holds the key to the osteolytic process. In fact, a new drug, denosumab (Prolia™), a fully human monoclonal antibody to RANKL, has been approved by the US Food and Drug Administration (FDA) for the treatment of postmenopausal women with high risk of osteoporotic fractures, and is under priority review for patients with bone metastases. Osteoblasts and bone stromal cells can respond to a variety of substances that upregulate RANKL. PTH/PTHrP, TNF-α, prostaglandins (PGE2), IL-1, IL-11, FGF-2, and IGF-1 have been reported to increase RANKL production. Cells of the immune system, T cells and dendritic cells can also express RANKL. In this context, RANKL increases in the presence of inflammatory agents from infectious organisms, such as lipopolysaccharide, CpGpDNA and viral double-stranded DNA [41]. Several of these RANKL inducers merit further discussion with respect to metastatic breast cancer-induced osteolysis.
Parathyroid hormone-related protein
PTHrP, one of many proteins controlled by Runx2, is a major effector in breast cancer bone metastasis progression and bone loss. It is common to find increased PTHrP serum levels in breast cancer patients. PTHrP is expressed in the primary tumors of about 50% of patients and in more than 90% of breast cancer bone metastasis samples [18]. In the late 1980 s, PTHrP was linked to hypercalcemia in several cancers, providing evidence that PTHrP was involved in bone resorption. Guise [18] demonstrated that increasing the expression of PTHrP in cancer cells enhanced osteolytic lesions in vivo, while decreasing the expression reduced the number and size of lesions. However, PTHrP does not directly stimulate osteoclast differentiation, but rather stimulates other cells to increase RANKL and decrease OPG production. In addition, factors such as TGF-β and IGFs that are released from the bone matrix during degradation serve to increase PTHrP expression in breast cancer cells. All in all, PTHrP is an important mediator between breast cancer cells and cells of the bone microenvironment and, as such, is a major contributor to the bone degradation process.
COX-2 and prostaglandins
The cyclooxygenase enzymes COX-1 and COX-2 catalyze the conversion of arachidonic acid to prostaglandins and thromboxanes. While COX-1 is constitutively expressed in most tissues, COX-2 expression appears to be limited to brain, kidney, bone, reproductive organs and some neoplasms. PGs produced from this arachidonic acid conversion are both autocrine and paracrine factors that help to govern physiologic homeostasis. Of the many prostaglandins, PGE2 is known to play a critical role in cancer progression. PGE2 is associated with inflammation, cell growth, tumor development and metastasis [42].
In the early 1970 s it was reported that prostaglandins could resorb fetal bone in culture [43], and that aspirin, a COX-1 inhibitor, and indomethacin, a COX-2 inhibitor, could prevent osteolysis in tissue culture [44]. These findings led to a flurry of studies to develop COX and prostaglandin inhibitors as cures for bone metastasis. It is now known that PGE2 signaling through its receptor EP4 plays a crucial role in osteolysis by inducing monocytes to form mature osteoclasts. In a series of in vitro, ex vivo and in vivo experiments, Ohshiba and colleagues [45] demonstrated that direct cell-cell contact between breast cancer cells and osteoblasts caused an increase in COX-2 expression in the osteoblasts due to activation of the NFκB/mitogen-activated protein (MAP) kinase pathway. This increase in COX-2 results in increased secretion of PGE2, which binds to EP4 receptors on the surface of the osteoblasts. The receptor binding activity in turn causes an increase in production of RANKL. The PGE2-mediated production of RANKL induces osteoclastogenesis via RANK.
NF-κB/MAP-kinase inhibitors (SN50, PD98059 and SB203580), COX-2 inhibitors (indomethacin) and EP4 receptor decoy [46] all result in a down-regulation of RANKL production and a concomitant decrease in osteoclastogenesis. COX-2 activity in breast cancer cells has also been found to modulate the expression and activity of MMPs. In the highly metastatic, COX-2-expressing breast cancer cell line Hs578T, treatment with the selective COX-2 inhibitor Ns-398 markedly decreased the production of MMP1, 2, 3, and 13 in a dose-dependent manner. COX-2 inhibition also partially attenuated the ability of two breast cancer cell lines to degrade and invade extracellular matrix components such as laminin and collagen [47].
Extracellular matrix metalloproteinase inducer
A newly discovered molecule downstream of RANKL is extracellular matrix metalloproteinase inducer (EMMPRIN)/CD147, a cell surface glycoprotein that is known to induce MMPs and VEGF [48]. While EMMPRIN is produced normally during tissue remodeling, it increases during tumor progression and metastasis. This molecule is also produced by metastatic breast cancer cells [49]. Increased production of EMMPRIN in turn leads to increases in VEGF and MMPs. Both RANKL and VEGF can induce osteoclast formation [48], and MMPs play a role in bone matrix degradation.
Extracellular matrix degradation and released matrix factors
Matrix metalloproteinases/cathepsin K
The MMPs are considered to be important in the bone metastatic process. In a recent comprehensive review article, Lynch [50] presents the case that they are 'master regulators' of the vicious cycle. As might be expected from the nature of the osteolytic process, that is, the degradation of bone, the microenvironment contains many proteases. Among these are the MMPs. The MMP family, composed of more than 20 members, can collectively degrade all components of the extracelluar matrix. Nevertheless, they do not appear to function in the osteoclast resorption lacuna, probably due to the low pH in this compartment. Cathepsin K is believed to be the major protease in this capacity. However, the MMPs may be involved in matrix remodeling once the osteoclasts are finished. Orr and colleagues [5] have determined MMPs sufficient to resorb bone in vitro and to contribute to the process in vivo. Matrix degradation appears to be only one of the roles of MMPs. They also are regulators of other molecules important in the vicious cycle. Kang and colleagues [20] found that expression of two MMP genes, MMP1 and ADAMTS1, discriminated between a subline of osteotropic metastatic MDA-MB-231 cells and the parental line.
Where do the MMPs come from? Cancer cells, osteoblasts, osteoclasts and endothelial cells produce MMPs. In addition, other cells not specific for bone but likely to be found in the bone (macrophages, neutrophils and T lymphocytes) produce MMPs. As pointed out by Lynch, the spatial and temporal expression of these molecules is of utmost importance. This information is not easily obtained with in vitro studies.
Cathepsin K is the major mediator of bone resorption, controlling the osteoclast portion of the vicious cycle. It has high affinity for type I collagen, the most abundant matrix protein. However, cathepsin K is also produced by other cells in the bone microenvironment, such as macrophages and bone marrow stromal cells. One of its substrates is SPARC (secreted protein acidic and rich in cysteine; osteonectin/BM-40) [51]. Proteolytic cleavage of SPARC releases biologically active cleavage products that affect angiogenesis factors such as VEGF, platelet-derived growth factor (PDGF) and FGF-2. SPARC cleavage also coincides with an increase in inflammatory cytokines such as IL-6 and IL-8 [51]. Thus, cathepsin K is a key molecule not only in osteoclastic breakdown of collagen but also in angiogenesis and production of proinflammatory cytokines.
Transforming growth factor-β/insulin-like growth factors/vascular endothelial growth factor
At least three major growth factors sequestered in the matrix are activated by MMPs. TGF-β is one of the most prominent. Several MMPs (MMP2, 3, 9) can release TGF-β from the latent state, allowing it to become active. Active TGF-β is involved in tumor growth, osteoblast retraction from the bone surface, inhibition of osteoblast differentiation [52, 53] and promotion of osteoclast differentiation. Another growth factor sequestered in the matrix is IGF. IGF binding proteins keep this molecule latent. MMP1, 2, 3 process the binding factors and free IGF, allowing it to bind to its receptors found both on osteoblasts and osteoclasts. IGF binding initiates production of M-CSF and RANKL by osteoblasts and c-fms and RANK by osteoclasts [54]. VEGF also forms a complex with the extracellular matrix [31, 55]. MMP-9 is important in the cascade leading to activation of VEGFA. Studies with MMP9-null mice indicate its importance in tumor progression in ovarian cancer, prostate cancer and bone metastasis [56]. While the case for the importance of MMPs as metastasis regulators is strong, they themselves are regulated by tissue inhibitors of metalloproteinase (TIMPs). Furthermore, the molecules activated by MMPs also have counter molecules creating a network of accelerators and decelerators centered around MMPs.
Osteoblast and osteoclast differentiation factors
Platelet-derived growth factor
PDGF is a dimeric protein consisting of two of four possible subunits. It binds to two class III tyrosine kinase receptors, PDGFRα and PDGFRβ, leading to activation of several signaling molecules. PDGF can function as a mitogen for cells of mesenchymal origin and possesses chemoattractant properties, making it an important factor in cell proliferation and migration. At the tissue level, PDGF is involved in bone formation, wound healing, erythropoiesis and angiogenesis as well as tumor growth and lesion development [57].
In normal bone remodeling, osteoclasts secrete PDGF, which acts as a chemoattractant to recruit pre-osteoblasts to the site of bone repair [58]. Many metastatic breast cancer cell lines have been found to also secrete PDGF, which has a strong impact on osteoblast development. In a study by Mercer and Mastro [59], osteoblasts treated with conditioned media from MDA-MB-231 breast cancer cells displayed disorganized F-actin fibrils and reduced focal adhesion plaques. When treated with neutralizing antibody to PDGF, the osteoblasts assumed normal morphology. In addition, PDGF has been shown to inhibit osteoblast differentiation [60], making it an important factor in bone remodeling and the osteolytic bone metastasis.
Placental growth factor
Placental growth factor is a VEGF homologue that binds to the VEGF receptor VEGFR-1. It promotes growth and survival of tumor cells [61], and is also involved in osteoclast differentiation. The use of blocking antibodies to placental growth factor in two xenograft mouse/human models greatly decreased the numbers and size of osteolytic lesions [61]. Surprisingly, this treatment did not affect angiogenesis in the bone. The mechanisms are thought to be inhibition of tumor cell adhesion as well as osteoclast differentiation.
In summary, all of these factors contribute to propagating the vicious cycle and increasing osteolysis (Figure 1B). Osteomimetic factors driven by abnormal Runx2 activation in breast cancer cells may increase their survival in the bone microenvironment. Runx2 also promotes PTHrP expression in breast cancer cells, which in turn stimulates other cells, such as osteoblasts, to produce more RANKL, leading to further osteoclast activation. Meanwhile, COX-2 produced by breast cancer cells and osteoblasts increases the localized PGE2 concentration, which can directly bind to osteoblasts, promoting RANKL expression and further stimulating osteoclast differentiation. Once osteoclasts are activated, they degrade bone matrix through several proteolytic enzymes, including MMPs and cathepsin K. Although cathepsin K is the major bone resorbing protease, MMPs, which are secreted by many cells, may be the 'master regulator' of the entire mechanism. Their multifunctionality demonstrates their importance. MMPs are involved in the bone remodeling process after osteoclasts are finished. They activate latent molecules released from the matrix. At least three essential molecules, TGF-β, IGF, and VEGF, need to be activated by MMPs before they can function. These functional molecules complete the cycle and osteolysis continues. It should be noted that in addition to obvious members of the vicious cycle, other factors are produced during the process, including inflammatory cytokines, which significantly affect tumor cell survival, cell differentiation, and angiogenesis.
Physiological states that exacerbate osteolysis
While not directly responsible for osteolysis in metastatic breast cancer disease, there are physiological parameters that can amplify the degree of bone loss. Clinical studies of newly diagnosed breast cancer patients have revealed that high bone turnover correlates with a higher risk of skeletal complications [62]. For post-menopausal women, high bone turnover may be caused by estrogen deficiency. Estrogen profoundly affects bone remodeling by suppressing production of RANKL while increasing production of OPG. Estrogen also increases osteoblast pro-collagen synthesis and decreases osteoblast apoptosis [63]. In addition, production of inflammatory cytokines (that is, IL-6, TNF-α, M-CSF, IL-1) is suppressed by estrogen [64]. Estrogen has also been shown to promote osteoclast apoptosis and inhibit activation of mature osteoclasts. Unfortunately, some of the therapies used for breast cancer patients may exacerbate the problem. For example, the use of aromatase inhibitors increases the risk for osteoporosis. Chemotherapy may bring about ovarian failure and premature menopause [1].
As primary constituents in bone metabolism, calcium and vitamin D can not be overlooked as critical regulators of osteolysis in bone metastatic breast cancer. In middle aged and elderly women, calcium and/or vitamin D deficiencies are quite common, as is the incidence of breast cancer [65]. Epidemiological studies have also correlated the increase in breast cancer rates with decreasing sunlight exposure. It was recently reported that mice deficient in vitamin D or calcium showed increased metastatic tumor growth and accelerated rates of bone resorption [66, 67]. In light of these findings, correction of calcium and vitamin D deficiencies should be considered as adjuvant therapies in slowing or preventing osteolysis in breast cancer patients.
Chronic inflammation has long been considered a risk factor in cancer initiation [68]. Inflammation associated with bone fractures and arthritic joints has been anecdotally associated with the appearance of bone metastasis, often many years after the primary tumor has been treated. Recently, Roy and colleagues [69] investigated this association in a mouse model of autoimmune arthritis and found that arthritic mice had an increase in both lung and bone metastasis compared to the non-arthritic mice. Thus, inflammation is likely to be important in cancer initiation, metastasis and the resulting osteolysis.
Breaking the vicious cycle
Understanding the mechanisms of osteolysis should be the key to designing the cure. Of course, the best cure for bone metastasis is prevention. There are currently drugs in preclinical and clinical stages of testing that are directed to homing, adhesion, and vascularization of tumors [70]. However, once bone metastasis has occurred, the aim has been to break the osteolytic cycle by targeting osteoclasts. Drugs of the bisphosphonate family have been used for many years as the standard of care. Until recently they were the only FDA approved drugs for metastatic bone disease [71]. These molecules bind to hydroxyapatite of the bone matrix and are ingested by osteoclasts, which then undergo apoptosis. There is evidence that bisphosphonates also contribute to tumor cell death, especially in combination with chemotherapy [72]. There are conflicting reports regarding their effect on osteoblasts. At higher doses they may in fact prevent osteoblast differentiation [30]. Of the bisphosphonates, zoledronic acid is the most potent. Clinical evidence indicates that this drug can reduce the rate of bone loss, but is not curative. It improves the quality of life by preventing fractures but does not prolong life [73]. Denosumab (Prolia™), the latest drug to enter the field, is a monoclonal antibody to RANKL. It inhibits the differentiation of osteoclasts by competitive binding with RANKL. Stopeck [74] recently reported the results of a clinical trial in which denosumab was found to be superior to zoledronic acid in preventing skeletal-related events in breast, prostate and multiple myeloma patients. Denosumab has recently been approved by the FDA for treatment of osteoporosis in women with high risk of fractures and is being considered for treatment of bone metastasis. However, both drugs are associated with low incidence of osteonecrosis of the jaw [75]. Another drug, teriparatide (Forteo™), the amino-terminal 34 amino acids of parathyroid hormone, has been used for many years to treat osteoporosis. Teriparatide, in contrast to bisphosphonates and denosumab, acts on osteoblasts to stimulate bone formation. At first glance it would seem ideal to pair bisphosphonates or denosumab with teriparatide since the former two block bone resorption and the latter stimulates bone deposition. However, teriparatide is associated with an increased risk of osteosarcoma and exacerbation of skeletal metastases because of its effect on bone turnover [75]. Other drugs on the horizon target TGF-β, and cathepsin K. Various approaches, including kinase inhibitors, ligand-neutralizing antibodies and anti-sense molecules, are being investigated [33].
Conclusions and the future
Most breast cancer metastasis to bone results in osteolytic lesions. Despite the role of the osteoclasts in this process, the outcome is due in large part to the impact of cancer cells directly and indirectly on osteoblasts. Induction of aberrant osteoclastogenesis is only part of the equation. Breast cancer cells also cause inhibition of osteoblast differentiation and adhesion, downregulation of collagen synthesis and increased osteoblast apoptosis. Thus, bone loss is the result of excessive bone degradation and insufficient bone replacement. In the final stages of metastatic osteolytic breast cancer disease, the cancer cells, fueled by growth factors released from the degraded matrix, expand unchecked. Eventually, bone remodeling ceases as both osteoblasts and osteoclasts are lost.
What can be done to stop osteolytic metastasis? To date, osteoclasts have been the primary target of drug therapies. Current treatments can improve bone density, decrease skeletal related events and ease bone pain, yet existing bone lesions do not heal. While drugs that inhibit osteoclast differentiation or activity are vital to treating osteolysis, therapies designed to restore osteoblast number and function will be required to fully resolve osteolytic lesions. Part of this uncertainty is because we do not fully understand all of the cell, cytokine and growth factor interactions that occur in the bone microenvironment.
Identification of a stimulator or protector of osteoblasts would be a major improvement in treatment for osteolytic breast cancer as well as other diseases of bone loss. However, there is no guarantee that inhibition of osteolytic lesions would prevent the growth of cancer cells in the bone or their spread to other organs. It is interesting that cancer cells often remain dormant in bone for many years before they begin to grow. Continuing research into the mechanisms of cancer cell dormancy could result in a treatment that would prevent cancer cell proliferation in the bone and the chain of events that leads to osteolysis.
Since the discovery of RANKL and its role in bone remodeling, the field of bone metastasis has moved rapidly. It is now generally accepted that the bone microenvironment is critical to the colonization and growth or dormancy of metastases. Nevertheless, the inaccessibility, opacity and size of the skeleton make it difficult to study even in laboratory animals. Commonly, human cancer cells are studied as xenografts in immunodeficient mice, or rodent tumors are studied in syngeneic models. However, more accessible and defined [76] models are needed. Several groups have developed in vivo models in which bone or bone substitutes are implanted in animals. Retrieval of the bone at specific times gives a snapshot of the status of metastases. For example, a hydroxyapatite scaold pre-loaded with bone morphogenetic protein-2 enhanced the growth rate of mammary tumor cells in the scaold [77]. Fragments of human fetal bone implanted in SCID mice allow one to examine human cancer with human bone [76]. These approaches still rely on animals. Recently we have begun developing an in vitro bioreactor [78]. Using this device, we have been able to grow osteoblasts into a mineralized tissue. Metastastic human breast cancer cells (MDA-MB-231) added to this culture attach, penetrate the tissue and form single cell files characteristic of metastases seen in pathologic tissues. The cancer cells affect osteoblast morphology and extracellular matrix. We are in the process of adding osteoclasts to the system to create a rudimentary in vitro bone remodeling unit. This approach will allow testing of components and drugs in a model less complex than an animal but more relevant than standard tissue culture.
Authors' information
AMM, the senior investigator and corresponding author, has worked in the area of breast cancer metastasis to bone for over 12 years. Along with colleagues and students she has focused particularly on the fate of osteoblasts in the metastatic bone environment. DMS is a senior research technician with many years experience in the bone field. Y-CC is a senior graduate student completing work on the studies of selenium in breast cancer metastasis.
Note
This article is part of a review series on New pathways of metastasis, edited by Lewis Chodosh. Other articles in the series can be found online at http://breast-cancer-research.com/series/metastasis_pathway
Abbreviations
- COX:
-
cyclooxygenase
- EMMPRIN:
-
extracellular matrix metalloproteinase inducer
- FDA:
-
Food and Drug Administration
- FGF:
-
fibroblast growth factor
- IGF:
-
insulin-like growth factor
- IHH:
-
Indian hedgehog
- IL:
-
interleukin
- MAP:
-
mitogen-activated protein
- MCP:
-
monocyte chemotactic protein
- NF:
-
nuclear factor
- M-CSF:
-
macrophage colony-stimulating factor
- MMP:
-
matrix metalloproteinase
- OPG:
-
osteoprotegerin
- OPN:
-
osteopontin
- PDGF:
-
platelet-derived growth factor
- PG:
-
prostaglandin
- PTH:
-
parathyroid hormone
- PTHrP:
-
parathyroid hormone-related protein
- RANK:
-
receptor activator for NFκB
- RANKL:
-
receptor activator for NFκB ligand
- Runx2:
-
Runt-related transcription factor 2
- SPARC:
-
secreted protein acidic and rich in cysteine: osteonectin/BM-40
- TGF:
-
transforming growth factor
- TNF:
-
tumor necrosis factor
- VEGF:
-
vascular endothelial growth factor.
References
Lipton A, Uzzo R, Amato RJ, Ellis GK, Hakimian B, Roodman GD, Smith MR: The science and practice of bone health in oncology: managing bone loss and metastasis in patients with solid tumors. J Natl Compr Canc Netw. 2009, 7 (Suppl 7): S1-29. quiz S30
Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ: Cancer Statistics, 2007. CA Cancer J Clin. 2007, 57: 43-66. 10.3322/canjclin.57.1.43.
Mundy GR: Bone Remodeling and its Disorders. 1999, London: Martin Dunitz Ltd
Raisz LG, Mundy GR, Luben RA: Skeletal reactions to neoplasms. Ann N Y Acad Sci. 1974, 230: 473-475. 10.1111/j.1749-6632.1974.tb14480.x.
Lee J, Weber M, Mejia S, Bone E, Watson P, Orr W: A matrix metalloproteinase inhibitor, batimastat, retards the development of osteolytic bone metastases by MDA-MB-231 human breast cancer cells in Balb C nu/nu mice. Eur J Cancer. 2001, 37: 106-113. 10.1016/S0959-8049(00)00363-4.
Clohisy DR, Perkins SL, Ramnaraine ML: Review of cellular mechanisms of tumor osteolysis. Clin Orthop Relat Res. 2000, 373: 104-114. 10.1097/00003086-200004000-00013.
Lerner UH: Bone remodeling in post-menopausal osteoporosis. J Dent Res. 2006, 85: 584-595. 10.1177/154405910608500703.
Feng X, McDonald JM: Disorders of bone remodeling. Annu Rev Pathol. 2010,
Proff P, Romer P: The molecular mechanism behind bone remodelling: a review. Clin Oral Investig. 2009, 13: 355-362. 10.1007/s00784-009-0268-2.
Zambonin Zallone A, Teti A, Primavera MV: Resorption of vital or devitalized bone by isolated osteoclasts in vitro. The role of lining cells. Cell Tissue Res. 1984, 235: 561-564.
Corisdeo S, Gyda M, Zaidi M, Moonga BS, Troen BR: New insights into the regulation of cathepsin K gene expression by osteoprotegerin ligand. Biochem Biophys Res Commun. 2001, 285: 335-339. 10.1006/bbrc.2001.5127.
Hadjidakis DJ, Androulakis II: Bone remodeling. Ann N Y Acad Sci. 2006, 1092: 385-396. 10.1196/annals.1365.035.
Kozlow W, Guise TA: Breast cancer metastasis to bone: mechanisms of osteolysis and implications for therapy. J Mammary Gland Biol Neoplasia. 2005, 10: 169-180. 10.1007/s10911-005-5399-8.
Guise TA, Mundy GR: Cancer and bone. Endocr Rev. 1998, 19: 18-54. 10.1210/er.19.1.18.
Guise TA, Kozlow WM, Heras-Herzig A, Padalecki SS, Yin JJ, Chirgwin JM: Molecular mechanisms of breast cancer metastases to bone. Clin Breast Cancer. 2005, 5 (Suppl): S46-53. 10.3816/CBC.2005.s.004.
Mundy GR, Sterling JL: Metastatic solid tumors to bone. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Edited by: Rosen CL. 2008, Washington, DC: American Society for Bone and Mineral Research, 374-378. full_text. 7
Mundy GR: Mechanisms of bone metastasis. Cancer. 1997, 80 (8 Suppl): 1546-1556. 10.1002/(SICI)1097-0142(19971015)80:8+<1546::AID-CNCR4>3.0.CO;2-I.
Guise TA: Parathyroid hormone-related protein and bone metastases. Cancer. 1997, 80 (8 Suppl): 1572-1580. 10.1002/(SICI)1097-0142(19971015)80:8+<1572::AID-CNCR7>3.0.CO;2-M.
Karaplis AC, Goltzman D: PTH and PTHrP effects on the skeleton. Rev Endocr Metab Disord. 2000, 1: 331-341. 10.1023/A:1026526703898.
Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, Massague J: A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003, 3: 537-549. 10.1016/S1535-6108(03)00132-6.
Bendre M, Montague DC, Peery T, Akel NS, Gaddy D, Suva LJ: Interleukin-8 stimulation of osteoclastogenesis and bone resorption is a mechanism for the increased osteolysis of metastatic bone disease. Bone. 2003, 33: 28-37. 10.1016/S8756-3282(03)00086-3.
Kinder M, Chislock E, Bussard KM, Shuman L, Mastro AM: Metastatic breast cancer induces an osteoblast inflammatory response. Exp Cell Res. 2008, 314: 173-183. 10.1016/j.yexcr.2007.09.021.
Kingsley LA, Fournier PG, Chirgwin JM, Guise TA: Molecular biology of bone metastasis. Mol Cancer Ther. 2007, 6: 2609-2617. 10.1158/1535-7163.MCT-07-0234.
Ganapathy V, Ge R, Grazioli A, Xie W, Banach-Petrosky W, Kang Y, Lonning S, McPherson J, Yingling JM, Biswas S, Mundy GR, Reiss M: Targeting the transforming growth factor-beta pathway inhibits human basal-like breast cancer metastasis. Mol Cancer. 2010, 9: 122-10.1186/1476-4598-9-122.
Khosla S: Minireview: the OPG/RANKL/RANK system. Endocrinology. 2001, 142: 5050-5055. 10.1210/en.142.12.5050.
Coleman RE, Lipton A, Roodman GD, Guise TA, Boyce BF, Brufsky AM, Clézardin P, Croucher PI, Gralow JR, Hadji P, Holen I, Mundy GR, Smith MR, Suva LJ: Metastasis and bone loss: Advancing treatment and prevention. Cancer Treat Rev. 2010, 36: 615-620. 10.1016/j.ctrv.2010.04.003.
McHayleh W, Ellerman J, Roodman D: Hematologic malignancies and bone. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Edited by: Rosen CL. 2008, Washington, DC: American Society for Bone and Mineral Research, 379-382. full_text. 7
Yang Y, Ren Y, Ramani VC, Nan L, Suva LJ, Sanderson RD: Heparanase enhances local and systemic osteolysis in multiple myeloma by upregulating the expression and secretion of RANKL. Cancer Res. 2010, 70: 8329-8338. 10.1158/0008-5472.CAN-10-2179.
Tian E, Zhan F, Walker R, Rasmussen E, Ma Y, Barlogie B, Shaughnessy JD: The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med. 2003, 349: 2483-2494. 10.1056/NEJMoa030847.
Pozzi S, Vallet S, Mukherjee S, Cirstea D, Vaghela N, Santo L, Rosen E, Ikeda H, Okawa Y, Kiziltepe T, Schoonmaker J, Xie W, Hideshima T, Weller E, Bouxsein ML, Munshi NC, Anderson KC, Raje N: High-dose zoledronic acid impacts bone remodeling with effects on osteoblastic lineage and bone mechanical properties. Clin Cancer Res. 2009, 15: 5829-5839. 10.1158/1078-0432.CCR-09-0426.
Bussard KM, Venzon DJ, Mastro AM: Osteoblasts are a major source of inflammatory cytokines in the tumor microenvironment of bone metastatic breast cancer. J Cell Biochem. 2010,
Phadke PA, Mercer RR, Harms JF, Jia Y, Frost AR, Jewell JL, Bussard KM, Nelson S, Moore C, Kappes JC, Gay CV, Mastro AM, Welch DR: Kinetics of metastatic breast cancer cell trafficking in bone. Clin Cancer Res. 2006, 12: 1431-1440. 10.1158/1078-0432.CCR-05-1806.
Juarez P, Guise TA: TGF-beta in cancer and bone: Implications for treatment of bone metastases. Bone. 2010,
Akech J, Wixted JJ, Bedard K, van der Deen M, Hussain S, Guise TA, van Wijnen AJ, Stein JL, Languino LR, Altieri DC, Pratap J, Keller E, Stein GS, Lian JB: Runx2 association with progression of prostate cancer in patients: mechanisms mediating bone osteolysis and osteoblastic metastatic lesions. Oncogene. 2010, 29: 811-821. 10.1038/onc.2009.389.
Morrissey C, Lai JS, Brown LG, Wang YC, Roudiffer MP, Coleman IM, Gulati R, Vakar-Lopez F, True LD, Corey E, Nelson PS, Vessella RL: The expression of osteoclastogenesis-associated factors and osteoblast response to osteolytic prostate cancer cells. Prostate. 2010, 70: 412-424.
Rucci N, Teti A: Osteomimicry: how tumor cells try to deceive the bone. Front Biosci (Schol Ed). 2010, 2: 907-915. 10.2741/S110.
Standal T, Borset M, Sundan A: Role of osteopontin in adhesion, migration, cell survival and bone remodeling. Exp Oncol. 2004, 26: 179-184.
Elazar V, Adwan H, Bauerle T, Rohekar K, Golomb G, Berger MR: Sustained delivery and efficacy of polymeric nanoparticles containing osteopontin and bone sialoprotein antisenses in rats with breast cancer bone metastasis. Int J Cancer. 2010, 126: 1749-1760.
Marie PJ: Transcription factors controlling osteoblastogenesis. Arch Biochem Biophys. 2008, 473: 98-105. 10.1016/j.abb.2008.02.030.
Pratap J, Wixted JJ, Gaur T, Zaidi SK, Dobson J, Gokul KD, Hussain S, van Wijnen AJ, Stein JL, Stein GS, Lian JB: Runx2 transcriptional activation of Indian Hedgehog and a downstream bone metastatic pathway in breast cancer cells. Cancer Res. 2008, 68: 7795-7802. 10.1158/0008-5472.CAN-08-1078.
Troen BR: Molecular mechanisms underlying osteoclast formation and activation. Exp Gerontol. 2003, 38: 605-614. 10.1016/S0531-5565(03)00069-X.
Lerner UH: Inflammation-induced bone remodeling in periodontal disease and the influence of post-menopausal osteoporosis. J Dent Res. 2006, 85: 596-607. 10.1177/154405910608500704.
Klein DC, Raisz LG: Prostaglandins: stimulation of bone resorption in tissue culture. Endocrinology. 1970, 86: 1436-1440. 10.1210/endo-86-6-1436.
Powles TJ, Clark SA, Easty DM, Easty GC, Neville AM: The inhibition by aspirin and indomethacin of osteolytic tumor deposits and hypercalcaemia in rats with Walker tumour, and its possible application to human breast cancer. Br J Cancer. 1973, 28: 316-321.
Ohshiba T, Miyaura C, Ito A: Role of prostaglandin E produced by osteoblasts in osteolysis due to bone metastasis. Biochem Biophys Res Commun. 2003, 300: 957-964. 10.1016/S0006-291X(02)02937-6.
Takahashi T, Uehara H, Bando Y, Izumi K: Soluble EP2 neutralizes prostaglandin E2-induced cell signaling and inhibits osteolytic tumor growth. Mol Cancer Ther. 2008, 7: 2807-2816. 10.1158/1535-7163.MCT-08-0153.
Larkins TL, Nowell M, Singh S, Sanford GL: Inhibition of cyclooxygenase-2 decreases breast cancer cell motility, invasion and matrix metalloproteinase expression. BMC Cancer. 2006, 6: 181-10.1186/1471-2407-6-181.
Aldridge SE, Lennard TW, Williams JR, Birch MA: Vascular endothelial growth factor acts as an osteolytic factor in breast cancer metastases to bone. Br J Cancer. 2005, 92: 1531-1537. 10.1038/sj.bjc.6602417.
Rucci N, Millimaggi D, Mari M, Del Fattore A, Bologna M, Teti A, Angelucci A, Dolo V: Receptor activator of NF-kappaB ligand enhances breast cancer-induced osteolytic lesions through upregulation of extracellular matrix metalloproteinase inducer/CD147. Cancer Res. 2010, 70: 6150-6160. 10.1158/0008-5472.CAN-09-2758.
Lynch CC: Matrix metalloproteinases as master regulators of the vicious cycle of bone metastasis. Bone. 2010,
Podgorski I, Linebaugh BE, Koblinski JE, Rudy DL, Herroon MK, Olive MB, Sloane BF: Bone marrow-derived cathepsin K cleaves SPARC in bone metastasis. Am J Pathol. 2009, 175: 1255-1269. 10.2353/ajpath.2009.080906.
Kang JS, Alliston T, Delston R, Derynck R: Repression of Runx2 function by TGF-beta through recruitment of class II histone deacetylases by Smad3. EMBO J. 2005, 24: 2543-2555. 10.1038/sj.emboj.7600729.
Mercer RR, Miyasaka C, Mastro AM: Metastatic breast cancer cells suppress osteoblast adhesion and differentiation. Clin Exp Metastasis. 2004, 21: 427-435. 10.1007/s10585-004-1867-6.
Wang Y, Nishida S, Elalieh HZ, Long RK, Halloran BP, Bikle DD: Role of IGF-I signaling in regulating osteoclastogenesis. J Bone Miner Res. 2006, 21: 1350-1358. 10.1359/jbmr.060610.
Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D: Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000, 2: 737-744. 10.1038/35036374.
Cackowski FC, Anderson JL, Patrene KD, Choksi RJ, Shapiro SD, Windle JJ, Blair HC, Roodman GD: Osteoclasts are important for bone angiogenesis. Blood. 2010, 115: 140-149. 10.1182/blood-2009-08-237628.
Raica M, Anca M: Platelet-derived growth factor (PDGF)/PDGF receptors (PDGFR) axis as target for antitumor and antiangiogenic therapy. Pharmaceuticals. 2010, 3: 572-599. 10.3390/ph3030572.
Sanchez-Fernandez MA, Gallois A, Riedl T, Jurdic P, Hoflack B: Osteoclasts control osteoblast chemotaxis via PDGF-BB/PDGF receptor beta signaling. PloS one. 2008, 3: e3537-10.1371/journal.pone.0003537.
Mercer RR, Mastro AM: Cytokines secreted by bone-metastatic breast cancer cells alter the expression pattern of f-actin and reduce focal adhesion plaques in osteoblasts through PI3K. Exp Cell Res. 2005, 310: 270-281. 10.1016/j.yexcr.2005.07.029.
Kubota K, Sakikawa C, Katsumata M, Nakamura T, Wakabayashi K: PDGF BB purified from osteoclasts acts as osteoblastogenesis inhibitory factor (OBIF). J Biomol Tech. 2002, 13: 62-71.
Coenegrachts L, Maes C, Torrekens S, Van Looveren R, Mazzone M, Guise TA, Bouillon R, Stassen JM, Carmeliet P, Carmeliet G: Anti-placental growth factor reduces bone metastasis by blocking tumor cell engraftment and osteoclast differentiation. Cancer Res. 2010, 70: 6537-6547. 10.1158/0008-5472.CAN-09-4092.
Brown JE, Thomson CS, Ellis SP, Gutcher SA, Purohit OP, Coleman RE: Bone resorption predicts for skeletal complications in metastatic bone disease. Br J Cancer. 2003, 89: 2031-2037. 10.1038/sj.bjc.6601437.
Clarke BL, Khosla S: Physiology of bone loss. Radiol Clin North Am. 2010, 48: 483-495. 10.1016/j.rcl.2010.02.014.
Carlsten H: Immune responses and bone loss: the estrogen connection. Immunol Rev. 2005, 208: 194-206. 10.1111/j.0105-2896.2005.00326.x.
Ooi LL, Zheng Y, Stalgis-Bilinski K, Dunstan CR: The bone remodeling environment is a factor in breast cancer bone metastasis. Bone. 2010, [Epub ahead of print]
Ooi LL, Zhou H, Kalak R, Zheng Y, Conigrave AD, Seibel MJ, Dunstan CR: Vitamin D deficiency promotes human breast cancer growth in a murine model of bone metastasis. Cancer Res. 2010, 70: 1835-1844. 10.1158/0008-5472.CAN-09-3194.
Zheng Y, Zhou H, Modzelewski JR, Kalak R, Blair JM, Seibel MJ, Dunstan CR: Accelerated bone resorption, due to dietary calcium deficiency, promotes breast cancer tumor growth in bone. Cancer Res. 2007, 67: 9542-9548. 10.1158/0008-5472.CAN-07-1046.
Balkwill F, Mantovani A: Cancer and inflammation: implications for pharmacology and therapeutics. Clin Pharmacol Ther. 2010, 87: 401-406. 10.1038/clpt.2009.312.
Roy DL, Pathangey LB, Tinder TL, Schettini JL, Gruber HE, Mukherjee P: Breast-cancer-associated metastasis is significantly increased in a model of autoimmune arthritis. Breast Cancer Res. 2009, 11: R56-10.1186/bcr2345.
Clezardin P, Teti A: Bone metastasis: pathogenesis and therapeutic implications. Clin Exp Metastasis. 2007, 24: 599-608. 10.1007/s10585-007-9112-8.
Lipton A: Bone continuum of cancer. Am J Clin Oncol. 2010, 33 (3 Suppl): S1-7. 10.1097/COC.0b013e3181deb9e5.
Lipton A: Emerging role of bisphosphonates in the clinic--antitumor activity and prevention of metastasis to bone. Cancer Treat Rev. 2008, 34 (Suppl 1): S25-30. 10.1016/j.ctrv.2008.03.008.
Coleman R, Gnant M: New results from the use of bisphosphonates in cancer patients. Curr Opin Support Palliat Care. 2009, 3: 213-218. 10.1097/SPC.0b013e32832f4149.
Stopeck A: Denosumab findings in metastatic breast cancer. Clin Adv Hematol Oncol. 2010, 8: 159-160.
Grey A: Teriparatide for bone loss in the jaw. N Engl J Med. 2010, 363: 2458-2459. 10.1056/NEJMe1010459.
Nemeth JA, Harb JF, Barroso U, He Z, Grignon DJ, Cher ML: Severe combined immunodeficient-hu model of human prostate cancer metastasis to human bone. Cancer Res. 1999, 59: 1987-1993.
Halpern J, Lynch CC, Fleming J, Hamming D, Martin MD, Schwartz HS, Matrisian LM, Holt GE: The application of a murine bone bioreactor as a model of tumor: bone interaction. Clin Exp Metastasis. 2006, 23: 345-356. 10.1007/s10585-006-9044-8.
Mastro AM, Vogler EA: A three-dimensional osteogenic tissue model for the study of metastatic tumor cell interactions with bone. Cancer Res. 2009, 69: 4097-4100. 10.1158/0008-5472.CAN-08-4437.
Acknowledgements
Research in the Mastro Laboratory has been funded by grants from the US Army Medical and Materiel Command Breast Cancer Research Program (DAMD 17-02-1-0358, W81XWH-06-1-0432, W81XWH-08-1-0488, W81XWH-06-0363), The Susan G Komen Breast Cancer Foundation (BCTR0601044 and BCTR104406), and with supplementary aid from the National Foundation for Cancer Research, Center for Metastasis Research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Chen, YC., Sosnoski, D.M. & Mastro, A.M. Breast cancer metastasis to the bone: mechanisms of bone loss. Breast Cancer Res 12, 215 (2010). https://doi.org/10.1186/bcr2781
Published:
DOI: https://doi.org/10.1186/bcr2781