
Abstract
Introduction
Certain rare, familial mutations in the ATM, BRCA1, BRCA2, CHEK2 or TP53 genes increase susceptibility to breast cancer but it has not, until now, been clear whether common polymorphic variants in the same genes also increase risk.
Methods
We have attempted a comprehensive, single nucleotide polymorphism (SNP)- and haplotype-tagging association study on each of these five genes in up to 4,474 breast cancer cases from the British, East Anglian SEARCH study and 4,560 controls from the EPIC-Norfolk study, using a two-stage study design. Nine tag SNPs were genotyped in ATM, together with five in BRCA1, sixteen in BRCA2, ten in CHEK2 and five in TP53, with the aim of tagging all other known, common variants. SNPs generating the common amino acid substitutions were specifically forced into the tagging set for each gene.
Results
No significant breast cancer associations were detected with any individual or combination of tag SNPs.
Conclusion
It is unlikely that there are any other common variants in these genes conferring measurably increased risks of breast cancer in our study population.
Similar content being viewed by others

Introduction
Four of the genes which lie in the DNA damage-recognition and repair pathway, ATM, BRCA1, BRCA2 and TP53, have mutations that are recognised to increase breast cancer susceptibility with moderate to high penetrance. Such mutations are very rare, and most probably of recent origin. A fifth gene, CHEK2, in the same pathway, has a deletion (1100delC) that reaches polymorphic frequencies (>0.01) in some European countries and doubles the risk of breast cancer in female carriers [1]. Together these mutations account for only a small proportion (2% to 5%) of all breast cancer incidences [2, 3]. Breast cancer is, however, a common disease and genetic epidemiological data suggest that there is a low-penetrance genetic contribution to most cases [4, 5]. It is likely that at least a part of breast cancer aetiology will fit the common disease-common variant hypothesis, which states that patients with a common, complex disease are likely to share some common, low-penetrance alleles that increase their susceptibility to that disease. This raises the question of whether such common, polymorphic susceptibility alleles exist within these five genes in addition to the rare, disease-causing mutations that are already known.
It is now possible to attempt a comprehensive exclusion of all common single nucleotide polymorphisms (SNPs) from association with breast cancer susceptibility, using an empirical tag SNP approach. Studies have already been attempted in some of these genes. In ATM, Tamimi and colleagues [6] examined 5 haplotype-tagging SNPs (htSNPs) in approximately 3,000 subjects from the Nurses Heath Study but found no associations with breast cancer risk. For BRCA1, Cox and colleagues [7] used 4 htSNPs in the Nurses Health Study and identified one haplotype associated with an odds ratio (OR) of 1.18 (95% confidence interval (CI) 1.02 to 1.37), while Freedman and colleagues [8] examined 9 SNP-tagging SNPs (stSNPs) in the approximately 900 Caucasian subjects from the Multi-Ethnic Cohort but found no significant effects. For BRCA2, Freedman and colleagues [9] also used 21 stSNPs in the Multi-Ethnic Cohort and found one (rs206340) to be associated with a homozygous increase in risk (OR = 1.59, 95% CI 1.18 to 2.16). In addition, numerous studies have examined the N372H amino acid substitution in BRCA2 with conflicting results [10–12]. In CHEK2, Einarsdottir and colleagues [13] studied 6 htSNPs in approximately 3,000 Swedish cases and controls but found no significant associations with breast cancer. No comprehensive tagging study has yet been carried out for TP53, but several studies have examined the association of the non-synonymous R72P change and again the results have been mixed (reviewed in [14]).
We have used a combined SNP- and haplotype-tagging approach in an attempt to mark all the common variants in these five genes. Selected tag SNPs were then evaluated in our East Anglian breast cancer case-control study. The principal hypothesis underlying this experiment is that one or more common variants in these five genes are associated with an altered risk of breast cancer. We therefore aimed to identify a set of tag SNPs that efficiently captures all the known common variation (minor allele frequency (MAF) > 0.05) and is, therefore, likely also to tag most of the unknown common variants. This approach is most reliable where the gene has been re-sequenced in a sample of individuals that is sufficiently large to identify all common variants. We thus preferentially used re-sequencing data from the Environmental Genome Project (EGP) [15], but in genes where data was not available at the commencement of our study, data from the HAPMAP project [16] was used. HAPMAP does not re-sequence genes but provides genotype data on a sufficiently dense set of SNPs in samples of subjects from different ethnicities.
Our goal was to identify any common variants that show evidence for association with breast cancer susceptibility or, failing that, to exclude the possibility that common variants in these five genes are associated with an altered risk of breast cancer.
Materials and methods
Patients and controls
Cases were drawn from the SEARCH (breast) collection, an ongoing population-based study of breast cancer, with cases ascertained through the Eastern Cancer Registry (formerly East Anglian Cancer Registry). All patients diagnosed with invasive breast cancer below age 55 years since 1991 and still alive in 1996 (prevalent cases, median age 48 years), together with all those diagnosed below age 70 years between 1996 and the present (incident cases, median age 54 years), were eligible to take part. Of the eligible breast cancer patients, 67% returned a questionnaire and 64% provided a blood sample for DNA analysis. Eligible patients who did not take part in the study were similar to participants except, as might be expected, the proportion of clinical stage III/IV cases was somewhat higher in non-participants (Additional file 1). Controls were randomly selected from the Norfolk component of EPIC (European Prospective Investigation of Cancer). EPIC is a prospective study of diet and cancer being carried out in nine European countries. The EPIC-Norfolk cohort comprises 25,000 individuals resident in Norfolk, East Anglia, the same region from which the cases have been recruited. Controls are not matched to cases, but are broadly similar in age, being aged 42 to 81 years. The ethnic background of both cases and controls, as reported on the questionnaires, is similar, with >98% being white. This study has been approved by the Eastern Region Multicentre Research Ethics Committee, and all participants gave written informed consent.
The total number of cases available for analysis was 4,474, of which 27% were prevalent cases. The samples have been split into two sets in order to save DNA and reduce genotyping costs: the first set (n = 2,271 cases and 2,280 controls) is genotyped for all SNPs and the second set (n = 2,203 cases and 2,280 controls) is then tested for those SNPs that show marginally significant associations in set 1 (P-heterogeneity or P-trend <0.1). Cases were randomly selected for set 1 from the first 3,500 recruited, with set 2 comprising the remainder of these plus the next 974 incident cases recruited. As the prevalent cases were recruited first, the proportion of prevalent cases was somewhat higher in set 1 than in set 2 (33% versus 20%). Median age at diagnosis was similar in both sets (51 and 52 years old, respectively). There was no significant difference in the morphology, histopathological grade or clinical stage of the cases by set or by prevalent/incident status.
Power
The statistical power of the study depends on the susceptibility allele frequency, the risks conferred and the genetic mode of action (dominant, recessive, co-dominant). The staged approach substantially reduces genotyping costs without significantly affecting statistical power – a comparison is shown in Additional file 2. For example, assuming that the causative SNP is tagged with a pairwise correlation coefficient (rp 2) of 0.8, a type I error rate of 0.0001 and a genotyping success rate of 0.95, the staged/full study has 86/88% power to detect a dominant allele with MAF = 0.05 that confers a relative risk of 1.5 or 87/89% power to detect a dominant allele with MAF = 0.25 that confers a relative risk of 1.3. Power to detect recessive alleles is less; 53/60% for an allele with MAF = 0.25 and risk 1.5 and 71/75% for an allele with MAF = 0.5 and risk 1.3.
Selection of tagging SNPs
We attempted to define a set of tag SNPs such that all known common SNPs in a gene had an estimated r2 > 0.8 with at least one tag SNP using the tagSNPs program [17]. The best measure of the extent to which one SNP tags another SNP is the pairwise correlation coefficient (rp 2), since the loss in power incurred by using a marker SNP in place of a true causal SNP is directly related to this measure. We aimed to define a set of tagging SNPs such that all known common SNPs had an estimated rp 2 of >0.8 with at least one tagging SNP. However, some SNPs are poorly correlated with other single SNPs but may be efficiently tagged by a haplotype defined by multiple SNPs, thus reducing the total number of tag SNPs needed. As an alternative, therefore, we aimed for a correlation between each SNP and a haplotype of tag SNPs (r2S) of >0.8. Since tag SNP selection is problematic if there is extensive haplotype diversity, where necessary we divided a gene into haplotype blocks and selected the tagging SNPs for each block separately. It is possible to use a variety of formal definitions of haplotype blocks, but we simply used the graphical representations of the pattern of linkage disequilibrium (LD) based on D' and selected blocks such that the common haplotypes in each block accounted for at least 80% of all haplotypes observed using the Haploview program.
This tag SNP approach is most reliable where the gene has been re-sequenced in a sample of individuals that is sufficiently large to identify all common variants. We preferentially used data from the EGP [15], which has been re-sequencing candidate genes for cancer across panels of individuals representative of US ethnicities. The original panel (P1-PDR90) of 90 individuals consisted of 24 European Americans, 24 African Americans, 12 Mexican Americans, 6 Native Americans and 24 Asian Americans, but the ethnic group identifiers were not available. It is known that there is greater genetic and haplotype diversity in individuals of African origin. To reduce this we have identified and excluded 28 of the samples with the greatest African ancestry by comparing the genotypes of the PDR-90 subjects with the genotypes of the National Heart Lung and Blood Institute Variation Discovery Resource Project African American Panel [15] for the same SNPs. Data from the remaining 62 individuals were used to identify tag SNPs. For CHEK2, where complete EGP data were not initially available, data from European (CEU) subjects in HAPMAP were used as an alternative [16].
The CHEK2 Del1100C mutation was too rare to be selected as a tag SNP, or to be tagged in the current study, but it had been previously assayed in the SEARCH cases and controls [1] and those data have been incorporated here.
Taqman genotyping
Genotyping was carried out using Taqman® (Applied Biosystems, Warrington, UK) according to the manufacturer's instructions. Primers and probes were supplied directly by Applied Biosystems as Assays-by-Design™. All assays were carried out in 384-well plates. Cases and controls were arranged in a chequerboard pattern on each plate to ensure even treatment during the assay procedure and each plate included negative controls (with no DNA) and positive controls duplicated on a separate quality control plate. Plates were read on the ABI Prism 7900 using the Sequence Detection Software (Applied Biosystems). Failed genotypes were not repeated. Assays in which the genotypes of duplicate samples did not show >95% concordance were discarded and replaced with alternative assays with the same tagging properties.
Statistical methods
For each SNP, deviation of genotype frequencies in controls from the Hardy-Weinberg equilibrium (HWE) was assessed by a χ2 test with one degree of freedom. Genotype frequencies in cases and controls were compared by χ2 test for heterogeneity (two degrees of freedom) and test for trend (one degree of freedom). Genotype specific risks were estimated as ORs using unconditional logistic regression. Genotype distributions were also compared between prevalent and incident cases and between subjects in set 1 and set 2 with χ2 tests (two degrees of freedom). No statistically significant differences were found (data not shown) and so the results have been combined. The tagSNPsv2 program [17] was used to impute all the haplotypes generated by the tag SNPs in each LD block and to estimate the probabilities of each subject carrying each of the common (>0.05) haplotypes. Rarer haplotype probabilities were pooled into a single category. Haplotype frequencies were compared between cases and controls and haplotype-specific risks were estimated as ORs with associated CIs using unconditional logistic regression.
Results and discussion
SNP-tagging
ATM
The EGP identified 75 common variants in their set of 90 mixed-ethnicity subjects. After the exclusion of the 28 subjects with most African ancestry, 69 SNPs suitable for study in our European population remained. These fall into a single LD block with no evidence for recombination hot-spots. A set of nine tag SNPs were identified using the tagSNPsv2 program [17] and all were successfully genotyped. Tagging details are shown in Table 1.
BRCA1
The EGP identified 123 common variants, of which 113 were suitable for study in our sets (Table 1). Again, these lie in a single block of LD. A set of eight tag SNPs was identified but three of these could not be made into Taqman® assays and no others could be found to provide the same information (that is, they are singletons). A further SNP appeared to be hyper-mutable and it was not further investigated. Eventually, four tag SNPs were successfully genotyped (Table 1). An additional SNP, Q356R (BRCA1-02), was not selected as a tag SNP as its MAF in EGP was below our threshold of 0.05, but it was genotyped here because we had previously found some evidence for its association with an increased risk of both ovarian and breast cancer.
BRCA2
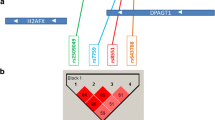
The EGP identified 113 common SNPs and 91 of these remained for study after exclusion of the African subjects. The BRCA2 block structure (Figure 1) is more complex than seen in the other genes; the first ten SNPs show no clear block structure and the remaining can be divided into two, largely separate, blocks. Twenty-two tag SNPs were initially identified but for six of these (all singletons) the local sequence precluded the manufacture of a working Taqman® assay and so these could not be analysed here (Table 1).

Haploview output showing linkage disequilibrium relationships between the 91 eligible BRCA2 SNPs. The matrix indicates the D' value between each pair of SNPs – darker colours indicate higher values.
CHEK2
At the commencement of this study this gene had not been re-sequenced by EGP and so HAPMAP phase 1 data for the Caucasians (CEPH trios) were used instead. HAPMAP had genotypes for 30 SNPs with MAF > 0.05 and these fell into a single LD block. Ten tag SNPs (Table 1), were successfully genotyped. Previously published [1] data on a subset of these subjects who were genotyped for the 1100delC variant are also included for comparison (Table 2).
TP53
The EGP identified 46 common SNPs, of which 39 were suitable for study after exclusion of the African subjects. These lie in a single LD block and are tagged with six SNPs. We set the tagging parameters to include two SNPs that had been selected for a previous study (TP53-01 (R72P, rs1042522) and TP53-02 (rs1625895)). One SNP (rs17880722) could not be made into a Taqman® assay and, since these were singletons, this left a final set of five tag SNPs that were genotyped (Table 1).
SNP associations
The genotype distributions for all SNPs genotyped in breast cancer case-control set 1 are shown in the left-hand columns of Table 2. In the BRCA1 and TP53 genes, no SNPs showed suggestive association (p < 0.1) at this stage and none were further investigated. One tag SNP in the ATM gene (ATM-08, rs3092991), four in BRCA2 (BRCA2-04,-11,-13 and -21/rs206118, rs11571686, rs1799955 and rs206343) and three in CHEK2 (CHEK2-09,-11 and -14/rs2236141, rs1076807 and rs9608698) provided sufficient evidence of association to merit further evaluation in case-control set 2. The genotype distributions of these SNPs in the entire study (sets 1 and 2) are shown in the right-hand columns of Table 2. After the completion of both stages, no tag SNP from ATM, BRCA1, BRCA2 or TP53 was associated with a significant difference in breast cancer risk.
The only SNP that was significantly associated (nominal threshold of p < 0.05) after completion of stage 2 was CHEK2-14 (rs9608698; P-trend = 0.02). This SNP was associated with an increased risk of breast cancer in both heterozgotes (OR = 1.10; 95% CI 0.99 to 1.21) and rare homozygotes (OR = 1.15; 95% CI 1.02 to 1.29) relative to the common homozygotes.
We specifically chose to study SNPs that generate amino acid substitutions and had been included in previously published association studies. BRCA1 P271L (BRCA1-01, rs1799917), BRCA2 N372H (BRCA2-22, rs144848) and TP53 R72P (TP53-01, rs1042522) were included in the set of tagging SNPs for their respective genes and it can be seen that, in this study, none of these SNPs show any significant association with breast cancer risk. A recent collaborative study [18] has also investigated BRCA2 N372H in more than 31,000 subjects (cases and controls) as well as TP53 R72P in almost 20,000 subjects. That study, similarly, found no main effect with either amino acid substitution, although it did report some evidence of BRCA2 HH homozygotes in the youngest age groups having an increased risk of breast cancer. This finding is compatible with the original two publications, which reported a significantly increased risk of breast cancer in HH homozygotes, since both had concentrated on very young onset breast cancer cases [11, 12].
We were unable to confirm the previously reported breast cancer association of BRCA1 Q356R (BRCA1-02, rs1799950). The OR for the rare, RR, homozygote relative to the common, QQ, group is 0.63 (95% CI 0.23 to 1.23; P-trend = 0.20, P-recessive test = 0.18). This estimate is comparable with our previous estimate, which was based on only 1,400 subjects [19], but even this study, which is larger by 6 times, has <50% power to test the hypothesis that this SNP is associated with such a recessive protective effect (Additional file 2).
Haplotype associations
Some of the variants within these genes have not been well tagged by an individual tag SNP, but a number of these have been better covered by a combination (haplotype) of several SNPs. Thus, the haplotypes predicted from the genotypes of tag SNPs were also tested for association with breast cancer risk; each common haplotype (predicted frequency >0.05) in each LD block was tested individually, in addition to the effect of the combined rarer haplotypes. The results are shown in Table 3.
For TP53 four common haplotypes are predicted to be formed from the five tag SNPS and none are significantly associated with differences in breast cancer risk. For BRCA1 the five genotyped SNPs generate five common haplotypes. Four of these are not associated with differences in breast cancer risk. The remaining one uniquely carries the R356 allele of the Q356R polymorphism and its OR is thus very similar to that calculated on the Q356R SNP analysis. For ATM, the eight tag SNPs generate five common haplotypes. Four show no significant association with breast cancer risk and the remaining one, which uniquely carries the rare allele of ATM-08 (rs3092991), displays a similar OR to that calculated by examination of this SNP alone. Both the potential associations with BRCA1 Q356R and ATM rs3092992 were found to be false positives after examination in stage 2 and the same conclusion can be drawn about their haplotypes.
For BRCA2 the pattern is more complex and haplotypes were analysed separately in the three LD blocks. In each block certain haplotypes gave results in set 1 that merited further investigation in set 2 (Table 3). Three further tag SNPs, in addition to the five described in the previous section, were genotyped in set 2 to enable the discrimination of these haplotypes. However, after stage 2, the potential associations with these haplotypes had reduced in significance and so we can conclude that they were also false positives.
Freedman and colleagues [9] reported that a haplotype of BRCA2 tagged by SNP rs206340 was associated with a significant increase in breast cancer risk among homozygotes (OR aa/gg = 1.59; 95% CI 1.18 to 2.16)). In our study the equivalent tag SNP was BRCA2-21 (rs206343; rp 2 with rs206340 = 0.89), which contrarily showed a non-significant trend for the minor allele to decrease, rather than increase, risk (Table 2; OR gg/aa = 0.89; 95% CI 0.73 to 1.09)].
Two haplotypes of CHEK2, both carrying the minor allele of CHEK2-14 (rs9608698), showed potential associations but with risks in opposing directions. These were evaluated further by generating haplotypes from the SNP markers that had already been taken into stage 2 as well as the 1100delC mutation that had been typed for a previous study. The 1100delC mutation, which has a frequency of 0.4% in these East Anglian controls, is carried on a single haplotype that also carries CHEK2-14. This rare haplotype is, as expected, significantly associated with breast cancer susceptibility (OR = 2.66; 95% CI 1.40 to 5.05) for the carrier versus the non-carrier, but other, more common haplotypes tagged by CHEK2-14 but not 1100delC also show marginal evidence for association with differences in risk
Deviation from Hardy-Weinberg equilibrium
The coding region of the ATM gene has been reported to be less polymorphic than other comparable genes, indicative of constraint by Darwinian selection pressure [20]. The genotype distribution of the BRCA2 N372H SNP has also previously been noted to deviate from HWE [11, 21], again indicating possible selection. Here we note that there are more deviations from HWE than would be expected by chance. The genotype distributions of 11 of the 45 tag SNPs (24%) deviate from HWE below the 10% significance level, and 5 (11%) deviate below the 5% level, and 1 of these (BRCA2-18, rs11571789) deviates with p = 6 × 10-5. All significant deviations from HWE fall in just three of the five genes, ATM, BRCA2 and TP53. Examination of the genotype clusters and other quality control measures gives no indication that these deviations are the result of genotyping artefacts. This leaves open the possibility that common variants in these three genes are subject to selection, acting either directly or indirectly via selective sweep across other variants in LD with them.
Limitations
There are three classes of variant that we cannot be certain we have excluded from association. The first class comprises dominant alleles with MAF below 0.01 – these include the known rare, disease-causing mutations in these five genes. The BIC database [22] lists 27 mis-sense mutations in BRCA1 and 20 in BRCA2 (as well as many more variants of unknown function), which, like SNPs, all result from single-base substitutions, but are present only in single families. Similarly, there are as yet unknown numbers of rare mutations in the ATM gene that increase risk of breast cancer in heterozygous carriers [23]. Some of these result from single base substitutions and could be expected to be present in up to 3% of our cases (for example, S49C) [24].
The second class comprises those SNPs that have MAF = 0.05 to 0.25 but are recessive in effect. For both the above classes, we had insufficient statistical power, even with a sample size of more than 9,000 subjects, for exclusion of all variants by genetic association.
The third class comprises variants that are not well tagged by any of our genotyped tag SNPs or their combined haplotypes. As can be seen from Table 1, there are several of these, predominantly in the BRCA2 and CHEK2 genes. Such hard-to-tag variants often lie outside LD blocks or are hyper-mutable. The minor alleles of these SNPs do not share identity-by-descent with other SNPs and so cannot be detected by association; they will only have an effect on breast cancer risk if they are the functional and directly increase cancer susceptibility.
Conclusion
There is ample evidence that rare mutations in each of the five genes ATM, BRCA1, BRCA2, CHEK2 and TP53 cause increased susceptibility to breast cancer in the families who carry them, but we find no evidence for the existence of common, polymorphic susceptibility alleles in these genes. However, there remains good evidence that such alleles, conforming to the common disease-common variant hypothesis, do exist in other breast cancer susceptibility genes.
Abbreviations
- CI:
-
confidence interval
- EGP:
-
Environmental Genome Project
- ht-SNP:
-
haplotype tagging SNP
- HWE:
-
Hardy-Weinberg equilibrium
- LD:
-
linkage disequilibrium
- MAF:
-
minor allele frequency
- OR:
-
odds ratio
- SNP:
-
single nucleotide polymorphism
- st-SNP:
-
SNP tagging SNP.
References
CHEK2 Breast Cancer Case-Control Consortium: CHEK2*1100delC and susceptibility to breast cancer: a collaborative analysis involving 10,860 breast cancer cases and 9,065 controls from 10 studies. Am J Hum Genet. 2004, 74: 1175-1182. 10.1086/421251.
Anglian Breast Cancer Study Group: Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Br J Cancer. 2000, 83: 1301-1308. 10.1054/bjoc.2000.1407.
Peto J, Collins N, Barfoot R, Seal S, Warren W, Rahman N, Easton DF, Evans C, Deacon J, Stratton MR: Prevalence of BRCA1 and BRCA2 gene mutations in patients with early-onset breast cancer. J Natl Cancer Inst. 1999, 91: 943-949. 10.1093/jnci/91.11.943.
Antoniou AC, Pharoah PD, McMullan G, Day NE, Stratton MR, Peto J, Ponder BJ, Easton DF: A comprehensive model for familial breast cancer incorporating BRCA1, BRCA2 and other genes. Br J Cancer. 2002, 86: 76-83. 10.1038/sj.bjc.6600008.
Pharoah PD, Antoniou A, Bobrow M, Zimmern RL, Easton DF, Ponder BA: Polygenic susceptibility to breast cancer and implications for prevention. Nat Genet. 2002, 31: 33-36. 10.1038/ng853.
Tamimi Y, Lines M, Coca-Prados M, Walter MA: Identification of target genes regulated by FOXC1 using nickel agarose-based chromatin enrichment. Invest Ophthalmol Vis Sci. 2004, 45: 3904-3913. 10.1167/iovs.04-0628.
Cox DG, Kraft P, Hankinson SE, Hunter DJ: Haplotype analysis of common variants in the BRCA1 gene and risk of sporadic breast cancer. Breast Cancer Res. 2005, 7: R171-R175. 10.1186/bcr973.
Freedman ML, Penney KL, Stram DO, Riley S, McKean-Cowdin R, Le Marchand L, Altshuler D, Haiman CA: A haplotype-based case-control study of BRCA1 and sporadic breast cancer risk. Cancer Res. 2005, 65: 7516-7522. 10.1158/0008-5472.CAN-05-0132.
Freedman ML, Penney KL, Stram DO, Le Marchand L, Hirschhorn JN, Kolonel LN, Altshuler D, Henderson BE, Haiman CA: Common variation in BRCA2 and breast cancer risk: a haplotype-based analysis in the Multiethnic Cohort. Hum Mol Genet. 2004, 13: 2431-2441. 10.1093/hmg/ddh270.
Cox DG, Hankinson SE, Hunter DJ: No association between BRCA2 N372H and breast cancer risk. Cancer Epidemiol Biomarkers Prev. 2005, 14: 1353-1354. 10.1158/1055-9965.EPI-04-0848.
Healey CS, Dunning AM, Teare MD, Chase D, Parker L, Burn J, Chang-Claude J, Mannermaa A, Kataja V, Huntsman DG, et al: A common variant in BRCA2 is associated with both breast cancer risk and prenatal viability. Nat Genet. 2000, 26: 362-364. 10.1038/81691.
Spurdle AB, Hopper JL, Chen X, Dite GS, Cui J, McCredie MR, Giles GG, Ellis-Steinborner S, Venter DJ, Newman B, et al: The BRCA2 372 HH genotype is associated with risk of breast cancer in Australian women under age 60 years. Cancer Epidemiol Biomarkers Prev. 2002, 11: 413-416.
Einarsdottir K, Humphreys K, Bonnard C, Palmgren J, Iles MM, Sjolander A, Li Y, Chia KS, Liu ET, Hall P, et al: Linkage disequilibrium mapping of CHEK2: common variation and breast cancer risk. PLoS Med. 2006, 3: e168-10.1371/journal.pmed.0030168.
Goode EL, Ulrich CM, Potter JD: Polymorphisms in DNA repair genes and associations with cancer risk. Cancer Epidemiol Biomarkers Prev. 2002, 11: 1513-1530.
NIEHS Environmental Genome Project. [http://egp.gs.washington.edu/]
HAPMAP Project. [http://www.HAPMAP.org]
Stram DO, Haiman CA, Hirschhorn JN, Altshuler D, Kolonel LN, Henderson BE, Pike MC: Choosing haplotype-tagging SNPS based on unphased genotype data using a preliminary sample of unrelated subjects with an example from the Multiethnic Cohort Study. Hum Hered. 2003, 55: 27-36. 10.1159/000071807.
Breast Cancer Association Consortium: Commonly studied single-nucleotide polymorphisms and breast cancer: results from the Breast Cancer Association Consortium. J Natl Cancer Inst. 2006, 98: 1382-1396.
Dunning AM, Chiano M, Smith NR, Dearden J, Gore M, Oakes S, Wilson C, Stratton M, Peto J, Easton D, et al: Common BRCA1 variants and susceptibility to breast and ovarian cancer in the general population. Hum Mol Genet. 1997, 6: 285-289. 10.1093/hmg/6.2.285.
Thorstenson YR, Shen P, Tusher VG, Wayne TL, Davis RW, Chu G, Oefner PJ: Global analysis of ATM polymorphism reveals significant functional constraint. Am J Hum Genet. 2001, 69: 396-412. 10.1086/321296.
Teare MD, Cox A, Shorto J, Anderson C, Bishop DT, Cannings C: Heterozygote excess is repeatedly observed in females at the BRCA2 locus N372H. J Med Genet. 2004, 41: 523-528. 10.1136/jmg.2003.017293.
BIC Database. [http://research.nhgri.nih.gov/bic/]
Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, North B, Jayatilake H, Barfoot R, Spanova K, et al: ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006, 38: 873-875. 10.1038/ng1837.
Stredrick DL, Garcia-Closas M, Pineda MA, Bhatti P, Alexander BH, Doody MM, Lissowska J, Peplonska B, Brinton LA, Chanock SJ, et al: The ATM missense mutation p.Ser49Cys (c.146C>G) and the risk of breast cancer. Hum Mutat. 2006, 27: 538-544. 10.1002/humu.20323.
Acknowledgements
We thank all the subjects who participated in these studies, the EPIC Management team (K-T Khaw, S Bingham and N Wareham) for access to control DNA, the Eastern Cancer Registry and Intelligence Unit. This work was funded by Cancer Research-UK. BAJP is a Gibb Fellow, DFE is a Principal Fellow and PDPP is a Senior Clinical Research Fellow of Cancer Research-UK.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
CB, CSH, KAP and SS chose the tagging set of SNPs, carried out the assays, analysed the data and helped prepare the manuscript. RNL provided data on the EPIC subjects. DJT chose and developed a suite of tag SNP and haplotype deduction programs for SEARCH and supervised their appropriate use. AMD, PDPP, DFE and BAJP are principle investigators within SEARCH. AMD managed this project and prepared the manuscript. CB, CSH and KAP contributed equally to this study. The SEARCH study team are currently: Jean Abraham, Shahana Ahmed, Antonis Antoniou, Patrick Benusiglio, Fiona Blows, Arancha Cebrian, Don Conroy, Bridget Curzon, Gary Dew, Kristy Driver, Helen Field, Patricia Harrington, Clare Jordan, Fabienne Lesueur, Craig Luccarini, Rebecca Mayes, Hannah Munday, Barbara Perkins, Karen Redman, Mitul Shah, Jonathan Tyrer, Paula Smith and Judy West.
Electronic supplementary material
13058_2007_1634_MOESM1_ESM.doc
Additional file 1: A table listing the basic epidemiological details of the breast cancer cases and controls in set 1 and set 2. (DOC 32 KB)
13058_2007_1634_MOESM2_ESM.doc
Additional file 2: A table showing a statistical power comparison of the two-stage versus the single-stage study design. (DOC 28 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Baynes, C., Healey, C.S., Pooley, K.A. et al. Common variants in the ATM, BRCA1, BRCA2, CHEK2 and TP53 cancer susceptibility genes are unlikely to increase breast cancer risk. Breast Cancer Res 9, R27 (2007). https://doi.org/10.1186/bcr1669
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/bcr1669