
Chapter summary
Regulation of osteoclast differentiation is an aspect central to the understanding of the pathogenesis and the treatment of bone diseases such as autoimmune arthritis and osteoporosis. In fact, excessive signaling by RANKL (receptor activator of nuclear factor κB ligand), a member of the tumor necrosis factor (TNF) family essential for osteoclastogenesis, may contribute to such pathological conditions. Here we summarize our current work on the negative regulation of osteoclastogenesis by unique signaling crosstalk between RANKL and interferons (IFNs). First, activated T cells maintain bone homeostasis by counterbalancing the action of RANKL through production of IFN-γ. This cytokine induces rapid degradation of the RANK (receptor activator of nuclear factor κB) adapter protein TRAF6 (TNF-receptor-associated factor 6), resulting in strong inhibition of the RANKL-induced activation of NF-κB and JNK (c-Jun N-terminal kinase). Second, RANKL induces the IFN-β gene but not IFN-α genes, in osteoclast precursor cells, and that IFN-β strongly inhibits the osteoclast differentiation by interfering with the RANKL-induced expression of c-Fos. The series of in vivo experiments revealed that these two distinct IFN-mediated regulatory mechanisms are both important to maintain homeostasis of bone resorption. Collectively, these studies revealed novel aspects of the two types of IFN, beyond their original roles in the immune response, and may offer a molecular basis for the treatment of bone diseases.
Similar content being viewed by others


Historical background
A delicate balance between bone resorption and bone formation is critical for the maintenance of bone strength and integrity, wherein bone-resorbing osteoclasts and bone-forming osteoblasts play central roles [1]. In fact, this physiologic process, termed bone remodeling, must be regulated strictly, and tipping the balance in favor of osteoclasts causes bone destruction observed in pathological conditions such as autoimmune arthritis, periodontitis, postmenopausal osteoporosis, Paget's disease and bone tumors [2, 3].
RANKL (receptor activator of nuclear factor κB ligand), a member of the TNF (tumor necrosis factor) family, is an essential cytokine for the differentiation of monocyte/macrophage precursors to osteoclasts [4–6]. Briefly, binding of RANKL to its receptor, RANK (receptor activator of nuclear factor κB), results in the recruitment of proteins of the TRAF (TNF-receptor-associated factor) family, such as TRAF6, which activates NF-κB and JNK (c-Jun N-terminal kinase) pathways [7–9]. By a yet unknown mechanism, RANKL also induces expression of c-Fos [10, 11]. The essential role of these TRAF6 and c-Fos pathways in osteoclastogenesis has been well documented by gene disruption studies [12–15]. To maintain normal bone homeostasis, this RANKL signaling must be properly regulated. In this context, osteoprotegerin (OPG), a soluble 'decoy' receptor for RANKL, is known as a negative regulator of osteoclastogenesis [16]. In fact, mice lacking OPG develop osteoporosis, suggesting that the balance between RANKL and OPG dictates the levels of bone resorption [17].
It has been reported that prolonged or aberrant activation of the immune system in certain autoimmune conditions often results in tissue destruction mediated by effector cells. In fact, enhanced osteoclastic bone resorption causes severe bone damage leading to progressive joint destruction in autoimmune arthritis [18, 19], wherein T-cell expression of RANKL may play a critical role. In this context, it has been reported that activated T cells promote osteoclastogenesis through expression of RANKL in vitro[18, 20]. However, it is not known whether activated T cells maintain bone homeostasis by counterbalancing the action of RANKL.
One may infer that the process of osteolastogenesis is under negative regulation by factors in addition to OPG, at multiple levels, in order to maintain bone homeostasis.
Regulation of osteoclastogenesis by T cells via IFN-γ
Involvement of the T-cell-produced IFN-γ in negative regulation of osteoclastogenesis
To study whether activated T cells contribute in the regulation of osteoclastogenesis, we used a well-established model of endotoxin-induced bone resorption, in which the essential involvement of T cells has been documented [21, 22]. A notable exacerbation of osteoclast formation and bone destruction was observed in mice lacking IFNGR1, one of the components of IFN-γ receptor (IFNGR) (IFN-γR-/- mice) [8]. This observation is also consistent with an enhanced severity in the T-cell-mediated collagen-induced model of autoimmune arthritis observed in IFN-γR-/- mice [23, 24]. When the effect of IFN-γ producing T cells on osteoclastogenesis was examined in vitro, osteoclast formation from RANKL-stimulated bone-marrow-derived monocyte/macrophage precursor cells (BMMs) was strongly inhibited by the coculture with T cells activated with the anti-CD3 antibody but not with resting T cells. When activated T cells were cocultured with IFN-γR-/- BMMs, however, the inhibitory effect of activated T cells was completely abrogated. These IFN-γR-/- BMMs differentiated into osteoclasts, albeit with low efficiency, when cocultured with activated T cells, even in the absence of recombinant RANKL, suggesting that the effect of T cells on osteoclastogenesis depends on the balance between RANKL and IFN-γ. Activated T-cell supernatant also suppressed osteoclastogenesis induced by recombinant RANKL, and this suppressive activity was inhibited by a neutralizing antibody against IFN-γ. These results collectively indicate that IFN-γ is critical for T-cell suppression of RANKL-induced osteoclastogenesis.
Mechanism of action of IFN-γ
A marked inhibitory effect of IFN-γ was observed by an in vitro assay for osteoclast formation in RANKL-stimulated BMMs, and a very low level of IFN-γ strongly inhibited osteoclast formation even in the presence of an excess amount of RANKL. The cytokine signals the cell through activation of the transcription factor STAT1 (signal transducer and activator of transcription 1) [25]. The active form of this transcription factor, termed IFN-γ-activated factor (GAF), induces target genes of IFN-γ either directly or through the induction of the transcription factor IRF-1 (interferon regulatory factor 1) [26]. The inhibitory effect of IFN-γ on osteoclastogenesis was completely abrogated in STAT1-/- mice but not in IRF-1-/- mice. Thus, the STAT1 (GAF)-mediated, IRF-1-independent gene induction pathway is critical for interfering with the RANKL signaling pathway.
To investigate the target of IFN-γ inhibition of osteoclastogenesis, RANKL-induced activation of NF-κB and JNK was examined in IFN-γ-treated BMMs. It was found that RANKL-induced activation of NF-κB and JNK was markedly inhibited in IFN-γ-treated BMMs and that this inhibition was accompanied by strong suppression of TRAF6 expression. Notably, overexpression of TRAF6 in BMMs by retrovirus-mediated gene transfer rendered them resistant to IFN-γ-mediated inhibition of osteoclastogenesis. In these cells, RANKL-induced activation of NF-κB and JNK was also restored, indicating further that TRAF6 is the critical target of the action of IFN-γ. Thus, downregulation of TRAF6 expression accounts, at least in part, for the IFN-γ inhibition of osteoclastogenesis.
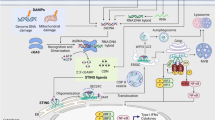
The mechanism by which TRAF6 expression is downregulated by IFN-γ was investigated further. It was found that TRAF6 protein levels, rather than its mRNA levels, were suppressed after RANKL stimulation in the presence of IFN-γ. Interestingly, stimulation by RANKL per se promoted TRAF6 degradation, which was markedly accelerated by IFN-γ. Since the TRAF6 level is upregulated during stimulation by RANKL, the equilibrium of TRAF6 protein is apparently shifted toward its synthesis rather than its degradation in the absence of IFN-γ, but the addition of IFN-γ reverses the equilibrium. Notably, IFN-γ alone had no effect on TRAF6 expression, suggesting that accelerated TRAF6 degradation by IFN-γ requires RANKL signaling. Further studies revealed that TRAF6 is a direct target for the ubiquitin-proteasome system, which is enhanced by IFN-γ [8] (Fig. 1).

Signaling crosstalk between RANKL and IFNs. IFN-γ and IFN-β inhibit RANKL signaling by downregulating essential mediators of osteoclastogenesis (TRAF6 and c-Fos, respectively). (See [8, 30], and text for the details). Fra, fos-related antigen; IRF, interferon regulatory factor; ISGF3, interferon-stimulated gene factor-3; JNK, c-Jun N-terminal kinase; NF-κB, nuclear factor κB; OPG, osteoprotegerin; RANK, receptor activator of nuclear factor κB; Stat, signal transducer and activator of transcription; TRAF, TNF receptor-associated factor.
Physiological significance of the IFN-γ-mediated regulation of RANKL signaling
It has recently been found that activated T cells promote osteoclastogenesis through expression of RANKL and regulate bone loss in autoimmune arthritis [18]. The results presented above revealed that activated T cells can also negatively affect osteoclastogenesis through IFN-γ production. It is likely that the balance between the actions of RANKL and of IFN-γ may regulate osteoclast formation. For example, during acute immune reactions, an enhanced production of IFN-γ counterbalances the augmentation of RANKL expression and reduces aberrant formation of osteoclasts. On the other hand, in chronic synovitis of rheumatoid arthritis, this balance may be skewed in favor of RANKL expression. In this context, it is noteworthy that expression of IFN-γ is suppressed, despite a significant infiltration of T cells [27, 28], in the arthritic joints in which RANKL expression is enhanced. Thus, the paucity of IFN-γ and the enhanced expression of RANKL may contribute to the activation of osteoclastogenesis in arthritis. It is clear that other factors must be taken into consideration in the control of osteoclastogenesis in arthritis, e.g. RANKL induction in IL-1-stimulated or TNF-α-stimulated synovial fibroblasts [3, 29], and local cytokine production pattern regulated by differentiation status of T cells into Th1 or Th2 type.
Autoregulation of RANKL signaling by IFN-β induction and the critical role of IFN-β in bone metabolism
Involvement of the IFN-γ system in the regulation of bone remodeling
During our study to analyze genes induced by RANKL, we noted induction of mRNAs known to be commonly induced by IFN-α/β. Although the integral role of the IFN-α/β system in the immune system has been extensively documented, it was unknown whether this system is linked to RANKL signaling. To investigate the physiological role of the IFN system in the control of bone homeostasis, we studied the skeletal system of mice deficient in one of the IFN receptor components, IFNAR1 (IFNAR1-/- mice) [30]. Surprisingly, a notable reduction of trabecular bone mass, a characteristic feature of osteoporosis, was observed in the mutant mice. Concomitantly, the number of osteoclasts was notably greater in the bone of the IFNAR1-/- mice than in the wild-type mice. Quantitative bone morphometric analyses further revealed a significant decrease in the trabecular bone volume, a feature that is diagnostic for enhanced osteoclastic bone resorption, without any remarkable difference in the markers of osteoblastic bone formation, indicating that the IFN-α/β signaling is physiologically critical for maintaining the normal bone mass by regulating osteoclastic bone resorption. RANKL-induced osteoclast formation in vitro is remarkably enhanced in BMMs from IFNAR1-/- mice, whereas no difference was seen in the osteoblast differentiation between cultured calvarial cells from wild-type mice and those from IFNAR1-/- mice. We deduce that the enhanced osteoclastogenesis in IFNAR1-/- mice is due to a cell-autonomous abnormality of BMMs.
Induction of the IFN-β gene by RANKL and the role of IFN-β in bone homeostasis
Stimulation by RANKL resulted in the induction of mRNA of IFN-β, but not of IFN-α, in BMMs. The findings suggest that, unlike the common action of these two IFN subtypes against viral infection, IFN-β is selectively involved in this osteoclast regulation (although the level of induction of mRNA was much lower than in virus-induced cells). When the skeletal system of the mice lacking IFN-β (IFN-β-/- mice) was examined, these mice also exhibited severe osteopenia resulting from enhanced osteoclastogenesis, an observation that indicates the role of RANKL-induced IFN-β in bone homeostasis.
Recombinant mouse IFN-β has a remarkably strong inhibitory effect on osteoclastogenesis induced by RANKL in combination with macrophage-colony-stimulating factor: the effect was observed at a concentration as low as one unit ml-1. IFN-β (as well as IFN-α) invokes cellular responses through activation/induction of transcription factors; these include ISGF3 [a heteromeric complex consisting of signal transducer and activator of transcription 1 (STAT1), STAT2 and interferon regulatory factor 9 (IRF-9)], STAT1 homodimer and IRF-1 [25, 26]. It was found that the inhibitory action of IFN-β is abrogated in BMMs from mice lacking STAT1 or IRF-9, indicating that the inhibitory action is linked to the ISGF3-mediated gene induction pathway (see Fig. 1).
To assess the efficacy of IFN-β for the suppression of osteoclast-mediated pathological conditions, an endotoxin-induced model of inflammatory bone destruction in mice was used [30]. Daily administration of IFN-β into the inflamed site markedly inhibited the osteoclast formation and bone resorption, indicating that IFN-β does indeed have a beneficial effect on bone destruction, most likely by downregulating osteoclastogenesis.
Signaling crosstalk between RANKL and IFN-β vying for c-Fos
To identify the downstream effector(s) of RANKL signaling affected by the IFN-β signaling, we first carried out immunoblot analysis of the effector molecules of RANKL signaling in IFN-β-treated BMMs and found that expression of c-Fos was selectively and dramatically downregulated. To examine further whether the inhibitory action of IFN-β is mediated through downregulation of c-Fos expression, c-Fos was artificially expressed by retrovirus-mediated gene transfer in BMMs. More than 50% of the virus-infected cells underwent differentiation in the presence of IFN-β, under condition in which fewer than 5% of cells did so when infected by either the control virus or virus expressing c-Jun.
What is the mechanism underlying the inhibition of c-Fos expression by IFN-β? Interestingly, the RANKL-induced Fos mRNA was not significantly altered by IFN-β, suggesting a post-transcriptional control mechanism(s). To gain further insights into the mechanism of c-Fos suppression by IFN-β, the synthesis of the c-Fos protein was studied by pulse-chase experiments [30], and it was found that the synthesis of c-Fos protein is inhibited in IFN-β-treated BMMs. It is not yet fully understood which target genes are responsible for suppression of c-Fos expression by IFN-β, but we have evidence that the ISGF3-inducible dsRNA-activated protein kinase (PKR) is responsible, albeit partly, for the suppression.
Requirement of c-Fos itself in the RANKL induction of the IFN-β gene
The induction of the IFN-α/β gene in virally infected cells requires two IRF transcription factors, IRF-3 and IRF-7 [31]. In the case of IFN-β, induction has also been known to depend on NF-κB (which is also activated by RANKL) [32]. Interestingly, when RANKL-induced expression of IFN-β mRNA was studied in BMMs from mice lacking c-Fos, or both IRF-3 and IRF-9, expression of this mRNA was no longer observed in the absence of c-Fos but was still detected in the absence of the IRFs critical for the virus-mediated induction. Furthermore, stimulation with RANKL induced a significant activation of the IFN-β promoter, as revealed by a reporter-gene transfection assay, and this activation was abrogated in cells from c-Fos-deficient mice. Thus, the RANKL-induced c-Fos per se induces its own inhibitor gene, IFN-β.
Conclusions and future prospects
Immune response is essential for host defense, but its prolonged or aberrant activation under certain autoimmune conditions often results in tissue destruction mediated by effector cells. As described here, a new biological function of IFN-γ was discovered: to protect against destruction of calcified tissue on T-cell activation [8]. The disease-limiting effect of IFN-γ in autoimmune arthritis may be explained, at least in part, by its potent suppressive effect on osteoclast development. Although the clinical application of IFN-γ has been difficult, probably because of its disease-promoting effect at the onset phase of autoimmunity [33], our results suggest that TRAF6, the critical target of IFN-γ-mediated suppression of osteoclastogenesis, could be a possible molecular target of pharmacological intervention in inflammatory bone diseases.
Our study also revealed a hitherto unknown signaling crosstalk between RANKL and IFN-β system, which is critical for bone homeostasis [30]. This crosstalk is unique in that RANKL signaling per se is responsible for the induction of IFN-β, and that c-Fos, the positive regulator of RANKL signaling, is required for the induction of its own inhibitor (see Fig. 1). In view of the osteoporotic phenotype found in mice lacking IFN-β or its receptor, we believe that this novel, negative-feedback regulation mechanism is physiologically important for maintaining bone mass. In this context, it would be interesting to find out if the negative regulation of osteoclastogenesis by IFN-β is modulated in osteopenic disease conditions.
Although further study is needed, the series of observations on the novel role of IFN-β may offer new therapeutic approaches to bone diseases such as inflammation-induced bone destruction and osteoporosis. In fact, exogenous application of IFN-β indeed has a beneficial effect against bone destruction in the lipopolysaccharide-induced model, most likely by downregulation of osteoclastogenesis. Our preliminary results also suggest that the systemic administration of IFN-β can reverse bone loss in an osteoporosis model of ovariectomized mice. In addition, identification of the critical target gene(s) of IFN-β responsible for the suppression of osteoclastogenesis may provide further insights into the regulation of bone remodeling.
Glossary of terms
BMM = bone-marrow-derived monocyte/macrophage precursor cell; GAF = IFN-γ-activated factor; IFNAR = interferon-α/β receptor; IFNGR = interferon-γ receptor; IRF = interferon regulatory factor; ISGF3 = interferon-stimulated gene factor 3; JNK = c-Jun N-terminal kinase; OPG = osteoprotegerin; RANK = receptor activator of nuclear factor κB; RANKL = receptor activator of nuclear factor κB ligand; TRAF = TNF receptor-associated factor.
References
Manolagas SC: Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. 2000, 21: 115-137. 10.1210/er.21.2.115.
Rodan GA, Martin TJ: Therapeutic approaches to bone diseases. Science. 2000, 289: 1508-1514. 10.1126/science.289.5484.1508.
Takayanagi H, Iizuka H, Juji T, Nakagawa T, Yamamoto A, Miyazaki T, Koshihara Y, Oda H, Nakamura K, Tanaka S: Involvement of receptor activator of nuclear factor κB ligand/osteoclast differentiation factor in osteoclastogenesis from synoviocytes in rheumatoid arthritis. Arthritis Rheum. 2000, 43: 259-269. 10.1002/1529-0131(200002)43:2<259::AID-ANR4>3.0.CO;2-W.
Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T: Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998, 95: 3597-3602. 10.1073/pnas.95.7.3597.
Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ: Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998, 93: 165-176. 10.1016/S0092-8674(00)81569-X.
Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, Khoo W, Wakeham A, Dunstan CR, Lacey DL, Mak TW, Boyle WJ, Pen-ninger JM: OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999, 397: 315-323. 10.1038/16852.
Wong BR, Josien R, Lee SY, Vologodskaia M, Steinman RM, Choi Y: The TRAF family of signal transducers mediates NF-κB activation by the TRANCE receptor. J Biol Chem. 1998, 273: 28355-28359. 10.1074/jbc.273.43.28355.
Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, Akinori T, Yokochi T, Oda H, Tanaka K, Nakamura K, Taniguchi T: T cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN. Nature. 2000, 408: 600-605. 10.1016/S0168-9002(98)00274-5.
Kobayashi N, Kadono Y, Naito A, Matsumoto K, Yamamoto T, Tanaka S, Inoue J: Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J. 2001, 20: 1271-1280. 10.1093/emboj/20.6.1271.
Matsuo K, Owens JM, Tonko M, Elliott C, Chambers TJ, Wagner EF: Fosl1 is a transcriptional target of c-Fos during osteoclast differentiation. Nat Genet. 2000, 24: 184-187. 10.1038/72855.
Wagner EF, Karsenty G: Genetic control of skeletal development. Curr Opin Genet Dev. 2001, 11: 527-532. 10.1016/S0959-437X(00)00228-8.
Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, Morony S, Capparelli C, Van G, Kaufman S, van der Heiden A, Itie A, Wakeham A, Khoo W, Sasaki T, Cao Z, Penninger JM, Paige CJ, Lacey DL, Dunstan CR, Boyle WJ, Goeddel DV, Mak TW: TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999, 13: 1015-1024.
Naito A, Azuma S, Tanaka S, Miyazaki T, Takaki S, Takatsu K, Nakao K, Nakamura K, Katsuki M, Yamamoto T, Inoue J: Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells. 1999, 4: 353-362. 10.1046/j.1365-2443.1999.00265.x.
Wang ZQ, Ovitt C, Grigoriadis AE, Mohle-Steinlein U, Ruther U, Wagner EF: Bone and haematopoietic defects in mice lacking c-fos. Nature. 1992, 360: 741-745. 10.1038/360741a0.
Grigoriadis AE, Wang ZQ, Cecchini MG, Hofstetter W, Felix R, Fleisch HA, Wagner EF: c-Fos: a key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science. 1994, 266: 443-448.
Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ: Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997, 89: 309-319. 10.1016/S0092-8674(00)80209-3.
Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, Boyle WJ, Simonet WS: Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12: 1260-1268.
Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe S, Wong T, Campagnuolo G, Moran E, Bogoch ER, Van G, Nguyen LT, Ohashi PS, Lacey DL, Fish E, Boyle WJ, Penninger JM: Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999, 402: 304-309. 10.1038/46303.
Takayanagi H, Juji T, Miyazaki T, Iizuka H, Takahashi T, Isshiki M, Okada M, Tanaka Y, Koshihara Y, Oda H, Kurokawa T, Nakamura K, Tanaka S: Suppression of arthritic bone destruction by adenovirus-mediated csk gene transfer to synoviocytes and osteoclasts. J Clin Invest. 1999, 104: 137-146.
Horwood NJ, Kartsogiannis V, Quinn JM, Romas E, Martin TJ, Gillespie MT: Activated T lymphocytes support osteoclast formation in vitro. Biochem Biophys Res Commun. 1999, 265: 144-150. 10.1006/bbrc.1999.1623.
Ukai T, Hara Y, Kato I: Effects of T cell adoptive transfer into nude mice on alveolar bone resorption induced by endotoxin. J Periodont Res. 1996, 31: 414-422.
Chiang CY, Kyritsis G, Graves DT, Amar S: Interleukin-1 and tumor necrosis factor activities partially account for calvarial bone resorption induced by local injection of lipopolysaccharide. Infect Immun. 1999, 67: 4231-4236.
Manoury-Schwartz B, Chiocchia G, Bessis N, Abehsira-Amar O, Batteux F, Muller S, Huang S, Boissier MC, Fournier C: High susceptibility to collagen-induced arthritis in mice lacking IFN-γ receptors. J Immunol. 1997, 158: 5501-5506.
Vermeire K, Heremans H, Vandeputte M, Huang S, Billiau A, Matthys P: Accelerated collagen-induced arthritis in IFN-γ receptor-deficient mice. J Immunol. 1997, 158: 5507-5513.
Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD: How cells respond to interferons. Annu Rev Biochem. 1998, 67: 227-264. 10.1146/annurev.biochem.67.1.227.
Taniguchi T, Ogasawara K, Takaoka A, Tanaka N: IRF family of transcription factors as regulators of host defense. Annu Rev Immunol. 2001, 19: 623-655. 10.1146/annurev.immunol.19.1.623.
Firestein GS, Zvaifler NJ: How important are T cells in chronic rheumatoid synovitis?. Arthritis Rheum. 1990, 33: 768-773.
Kinne RW, Palombo-Kinne E, Emmrich F: T-cells in the pathogenesis of rheumatoid arthritis: villains or accomplices?. Biochim Biophys Acta. 1997, 1360: 109-141. 10.1016/S0925-4439(96)00079-8.
Takayanagi H, Oda H, Yamamoto S, Kawaguchi H, Tanaka S, Nishikawa T, Koshihara Y: A new mechanism of bone destruction in rheumatoid arthritis: synovial fibroblasts induce osteoclastogenesis. Biochem Biophys Res Commun. 1997, 240: 279-286. 10.1006/bbrc.1997.7404.
Takayanagi H, Kim S, Mastuo K, H S, T S, Sato K, Yokochi T, Oda H, Tanaka K, Nakamura K, Ida N, Wagner EF, Taniguchi T: RANKL maintains bone homeostasis through c-Fos-dependent induction of IFN-β. Nature. 2002, 416: 744-749. 10.1038/416744a.
Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T: Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-α/β gene induction. Immunity. 2000, 13: 539-548. 10.1016/S1074-7613(00)00053-4.
Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Mani-atis T: Virus infection induces the assembly of coordinately activated transcription factors on the IFN-β enhancer in vivo. Mol Cell. 1998, 1: 507-518.
Billiau A: Interferon-γ: biology and role in pathogenesis. Adv Immunol. 1996, 62: 61-130.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Takayanagi, H., Kim, S. & Taniguchi, T. Signaling crosstalk between RANKL and interferons in osteoclast differentiation. Arthritis Res Ther 4 (Suppl 3), S227 (2002). https://doi.org/10.1186/ar581
Published:
DOI: https://doi.org/10.1186/ar581