
Chapter summary
The prevalence of rheumatoid arthritis (RA) is relatively constant in many populations, at 0.5–1.0%. However, a high prevalence of RA has been reported in the Pima Indians (5.3%) and in the Chippewa Indians (6.8%). In contrast, low occurrences have been reported in populations from China and Japan. These data support a genetic role in disease risk. Studies have so far shown that the familial recurrence risk in RA is small compared with other autoimmune diseases. The main genetic risk factor of RA is the HLA DRB1 alleles, and this has consistently been shown in many populations throughout the world. The strongest susceptibility factor so far has been the HLA DRB1*0404 allele. Tumour necrosis factor alleles have also been linked with RA. However, it is estimated that these genes can explain only 50% of the genetic effect. A number of other non-MHC genes have thus been investigated and linked with RA (e.g. corticotrophin releasing hormone, oestrogen synthase, IFN-γ and other cytokines). Environmental factors have also been studied in relation to RA. Female sex hormones may play a protective role in RA; for example, the use of the oral contraceptive pill and pregnancy are both associated with a decreased risk. However, the postpartum period has been highlighted as a risk period for the development of RA. Furthermore, breastfeeding after a first pregnancy poses the greatest risk. Exposure to infection may act as a trigger for RA, and a number of agents have been implicated (e.g. Epstein–Barr virus, parvovirus and some bacteria such as Proteus and Mycoplasma). However, the epidemiological data so far are inconclusive. There has recently been renewed interest in the link between cigarette smoking and RA, and the data presented so far are consistent with and suggestive of an increased risk.
Similar content being viewed by others

Introduction
This chapter reviews recent epidemiological data on the relative contributions of genetic and environmental risk factors for the development of RA. It considers and proposes the direct and indirect evidence to the contribution of various risk factors for disease susceptibility. The quality of the evidence varies and, where appropriate, this is highlighted.
Genetic factors
Descriptive epidemiology of RA
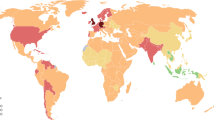
The descriptive epidemiology of RA is suggestive of a genetic effect. The occurrence of RA is relatively constant with a prevalence of between 0.5 and 1.0%, a frequency that has been reported from several European [1–8] and North-American populations [9, 10]. However, there are some interesting exceptions (Fig. 1).

Specifically, native American-Indian populations have the highest recorded occurrence of RA, with a prevalence of 5.3% noted for the Pima Indians [11] and of 6.8% for the Chippewa Indians [12]. By contrast, there are a number of groups with a very low occurrence. Studies in rural African populations, both in South Africa [13] and in Nigeria [14], failed to find any RA cases in studies of 500 and 2000 adults, respectively. Studies in populations from Southeast Asia [15], including China and Japan [16, 17], have similarly shown very low occurrences (0.2–0.3%).
'Migrant' studies
It is clearly difficult from a review of the descriptive epidemiological data to know whether environmental or genetic effects explain the differences between countries. One handle on this is to consider the occurrence in populations presumed to be of the same genetic origin but living in different environments. Such a situation arises by studying populations that have moved from one environment to another.
There are a few studies addressing this with respect to RA. A low occurrence of disease was found in one study of a Caribbean population of African origin living in Manchester, UK, suggesting that the protection to this group was indeed genetically determined [18]. Similarly, the investigation of a Chinese population living in an urban environment in Hong Kong showed the same consistent low frequency [19]. Recent data have shown, however, that Pakistanis living in England had a higher prevalence than those in Pakistan, but it is not as high as the prevalence in ethnic English populations [20]. In general, the data on the geographical occurrence of RA would support the existence of genetic factors being important and explaining differences in disease risk.
Familial clustering
The next stage in accruing evidence for genetic risk is to document an increased occurrence of disease in relatives of probands compared with the background population prevalence, so-called familial recurrence risk. Studies of hospital attendees are subject to bias as there may be a selection process whereby individuals would more probably be referred to hospital if they have an affected family member. Furthermore, several studies rely on family history as elicited by the proband, which again is subject to bias.
Few studies have been performed comparing familial recurrence risk in relatives of cases derived from population samples with those of controls. Indeed, such studies have only shown a modest increased risk [21, 22]. For example, a study from the Norfolk Arthritis Register in England showed only a twofold increased risk [23]. Such an observation does not negate the role of genetic factors, but underscores that their contribution to explain disease susceptibility may be modest. This is important as the familial recurrence risk is a key factor in determining the power of genetic linkage studies within affected family pairs. Indeed, in contrast to other autoimmune diseases such as insulin-dependent diabetes and multiple sclerosis, the familial recurrence risk in RA is certainly smaller, thereby making it harder for studies to identify new genetic factors.
Twin studies
A variant of studies of familial recurrence risk is the comparison of disease risk in the initially unaffected co-twin of monozygotic probands compared with dizygotic probands. The assumption is that the environmental sharing between these different twin pair types is the same and thus any increased disease concordance in the monozygotic twins confirms the genetic effect. It is important in such studies to ensure that it is only like-sexed dizygotic twin pairs that are compared with the monozygotic twin pairs. However, there may be a greater environmental concordance in monozygotic twins due, for example, to psychological and other factors.
Twin studies have consistently showed a fourfold increased concordance in monozygotic twins compared with dizygotic twins [24]. This increased risk, however, is of little value in attempting to quantify the genetic contribution to disease risk. The concordance between twins is dependent on the prevalence of disease. As the population prevalence approaches 100%, the concordance will increase accordingly, independent of the true genetic effect. The appropriate way of quantifying genetic risk is to assess the heritability based on a series of assumptions of environmental sharing and genetic sharing between twin types. Such a study has recently been attempted using data from both Finnish and English twins [24]. The results suggest that approximately 50–60% of the occurrence of disease in the twins is explained by shared genetic effects.
Genetic susceptibility factors: human leukocyte antigen
The role of HLA DRB1 alleles as a risk factor of RA has been known for 25 years. Associations between different HLA DRB1 alleles have been demonstrated in several populations across the world [25–31]. Indeed, there have been few populations where associations have not been demonstrated.
Interestingly, there do appear to be differences in the strength of association between different alleles. For example, HLA DRB1*0404 is a much stronger susceptibility factor than HLA DRB1*0101 [32] (Table 1). Although it has been suggested that the susceptibility alleles all share a single epitope [33], it is difficult to explain the variable risk under this model. Furthermore, the risk of disease is related not only to the presence of one single allele, but also to the full HLA DRB1 genotype [34]. Individuals who carry the so-called 'compound heterozygote' genotype HLA DRB*0401/*0404 thus have a substantially greater risk than, for example, individuals who carry single HLA DRB*0101 alleles.
There is some suggestion that the relationship between human leukocyte antigen (HLA) and RA may be more related to the severity of disease, and that the development of arthritis per se is only weakly related. Support for this comes from studies from the Norfolk Arthritis Register population-based study of inflammatory joint disease in England [32]. Data from this study show only a weak relationship between susceptibility to the disease and the HLA DRB1 genotype (Table 1). The association is fairly strong in those individuals who satisfy the criteria for RA. The data show there is an influence of genotype, with some genotypes having a stronger association as shown.
The HLA region on the short arm of chromosome 6 is a gene-rich area including several candidate genes that have an influence on the immune process. One of the most highly investigated is tumour necrosis factor (TNF). Studies have shown associations between TNF alleles and RA [35, 36], although one explanation may be linkage disequilibrium with HLA DRB1. Studies have also suggested, however, that the associations between HLA and TNF-c1 and TNF-b3 are independent of associations between HLA and the shared epitope [37]. Other studies have shown an extended haplotype stretching from HLA through to TNF that has been implicated in disease [38].
Genetic susceptibility factors: non-MHC genes
Data from twin studies in the HLA association and sharing studies have been used to estimate that only 50% of the genetic contribution to RA can be explained by HLA [24]. This has sparked a search for non-MHC genes.
The largest effort has been expanded in some whole genome screens on affected sibling pair families. Four such screens have now been undertaken in Europe [39], the United States [33], Japan [40] and the United Kingdom [41]. A number of markers emerge from these studies suggestive of a linkage with RA, although the linkage with HLA is by far the strongest. One problem is that such studies often have only a weak power to detect defects. By contrast, because such studies may be simultaneously testing the possibility that any one of 200 regions may be linked with disease, the likelihood of a false-positive result is also very high. It is for this reason that it is not surprising studies often fail to replicate results both between themselves and on further samples within the population. It is therefore necessary to undertake further validation studies and more in-depth investigations, using more closely spaced markers.
An alternative approach is to use a candidate gene screen where there is no prior reason for looking at a particular region. Such an approach has been productive, and evidence has shown that corticotrophin releasing hormone [42], CYP19 (oestrogen synthase) [43], IFN-γ [44–46] and other cytokines [47–49] are linked to RA. Other approaches have addressed the possibility that genetic regions linked to other autoimmune diseases, such as insulin-dependent diabetes [50], may also be linked to RA. Indeed, linkage to a locus on chromosome X was shown in one study [51]. A further strategy has been to use results and genome screens on animal models of arthritis to see whether syntenic regions are also linked to RA in humans. Such studies have suggested linkage to 17q22 [52].
Whether any of these positive findings discussed will result in the identification of a true disease susceptibility mutation remains to be seen. However, one clear problem is that RA itself is probably heterogeneous and studies that fail to take notice of this heterogeneity may make it possible to find a positive result.
Environmental factors
The term 'environment' is frequently used to describe all those susceptibility factors leading to disease that are not explicable on the basis of an identifiable genetic marker. In a strict sense, however, environment could be taken to refer to those factors external to the individual; for example, factors associated with diet, water or air-borne exposures. It is also important to consider factors implicated with diseases that are internal to the subject without an obvious genetic basis. An appropriate term for this group of factors is 'nongenetic host factors'.
Nongenetic host factors: hormonal and pregnancy factors
The increased risk of RA in females has lead to considerable effort in examining the role of hormonal and pregnancy factors in disease occurrence. In general, male sex hormones, particularly testosterone, are lower in men who have RA [25]. By contrast, levels of female sex hormones are not different between RA cases and controls [53].
Interestingly, exogenous hormonal influences are implicated in disease risk. The most widely studied of these is exposure to the oral contraceptive pill, based on an observation made over 20 years ago [54] (Fig. 2). There have been several studies [55] confirming that women who take the oral contraceptive pill are at reduced risk of developing RA [25]. There is no clear explanation for this and the association exists despite the formulation of the oral contraceptive pill varying enormously both between populations and over time. A follow-up of the original study was undertaken that suggested the oral contraceptive pill was protective. This showed that the initial protection was lost on follow-up [56]. One conclusion might therefore be that oral contraceptive use may postpone, rather than totally protect against, the development of RA.

The incidence of rheumatoid arthritis in relation to use of the oral contraceptive pill (OC). Data from the Royal College of General Practioners' oral contraception study [54].
Pregnancy itself has been investigated as a risk factor in RA development. Studies on the influence of pregnancy on RA have produced conflicting results. A number of studies [25] have suggested that women who are nulliparous are at increased risk of developing the disease, although there is no increased risk in women who are single [57]. It would thus appear that subfertility highlights a group at higher risk.
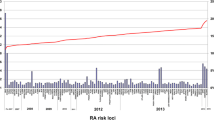
Recent studies have suggested that pregnancy might also be important with the interesting observation that the postpartum period, particularly after the first pregnancy, represents a strong risk period of disease development [58, 59] (Fig. 3). Subsequent investigations showed that much of this increased risk could be explained by exposure to breastfeeding and it is women who breastfeed after their first pregnancy who are at the greatest risk [60]. The suggestion then arose linking breastfeeding with sub-fertility in so far as RA may be related to either increased prolactin or abnormal response to prolactin, this latter hormone being proinflammatory [61].

Increased risk of rheumatoid arthritis onset in the postpartum period. * Relative to nonpregnant periods. Data from [59].
Nongenetic host factors: other
There have been a number of studies looking at other comorbidities that have an increased frequency in both subjects with RA and in their families. The most widely investigated has been the occurrence of other autoimmune diseases, particularly type 1 or insulin-dependent diabetes and autoimmune thyroid disease [62]. Other diseases, for example schizophrenia, have been shown to be negatively associated with RA development [63–65]. The significance of these findings is unclear.
There have been relatively few studies on anthropometric factors associated with RA, although one recent case–control study suggested that people who were obese were at higher risk [66]. The reason for this is unclear, and it is not certain whether this may represent a confounding factor of another exposure or whether people who are obese have, for example, increased production of oestrogens, which might pose a risk. A more recent case–control study found, however, after adjusting for age, smoking and marital status, that a link with obesity was nonsignificant [67].
Environmental factors: infection
Indirect evidence
There is much indirect evidence suggesting that exposure to infectious agents may be the trigger for RA. First, epidemiological data come from the observation of a decline in the incidence of RA in several populations [9, 16]. Many studies have indeed shown a halving in incidence over the past 30 years [68]. Given the genetically stable population, the most probable explanation is that of a decline in an infectious trigger. This effect of time on occurrence might also be related to the period of birth as well as to the current year of observations. The Pima Indians, for example, showed a decline in occurrence of disease, and an indepth study based on analysis of birth cohorts has shown a decline in the population occurrence of rheumatoid factor with increasingly recent birth cohorts [69].
There have been a few studies looking at clustering of RA in time and space, although there have been reports of nonrandom clusters occurring within the Norfolk Arthritis Register population [70]. Other indirect evidence regarding the role of an infectious agent has arisen from case–control studies suggesting that people who have had a blood transfusion, even some years prior to disease onset, may be at an increased risk of disease [66]. Recent practice has been to screen blood for a number of agents such as hepatitis, but the increased reporting of blood transfusion in older cohorts may indeed be explained by the increased likelihood of infection.
Possible infectious agents
There have been a large number of infectious agents that have been implicated in RA, including Epstein–Barr virus and parvovirus, as well as other agents, including bacteria such as Proteus and Mycoplasma. The epidemiological studies supporting or refuting these possible links are reviewed elsewhere [25] but, in general, such studies have been disappointing. One problem for the epidemiologist is that if RA represents the final common pathway of exposure to one of several different potential susceptibility organisms, many of which are also frequently observed in the general (i.e. nonarthritic population), it makes it more difficult to confirm a relationship with epidemiological studies.
Noninfectious environmental factors
There have been remarkably few studies on factors such as diet, although there is a theoretical basis for investigating the role of omega-3 fatty acids [71, 72]. Randomised trials suggest that diets high in eicosapentaenoic acid have a favourable effect on the outcome of RA [73–75]. This might be because such fatty acids compete with arachidonic acids, the latter of which are involved in inflammation. Whether such dietary factors have a role in RA onset is much less clear.
It is perhaps surprising, given how much this exposure has been investigated in other chronic diseases, that very little attention has been given to cigarette smoking until recently. However, findings from a number of recent studies showed that cigarette smoking is associated with an increased risk of RA [66, 67, 76–79] (Table 2). Studies have also suggested that smoking is related to development of rheumatoid factor independent of RA. Indeed, in many of the epidemiological studies showing a relationship between smoking and RA, the positive findings have been restricted to those with rheumatoid factor.
Future prospects
There has been considerable recent interest in understanding the epidemiology of RA. There have been several population studies in many different countries around the world, and observations of differential occurrence (with time, between populations and between the genders) has stimulated a number of analytical studies looking for both genetic and environmental risk factors. Future studies will benefit from advances in molecular biology techniques to aid with the identification and characterisation of potential new genes for RA susceptibility. These studies, as already described, have revealed some tantalising clues that will require further follow-up in years to come.
Concluding remarks
RA presents an epidemiological challenge and further elucidation of both genetic and environmental factors, together with interactions between them, are likely to be revealed.
Organisations supplying funds for research
Funds can be obtained from the Arthritis Research Campaign http://www.arc.org.uk and the Arthritis Foundation http://www.arthritis.org.
References
Carmona L, Villaverde V, Hernandez-Garcia C, Ballina J, Gabriel R, Laffon A: The prevalence of rheumatoid arthritis in the general population of Spain. Rheumatology. 2002, 41: 88-95. 10.1093/rheumatology/41.1.88.
Riise T, Jacobsen BK, Gran JT: Incidence and prevalence of rheumatoid arthritis in the county of Troms, northern Norway. J Rheumatol. 2000, 27: 1386-1389.
Aho K, Kaipiainen-Seppanen O, Heliovaara M, Klaukka T: Epidemiology of rheumatoid arthritis in Finland. Semin Arthritis Rheum. 1998, 27: 325-334.
Cimmino MA, Parisi M, Moggiana G, Mela GS, Accardo S: Prevalence of rheumatoid arthritis in Italy: the Chiavari study. Ann Rheum Dis. 1998, 57: 315-318.
Kvien TK, Glennas A, Knudsrod OG, Smedstad LM, Mowinckel P, Forre O: The prevalence and severity of rheumatoid arthritis in Oslo: results from a county register and a population survey. Scand J Rheumatol. 1997, 26: 412-418.
Power D, Codd M, Ivers L, Sant S, Barry M: Prevalence of rheumatoid arthritis in Dublin, Ireland: a population based survey. Ir J Med Sci. 1999, 168: 197-200.
Saraux A, Guedes C, Allain J, Devauchelle V, Valls I, Lamour A, Guillemin F, Youinou P, Le Goff P: Prevalence of rheumatoid arthritis and spondyloarthropathy in Brittany, France. J Rheumatol. 1999, 26: 2622-2627.
Simonsson M, Bergman S, Jacobsson LT, Petersson IF, Svensson B: The prevalence of rheumatoid arthritis in Sweden. Scand J Rheumatol. 1999, 28: 340-343. 10.1080/03009749950155319.
Gabriel SE, Crowson CS, O'Fallon WM: The epidemiology of rheumatoid arthritis in Rochester, Minnesota, 1955–1985. Arthritis Rheum. 1999, 42: 415-420. 10.1002/1529-0131(199904)42:3<415::AID-ANR4>3.0.CO;2-Z.
Gabriel SE: The epidemiology of rheumatoid arthritis. Rheum Dis Clin North Am. 2001, 27: 269-281.
del Puente A, Knowler WC, Pettit DJ, Bennett PH: High incidence and prevalence of rheumatoid arthritis in Pima Indians. Am J Epidemiol. 1989, 129: 1170-1178.
Harvey J, Lotze M, Stevens MB, Lambert G, Jacobson D: Rheumatoid arthritis in a Chippewa band. I. Pilot screening study of disease prevalence. Arthritis Rheum. 1981, 24: 717-721.
Brighton SW, de la Harpe AL, van Staden DJ, Badenhorst JH, Myers OL: The prevalence of rheumatoid arthritis in a rural African population. J Rheumatol. 1988, 15: 405-408.
Silman AJ, Ollier W, Holligan S, Birrell F, Adebajo A, Asuzu MC, Thomson W, Pepper L: Absence of rheumatoid arthritis in a rural Nigerian population. J Rheumatol. 1993, 20: 618-622.
Dans LF, TankehTorres S, Amante CM, Penserga EG: The prevalence of rheumatic diseases in a Filipino urban population: a WHO-ILAR COPCORD study. J Rheumatol. 1997, 24: 1814-1819.
Shichikawa K, Inoue K, Hirota S, Maeda A, Ota H, Kimura M, Ushiyama T, Tsujimoto M: Changes in the incidence and prevalence of rheumatoid arthritis in Kamitonda, Wakayama, Japan, 1965–1996. Ann Rheum Dis. 1999, 58: 751-756.
Zeng Q, Huang S, Chen R: 10-year epidemiological study on rheumatic diseases in Shantou area. Zhonghua Nei Ke Za Zhi. 1997, 36: 193-197.
MacGregor AJ, Riste LK, Hazes JMW, Silman AJ: Low prevalence of rheumatoid arthritis in black-Caribbeans compared with whites in inner city Manchester. Ann Rheum Dis. 1994, 53: 293-297.
Lau E, Symmons D, Bankhead C, MacGregor A, Donnan S, Silman A: Low prevalence of rheumatoid arthritis in the urbanized Chinese of Hong Kong. J Rheumatol. 1993, 20: 1133-1137.
Hameed K, Gibson T: A comparison of the prevalence of rheumatoid arthritis and other rheumatic diseases amongst Pakistanis living in England and Pakistan. Br J Rheumatol. 1997, 36: 781-785. 10.1093/rheumatology/36.7.781.
Lawrence JS, Ball J: Genetic studies on rheumatoid arthritis. Ann Rheum Dis. 1958, 17: 160-168.
Lawrence JS: Heberden oration, 1969. Rheumatoid arthritis – nature or nurture?. Ann Rheum Dis. 1970, 29: 357-379.
Jones MA, Silman AJ, Whiting S, Barrett EM, Symmons DPM: Occurrence of rheumatoid arthritis is not increased in the first degree relatives of a population based inception cohort of inflammatory polyarthritis. Ann Rheum Dis. 1996, 55: 89-93.
MacGregor AJ, Snieder H, Rigby AS, Koskenvuo M, Kaprio J, Aho K, Silman AJ: Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 2000, 43: 30-37. 10.1002/1529-0131(200001)43:1<30::AID-ANR5>3.0.CO;2-B.
Silman AJ, Hochberg MC: Rheumatoid arthritis. In Epidemiology of the Rheumatic Diseases. Edited by: Edited by AJ Silman, MC Hochberg. 2001, Oxford: Oxford University Press;, 31-71.
Pascual M, Nieto A, Lopez-Nevot MA, Ramal L, Mataran L, Caballero A, Alonso A, Martin J, Zanelli E: Rheumatoid arthritis in southern Spain – toward elucidation of a unifying role of the HLA class II region in disease predisposition. Arthritis Rheum. 2001, 44: 307-314. 10.1002/1529-0131(200102)44:2<307::AID-ANR47>3.3.CO;2-B.
Citera G, Padulo LA, Fernandez G, Lazaro MA, Rosemffet MG, Cocco JAM: Influence of HLA-DR alleles on rheumatoid arthritis: susceptibility and severity in Argentine patients. J Rheumatol. 2001, 28: 1486-1491.
Zanelli E, Breedveld FC, de Vries RRP: HLA class II association with rheumatoid arthritis – facts and interpretations. Hum Immunol. 2000, 61: 1254-1261. 10.1016/S0198-8859(00)00185-3.
Balsa A, Minaur NJ, Pascual-Salcedo D, McCabe C, Balas A, Fiddament B, Vicario JL, Cox NL, Martin-Mola E, Hall ND: Class II MHC antigens in early rheumatoid arthritis in Bath (UK) and Madrid (Spain). Rheumatology. 2000, 39: 844-849. 10.1093/rheumatology/39.8.844.
del Rincon I, Escalante A: HLA-DRB1 alleles associated with susceptibility or resistance to rheumatoid arthritis, articular deformities, and disability in Mexican Americans. Arthritis Rheum. 1999, 42: 1329-1338. 10.1002/1529-0131(199907)42:7<1329::AID-ANR5>3.0.CO;2-1.
Wakitani S, Murata N, Toda Y, Ogawa R, Kaneshige T, Nishimura Y, Ochi T: The relationship between HLA-DRB1 alleles and disease subsets of rheumatoid arthritis in Japanese. Br J Rheumatol. 1997, 36: 630-636. 10.1093/rheumatology/36.6.630.
Thomson W, Harrison B, Ollier B, Wiles N, Payton T, Barrett J, Symmons D, Silman A: Quantifying the exact role of HLA-DRB1 alleles in susceptibility to inflammatory polyarthritis: results from a large, population-based study. Arthritis Rheum. 1999, 42: 757-762. 10.1002/1529-0131(199904)42:4<757::AID-ANR20>3.3.CO;2-O.
Gregersen PK: The North American Rheumatoid Arthritis Consortium – bringing genetic analysis to bear on disease susceptibility, severity, and outcome. Arthritis Care Res. 1998, 11: 1-2.
Meyer JM, Evans TI, Small RE, Redford TW, Han JF, Singh R, Moxley G: HLA-DRB1 genotype influences risk for and severity of rheumatoid arthritis. J Rheumatol. 1999, 26: 1024-1034.
Mattey DL, Hassell AB, Dawes PT, Ollier WE, Hajeer A: Interaction between tumor necrosis factor microsatellite polymorphisms and the HLA-DRB1 shared epitope in rheumatoid arthritis: influence on disease outcome. Arthritis Rheum. 1999, 42: 2698-2704. 10.1002/1529-0131(199912)42:12<2698::AID-ANR28>3.0.CO;2-S.
Barton A, John S, Ollier WER, Silman A, Worthington J: Association between rheumatoid arthritis and polymorphism of tumor necrosis factor receptor II, but not tumor necrosis factor receptor I, in Caucasians. Arthritis Rheum. 2001, 44: 61-65. 10.1002/1529-0131(200101)44:1<61::AID-ANR9>3.0.CO;2-Q.
Hajeer AH, Dababneh A, Makki RF, Thomson W, Poulton K, Gay MA, Garcia-Porrua C, Mattey DL, Ollier WE: Different gene loci within the HLA-DR and TNF regions are independently associated with susceptibility and severity in Spanish rheumatoid arthritis patients. Tissue Antigens. 2000, 55: 319-325. 10.1034/j.1399-0039.2000.550405.x.
Hajeer AH, Worthington J, Silman AJ, Ollier WER: Association of tumor necrosis factor microsatellite polymorphisms with HLA-DRB1(*)04-bearing haplotypes in rheumatoid arthritis patients. Arthritis Rheum. 1996, 39: 1109-1114.
Cornelis F, Faure S, Martinez M, Prudhomme JF, Fritz P, Dib C, Alves H, Barrera P, de Vries N, Balsa A, Pascual-Salcedo D, Maenaut K, Westhovens R, Migliorini P, Tran TH, Delaye A, Prince N, Lefevre C, Thomas G, Poirier M, Soubigou S, Alibert O, Lasbleiz S, Fouix S, Bouchier C, Liote F, Loste MN, Lepage V, Charron D, Gyapay G, Lopes-Vaz A, Kuntz D, Bardin T, Weissenbach J: New susceptibility locus for rheumatoid arthritis suggested by a genome-wide linkage study. Proc Natl Acad Sci USA. 1998, 95: 10746-10750. 10.1073/pnas.95.18.10746.
Shiozawa S, Hayashi S, Tsukamoto Y, Goko H, Kawasaki H, Wada T, Shimizu K, Yasuda N, Kamatani N, Takasugi K, Tanaka Y, Shiozawa K, Imura S: Identification of the gene loci that predispose to rheumatoid arthritis. Int Immunol. 1998, 10: 1891-1895. 10.1093/intimm/10.12.1891.
Worthington J, Ollier WE, Leach MK, Smith I, Hay EM, Thomson W, Pepper L, Carthy D, Farhan A, Martin S, Dyer P, Davison J, Bamber S, Silman AJ: The Arthritis and Rheumatism Council's National Repository of Family Material: pedigrees from the first 100 rheumatoid arthritis families containing affected sibling pairs. Br J Rheumatol. 1994, 33: 970-976.
Fife MS, Fisher SA, John S, Worthington J, Shah CJ, Ollier WER, Panayi GS, Lewis CM, Lanchbury JS: Multipoint linkage analysis of a candidate gene locus in rheumatoid arthritis demonstrates significant evidence of linkage and association with the corticotropin-releasing hormone genomic region. Arthritis Rheum. 2000, 43: 1673-1678. 10.1002/1529-0131(200008)43:8<1673::AID-ANR2>3.0.CO;2-Y.
John S, Myerscough A, Eyre S, Roby P, Hajeer A, Silman AJ, Ollier WE, Worthington J: Linkage of a marker in intron D of the estrogen synthase locus to rheumatoid arthritis. Arthritis Rheum. 1999, 42: 1617-1620. 10.1002/1529-0131(199908)42:8<1617::AID-ANR8>3.0.CO;2-N.
Khani-Hanjani A, Lacaille D, Hoar D, Chalmers A, Horsman D, Anderson M, Balshaw R, Keown PA: Association between dinucleotide repeat in non-coding region of interferon-gamma gene and susceptibility to, and severity of, rheumatoid arthritis. Lancet. 2000, 356: 820-825. 10.1016/S0140-6736(00)02657-X.
Ollier WE: Role of interferon-gamma gene in rheumatoid arthritis?. Lancet. 2000, 356: 783-784. 10.1016/S0140-6736(00)02647-7.
Pokorny V, McLean L, McQueen F, Abu-Maree M, Yeoman S: Interferon-gamma microsatellite and rheumatoid arthritis. Lancet. 2001, 358: 122-123. 10.1016/S0140-6736(01)05342-9.
Hajeer AH, Lazarus M, Turner D, Mageed RA, Vencovsky J, Sinnott P, Hutchinson IV, Ollier WER: IL-10 gene promoter polymorphisms in rheumatoid arthritis. Scand J Rheumatol. 1998, 27: 142-145. 10.1080/030097498441029.
John S, Myerscough A, Marlow A, Hajeer A, Silman A, Ollier W, Worthington J: Linkage of cytokine genes to rheumatoid arthritis. Evidence of genetic heterogeneity. Ann Rheum Dis. 1998, 57: 361-365.
John S, Eyre S, Myerscough A, Barrett J, Silman A, Ollier W, Worthington J: Linkage and association analysis of candidate genes in rheumatoid arthritis. J Rheumatol. 2001, 28: 1752-1755.
Myerscough A, John S, Barrett JH, Ollier WE, Worthington J: Linkage of rheumatoid arthritis to insulin-dependent diabetes mellitus loci: evidence supporting a hypothesis for the existence of common autoimmune susceptibility loci. Arthritis Rheum. 2000, 43: 2771-2775. 10.1002/1529-0131(200012)43:12<2771::AID-ANR17>3.0.CO;2-V.
Myerscough A, John S, Barrett JH, Eyre S, Barton A, Brintnell B, Ollier WER, Worthington J: Linkage and linkage disequilibrium analysis of chromosome Xp11–p21 microsatellite markers with rheumatoid arthritis (RA) [abstract]. Arthritis Rheum. 2001, 44 (Suppl): 1299-
Barton A, Eyre S, Myerscough A, Brintnell B, Ward D, Ollier WE, Lorentzen JC, Klareskog L, Silman A, John S, Worthington J: High resolution linkage and association mapping identifies a novel rheumatoid arthritis susceptibility locus homologous to one linked to two rat models of inflammatory arthritis. Hum Mol Genet. 2001, 10: 1901-1906. 10.1093/hmg/10.18.1901.
Heikkila R, Aho K, Heliövaara M, Knekt P, Reunanen A, Aromaa A, Leino A, Palosuo T: Serum androgen-anabolic hormones and the risk of rheumatoid arthritis. Ann Rheum Dis. 1998, 57: 281-285.
Anonymous: Reduction in incidence of rheumatoid arthritis associated with oral contraceptives. Royal College of General Practioners' oral contraception study. Lancet. 1978, 1: 569-571.
Brennan P, Bankhead C, Silman A, Symmons D: Oral contraceptives and rheumatoid arthritis: results from primary care-based incident case–control study. Semin Arthritis Rheum. 1997, 26: 817-823.
Hannaford PC, Kay CR, Hirsch S: Oral contraceptives and rheumatoid arthritis: new data from the Royal College of General Practitioners' oral contraception study. Ann Rheum Dis. 1990, 49: 744-746.
Silman AJ: Epidemiology of rheumatoid arthritis. APMIS. 1994, 102: 721-728.
Nelson JL, Ostensen M: Pregnancy and rheumatoid arthritis. Rheum Dis Clin North Am. 1997, 23: 195-212.
Silman AJ, Kay A, Brennan P: Timing of pregnancy in relation to the onset of rheumatoid arthritis. Arthritis Rheum. 1992, 35: 152-155.
Brennan P, Silman A: Breast-feeding and the onset of rheumatoid arthritis. Arthritis Rheum. 1994, 37: 808-813.
Brennan P, Hajeer A, Ong KR, Worthington J, John S, Thomson W, Silman A, Ollier B: Allelic markers close to prolactin are associated with HLA-DRB1 susceptibility alleles among women with rheumatoid arthritis and systemic lupus erythematosus. Arthritis Rheum. 1997, 40: 1383-1386.
Silman AJ, Ollier WER, Bubel MA: Autoimmune thyroid disease and thyroid autoantibodies in rheumatoid arthritis patients and their families. Br J Rheumatol. 1989, 28: 18-21.
Oken RJ, Schulzer M: At issue: schizophrenia and rheumatoid arthritis: the negative association revisited. Schizophr Bull. 1999, 25: 625-638.
Mors O, Mortensen PB, Ewald H: A population-based register study of the association between schizophrenia and rheumatoid arthritis. Schizophr Res. 1999, 40: 67-74. 10.1016/S0920-9964(99)00030-4.
Rubinstein G: Schizophrenia, rheumatoid arthritis and natural resistance genes. Schizophr Res. 1997, 25: 177-181. 10.1016/S0920-9964(97)00023-6.
Symmons DPM, Bankhead CR, Harrison BJ, Brennan P, Barrett EM, Scott DGI, Silman AJ: Blood transfusion, smoking, and obesity as risk factors for the development of rheumatoid arthritis: results from a primary care-based incident case-control study in Norfolk, England. Arthritis Rheum. 1997, 40: 1955-1961.
Uhlig T, Hagen KB, Kvien TK: Current tobacco smoking, formal education, and the risk of rheumatoid arthritis. J Rheumatol. 1999, 26: 47-54.
Jacobsson LT, Hanson RL, Knowler WC, Pillemer S, Pettitt DJ, McCance DR, Bennett PH: Decreasing incidence and prevalence of rheumatoid arthritis in Pima Indians over a 25 year period. Arthritis Rheum. 1994, 37: 1158-1165.
Silman AJ, Enzer I, Knowler W, Dunn G, Jacobsson L: Strong influence of period of birth on the occurrence of rheumatoid factor: results from a 30 year follow-up study on Pima Indians [abstract]. Arthritis Rheum. 2000, 43 (Suppl): 605-
Silman AJ, Bankhead C, Rowlingson B, Brennan P, Symmons D, Gatrell A: Do new cases of rheumatoid arthritis cluster in time or in space?. Int J Epidemiol. 1997, 26: 628-634. 10.1093/ije/26.3.628.
Ariza-Ariza R, Mestanza-Peralta M, Cardiel MH: Omega-3 fatty acids in rheumatoid arthritis: an overview. Semin Arthritis Rheum. 1998, 27: 366-370.
James MJ, Cleland LG: Dietary n-3 fatty acids and therapy for rheumatoid arthritis. Semin Arthritis Rheum. 1997, 27: 85-97.
Volker D, Fitzgerald P, Major G, Garg M: Efficacy of fish oil concentrate in the treatment of rheumatoid arthritis. J Rheumatol. 2000, 27: 2343-2346.
Kremer JM: n-3 fatty acid supplements in rheumatoid arthritis. Am J Clin Nutr. 2000, 71 (Suppl): 349S-351S.
Hernandez-Cruz B, Alcocer-Varela J, Cardiel MH: Omega-3 fatty acids supplementation in Mexican patients with rheumatoid arthritis with standard treatment. A blinded, randomized, placebo controlled, one year, clinical trial [abstract]. Arthritis Rheum. 1998, 41 (Suppl): 738-
Albano SA, Santana-Sahagun E, Weisman MH: Cigarette smoking and rheumatoid arthritis. Semin Arthritis Rheum. 2001, 31: 146-159. 10.1053/sarh.2001.27719.
Harrison BJ, Silman AJ, Wiles NJ, Scott DG, Symmons DP: The association of cigarette smoking with disease outcome in patients with early inflammatory polyarthritis. Arthritis Rheum. 2001, 44: 323-330. 10.1002/1529-0131(200102)44:2<323::AID-ANR49>3.3.CO;2-3.
Hutchinson D, Shepstone L, Moots R, Lear JT, Lynch MP: Heavy cigarette smoking is strongly associated with rheumatoid arthritis (RA), particularly in patients without a family history of RA. Ann Rheum Dis. 2001, 60: 223-227. 10.1136/ard.60.3.223.
Karlson EW, Min Lee I, Cook NR, Manson JE, Buring JE, Hennekens CH: A retrospective cohort study of cigarette smoking and risk of rheumatoid arthritis in female health professionals. Arthritis Rheum. 1999, 42: 910-917. 10.1002/1529-0131(199905)42:5<910::AID-ANR9>3.3.CO;2-4.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Silman, A.J., Pearson, J.E. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res Ther 4 (Suppl 3), S265 (2002). https://doi.org/10.1186/ar578
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar578