
Abstract
Rheumatoid arthritis (RA) is an inflammatory autoimmune disease involving symmetric joints and is generally characterized by persistent pain, tenderness, and destruction of joints. The vast majority of RA patients produce autoantibodies, and immune cell involvement in disease development is well recognized, as is the contribution of other types of cells in synovial tissue, like fibroblasts. It is known that there are major genetic associations with the HLA locus, while multiple non-HLA genetic variants display relatively low risk of RA. Both HLA and non-HLA associations suggest that the profiles of genetic associations for autoantibody-positive vs. autoantibody-negative RA are different. Several alleles of HLA-DRB1 are associated with high risk for autoantibody-positive RA, with the strongest risk characterized by valine at position 11 of the protein sequence (HLA-DRB1*04 and *10 alleles). There is a strong protective effect for the risk of autoantibody-positive RA associated with HLA-DRB1*13 alleles. Although major genetic associations have been known for several years, understanding of the specific mechanisms in the development of increased risk of RA for these variations is work in progress. Current studies focus on the binding of immune receptors involved in recognition of putative peptides in activation of T cells, as well as investigation of cell signaling mechanisms. At least a part of RA risk could be explained by gene–gene and gene-environment interactions. There are currently more than 150 candidate loci with polymorphisms that associate with RA, mainly related to seropositive disease, and new discoveries are anticipated in the future from investigation of diverse human populations. This new research will help create a strong foundation for the continuing process of integrating genetic, epigenetic, transcriptomic, and proteomic data in studies of RA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Rheumatoid arthritis (RA) is a relatively common inflammatory disease in different world populations [1]. It does not generate immediate risk of death, such as with CVD or aggressive cancer, but significantly affects the everyday life of the patients and, together with unpleasant symptoms, may cause severe destruction of joints, multiple long-term side effects, and decreased life expectancy [2]. The cause of RA remains unknown, with the consensus of researchers being that multiple genetic and environmental factors are involved [3]. Therefore, large epidemiological studies to identify these factors have been performed, with a major focus on genetics. Historically, the first genome-wide association studies (GWAS) of RA were performed in 2007, the beginning of the GWAS era, in three parallel and separate studies: WTCCC, EIRA, and NARAC [4, 5]. During the following years, additional studies in several populations contributed to further discoveries of candidate genes, with the largest recent meta-analyses performed for 29,880 RA patients and 73,758 controls [6], for 22,628 RA patients and 288,664 controls [7], and recently for 35,871 RA patients and 240,149 controls [8], with notable overlap between these three studies. These data revealed a significant difference between the genetic effects of HLA haplotypes and non-HLA risk alleles, something which is an important attribute not only for RA, but of most autoimmune diseases.
RA is a phenotypically heterogeneous syndrome with diagnostic criteria based on clinical evaluation. The presence of autoantibody is of high value for diagnostical scores and may bias inclusion of patients to the study away from mild autoantibody-negative cases towards autoantibody-positive cases [9]. In a similar way to other complex diseases, sub-phenotyping of RA based on clinical and serological features is of great importance for interpretation of genetic associations. On the other hand, many genetic factors related to RA are also common for different autoimmune diseases, which strengthens the overall understanding of autoimmune mechanisms of human diseases and creates possibilities for future fine tuning of the diagnostic criteria.
Recent genetic analyses led to the discovery of more than 150 loci with association to RA, with HLA associations remaining most strong [6,7,8]. The HLA alleles were also found to be involved in multiple interactions with environmental and other genetic risk factors in the development of an increased risk of RA [10, 11]. A growing bulk of data from studies in the epigenetics, transcriptomics, and proteomics of RA requires proper integration with clinical data. It is also evident that the study of pre-disease is vital for understanding the early contribution of genetic risk factors in RA development, while studies of full-scale disease and more chronic cases may serve better for elucidating the involvement of genetic factors in treatment response and development of side effects and comorbidities.
One of the greatest challenges of genetic epidemiology studies is giving a functional interpretation from purely statistical evidence. Attribution of genetic associations to specific cellular or molecular phenotypes is not a straightforward process. Most previous genetic studies annotated associated SNPs by physical overlap with a particular gene or with a neighboring gene. With the development of our knowledge about human genome architecture, this type of annotation of signals has often been proven inaccurate and sometimes even misleading [12, 13]. Annotation requires broad assessment of the function of genes that are sometimes relatively distant from the association hit. The reasons for this confusion include linkage disequilibrium within genetic loci, but also the chromosome structure and complexity of chromatin function. With the advent of single-cell omics, a significantly more specific and robust annotation is expected in all areas of genetic research.
Disease classification issues and autoantibodies in rheumatoid arthritis
As with any complex disease, RA represents a collection of symptoms, and the diagnosis is consensus based [9]. This may generate some uncertainty at initial stages of the disease and may cause misclassification due to overlap of symptoms between different rheumatic diseases. On the other hand, the study of only well-established chronic disease is likely to be biased towards individuals with more severe RA and to non-responders to treatment. The availability of specific biomarkers for early and accurate diagnosis significantly improves the quality of phenotyping for genetic research.
Rheumatoid arthritis is a prototype autoimmune disease that, together with other multiple impairments of the immune system, is characterized by development of different types of autoantibodies. Rheumatoid factor (RF) and anti-citrullinated peptide/protein antibodies (ACPA, usually measured by anti-CCP ELISA) are most commonly detected [2] and due to high predictive scores are a part of the classification criteria for RA [9]. Other types of autoantibodies to modified proteins are detected mostly in high correlation to RF and ACPAs [14,15,16]. In systemic screening of human population biobanks for these autoantibodies in different populations, it was found that they may be detected in blood long before the first symptoms of RA [17, 18]. Later, with development of symptoms, the concentration of ACPAs increases significantly and in the majority of future RA patients, ACPAs are present at least a few months before the onset of symptoms [19]. In an attempt to dissect the importance of different ACPA reactivities, several studies have been performed on a peptide array with broad selection of citrullinated and non-citrullinated peptides [20]. There is a great degree of cross-reactivity between different types of autoantibodies, although some of them cross-correlate less strongly. It became an accepted practice in rheumatology research to consider autoantibody status as an important covariate in predictive models. However, it is also true that there is only a moderate difference between ACPA-positive and ACPA-negative RA in important clinical features such as disease activity scores at baseline, age of onset, and sex distribution. The greatest difference is probably in the risk of bone erosion [15]. Regarding ACPA-positive RA, it is also characteristic that clinical phenotypes are very similar in subgroups of patients with significantly different concentrations of autoantibodies [21]. There have also been several attempts to correlate the presence of different autoantibodies to clinical phenotypes. Discrimination between ACP-positive and ACPA-negative RA was especially efficient in genetic studies. It has become evident in recent years that there is only a moderate overlap between genetic risk factors between these two subgroups of RA.
Misclassification due to similar clinical phenotypes is not uncommon in rheumatology research and may cause patients with other rheumatic diseases to be included in a study group. This is more common for study designs that lack accurate information about autoantibody status and, in general, is more common for ACPA-negative RA when this autoantibody status information is available. In these cases, the diagnosis may sometimes convert, with time, to spondyloarthritis, which is known to have a different genetic association pattern [22]. On the other hand, multiple pleiotropic effects have been detected between rheumatic diseases [23, 24].
Discoveries of genetic associations in RA arose in the past through investigation of large cohorts with relatively poor sub-phenotyping of RA. Better clinical characterization of RA subgroups should now be pursued.
HLA-related genetic associations in rheumatoid arthritis
A genetic association between RA and variations at HLA was discovered early. With the growing understanding of the architecture of the HLA locus, in particular HLA-DRB1, it became evident that this association relates to a group of so named shared epitope (SE) alleles and is strongest for ACPA-positive RA [25]. The SE hypothesis was introduced in an attempt to find common features between different alleles or haplotypes within HLA-DRB1. Initially, a homologous amino acid sequence at positions 69–74 of the beta chain coded by the HLA-DRB1 gene was identified as the protein structure responsible for association with RA. By following this hypothesis, several DRB1 haplotypes were combined in a single SE allele group in multiple studies and compared with a reference group of other haplotypes. HLA haplotypes from most of the alleles of DRB1*01, *04, and *10 groups represent SE alleles, with *14:02 later suggested as an important contributor for American populations. In contrast, HLA-DRB1*13 alleles were found to bring strong protection against RA [26]. With further sub-phenotyping of RA using autoantibodies, first RF and later with ACPA, it was found that the strongest associations are mainly with autoantibody-positive RA [27, 28].
ACPA-positive RA
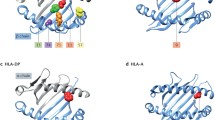
Recent revision of the shared epitope hypothesis revealed independent association hits in the HLA locus within and outside of HLA-DRB1 [29]. The most profound risk effect (OR = 3.8 with HLA-DRB1*15 as a reference) was detected for individuals with amino acid valine (Val) at position 11 of the HLA-DRB1 gene (or for highly linked histidine at position 13). The protein structure with Val at this position is characteristic both to HLA-DRB1*04 and HLA-DRB1*10 groups of alleles. Independent associations with lysine (Lys) at position 71 and alanine (Ala) at position 74 were also detected but could not be clearly explained by previously known associations of SE alleles since these amino acids are also common for several non-SE-related HLA-DRB1 haplotypes. Glutamic acid (Glu) at position 71 seems to be most protective (corresponds to *13:01, *13:02, *13:04, and *01:03 alleles), although in cumulative amino acid association, it does not give a strong effect; this is likely to be due to admixture by *04:02 allele, which also carries Glu at this position but does not demonstrate protection from RA in this study. Additional associations were also detected with HLA-B, HLA-DPB1, and HLA-A for ACPA-positive RA, with significantly lower effect size: 2.12 (95%CI 1.89–2.38) for aspartic acid (Asp) at position 9 of HLA-B; 1.40 (95%CI 1.31–1.50) for phenylalanine (Phe) at position 9 of HLA-DPB1; and 0.85 (95%CI 0.81–0.90) for asparagine (Asn) at position 77 of HLA-A. These associations are mirrored by a highest peak in Manhattan plots for GWAS in multiple studies, including the first published WTCCC study where this hit was a single genome-wide significant one for RA [4]. The most significant difference in association of ACPA-positive RA between Asian and European populations is in the type of amino acid at position 11 of HLA-DRB1 which generates the highest risk: in the Asian population, it is Asp, instead of Val, and this corresponds to the HLA-DRB1*09 group haplotype [30]. Additionally, in the Japanese population, a significant risk from allele of HLA-DO was found that was not seen in Europeans [30]. It is important to acknowledge that due to very high linkage disequilibrium within HLA, and specifically within HLA-DRB1, there are hundreds of correlated SNPs associated with RA. These associations are better explained by considering classical HLA-DRB1 haplotypes and/or subsequent amino acid polymorphisms as described above. Until now, classical HLA haplotypes and amino acid sequences corresponding to these haplotypes have also played important roles in the functional interpretation of HLA associations with autoimmunity, including RA.
A recent study by an international RA genetic consortium (RACI) focused on the association of 19 types of ACPAs in RA patients with variations in the HLA locus [31]. Occurrences of different ACPA reactivities were highly correlated and it was suggested that the detected ACPAs should be split into two groups, canonical and non-canonical, based on reactivity in the bulk anti-CCP (citrullinated circular peptide) test. This stratification of RA individuals by type of ACPAs revealed different genetic architectures for the two groups: association of non-canonical autoantibodies with Asp at position 9 in HLA-B; expression of canonical antibodies mainly associated with Val at position 11 in HLA-DRB1. However, this stratification was far from being completely contrast and almost every RA patient with canonical autoantibodies was also positive to at least one of the non-canonical ones. It is also indicative that non-canonical autoantibodies directed to peptides from the same proteins as canonical, although to the different protein epitope. Later, in a separate study, it was shown that a similar association with Asp at position 9 in HLA-B is characteristic for the anti-carbamylated protein antibodies (anti-CarP) in RA patients in the absence of anti-CCP antibodies [32].
ACPA-negative RA
Although association of ACPA-negative RA with the HLA locus is less pronounced, it was also recently studied in detail [33]. In this study, the authors performed additional filtering of patient clinical information to avoid misclassification through inclusion of patients with spondyloarthritis. It was shown that the profile of association of amino acids from HLA-DRB1 locus is very different from its association with ACPA-positive RA. Highest risk was found for amino acids leucine (Leu) and serine (Ser) at position 11 of HLA-DRB1, with odds 1.22 compared to healthy controls. Interestingly, most of the subsequent classical haplotypes that carry these amino acids at position 11 of HLA-DRB1 are either in linkage with HLA-DRB3 or do not link to other HLA-DRB genes (HLA-DRB1*01). A later study of independent European cohorts suggested association of ACPA-negative RA with Leu at position 67 of HLA-DRB1, with a risk effect of 1.38 [34]; this result is likely due to contamination of the RA group with unrecognized ACPA-positive individuals since this amino acid is characteristic for all SE allele-positive haplotypes, including *14:02, and should be regarded with caution. Data from a Japanese population supports an association of ACPA-negative RA with position 71 at HLA-DRB1, which may subsequently reflect a protective effect of all haplotypes that do not carry arginine (Arg) at this position [30].
An independent association with similar effect size to HLA-DRB1 was detected with asparagine (Asn) at position 9 of HLA-B in two independent studies; the association referred to classical allele HLA-B*08 [33, 34]. Association with HLA-DRB1*03 was previously found in minor studies for ACPA-negative RA, which also suggests a link to this haplotype with the HLA-DRB3 gene or more generally with ancestral haplotype (AH) 8.1, with HLA-B*08 and HLA-DRB1*03 involved [35]. Both studies referred to here (Han et al. and Bossini-Castillo et al.) employed imputation of genetic variations at the HLA locus based on reference data collected by the Type 1 Diabetes Genetics Consortium, where HLA-DRB paralog genes are not well represented. Therefore, multiple associations found in this study, together with previous findings in the HLA locus, may suggest that at least some of these hits are in linkage disequilibrium with unappreciated polymorphisms within the genetic loci contributing to the extended AH 8.1 haplotype, including copy number variations in C4A/C4B locus and variations in HLA-DRB3 gene.
Non-European populations
The findings described above reflect HLA association mainly with RA in European populations, though the situation for Asian and African populations is not completely identical. For the majority of Asian populations, the association of seropositive RA with SE alleles is more specifically related to the HLA-DRB1*04:05 allele, which is not totally absent but is relatively rare in African and European populations. On the other hand, there are additional contributions from this locus which need further investigation in large patient groups. In independent studies of Japanese (ACPA defined) [30] and Korean (mixed by ACPA status) [36] RA patients, HLA association generally followed a pattern of polymorphisms very similar to that in Europeans. Extension of the spectrum of amino acid variations characteristic to HLA-DRB paralogs did not bring additional association signals for the Korean population. However, Japanese-specific association with HLA-DRB1*09 haplotype was repeatedly shown [30, 37]. In a study of Han Chinese ACPA-positive RA patients, strong association with Asp at position 160 of the HLA-DQA1 gene was detected independent from the HLA-DRB1*04:05 allele and several other alleles from the HLA locus [38]. In an analysis of a multiethnic Malaysian cohort, a strong risk of ACPA-positive RA was associated with Val at position 11 of HLA-DRB1; it was found that HLA-DQB1*03:02 allele was inversely related to the risk of developing ACPA-positive RA in the Malay population [39].
Genetic association with RA in African populations remains significantly less investigated and awaits further research and evaluation. In relatively small studies with mixed serological phenotypes, a strong susceptibility in individuals with HLA-DRB1*04 alleles, or correlated SNPs, was confirmed, while other SE alleles were found less frequently in these population groups [40,41,42].
It is evident that genetic associations of HLA variants with different subgroups of RA and in different populations are the most important contributors to genetic risk of the disease. Further analysis of non-HLA genetic associations in the development of RA should be explicitly taken in the context of an understanding of this contribution.
Non-HLA-related genetic associations in rheumatoid arthritis
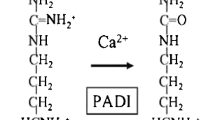
Several associations outside the HLA locus were detected before the GWAS era, including genetic variants at PTPN22, CTLA4, and PADI4 genes [43,44,45]. With expanding data from GWAS from different populations, it became evident that the effect size of non-HLA associations is far lower in comparison to the major associations with HLA-DRB1 SE alleles. Development of affordable genotyping methods, together with the use of transethnic meta-analysis, expanded this list to 151 loci and coverage of all human chromosomes except chromosome Y (Table 1, Fig. 1) [5,6,7,8, 34, 43, 46,47,48,49,50,51,52,53,54,55,56]. Not surprisingly, most associated hits (64% from the current list) could be annotated to genes with a known function in the immune system. The annotation to immune system is based on previous knowledge about specific function of the gene or a pathway in cells related to innate or adaptive immunity, or data on expression of this gene in immune cells. Many of these genes belong to immune system-specific or more ubiquitous cell signaling pathways. The involvement of these genes in cell function for non-immune cells, therefore, potentially extends the importance of genetic associations with RA to the function of cell types, tissues, and organs outside the immune system. One should keep in mind, however, that these annotations are usually based on the physical position of SNP within the gene location and that how gene function is linked to associated SNPs is a matter for future intensive research. Possible approaches will be discussed further in other articles in this issue.

Physical position of SNPs in association with rheumatoid arthritis in human chromosomes
As is the case with the HLA locus, genetic association studies that focus on autoantibody status reveal remarkable differences between ACPA-positive and ACPA-negative arthritis. However, previous investigations were most often performed in RA patients without known autoantibody status and the differences remain non-confirmed by direct testing in the subgroups of RA. According to follow-up analysis in different cohorts all over the world, it is likely that ACPA-positive RA is more prevalent and therefore most of the association hits in studies without discrimination by serology represent associations with autoantibody-positive RA. There have been several attempts to perform GWAS separately for these two groups that often end up with replication of hits for ACPA-positive RA and only few association signals for ACPA-negative RA (Table 1).
The number of known genetic associations with RA is ever growing due to an increase in the number of observations and the resulting increase of statistical power. To expand this further, the complementary approach is in the investigation of population groups to capture differences due to ancestral, most often ethnic, background. In a similar way to the situation for HLA genetics until recently, studies of Europeans have been dominant, and it is important to extend investigation of the scope of human polymorphisms specific for different populations in association studies. These studies are in progress for major Asian and African populations and will help to map true genetic associations for RA.
From the available data in European and East Asian populations, there are several genetic associations that show population differences. It was found that the second-best association with RA in the PTPN22 gene with exonic SNP rs2476601 is not detectable in East Asian populations due to very low frequency of the polymorphism in these populations [50]. Similarly, several other SNPs associated with RA in European populations close to the IL20RB, CUL5, TYK2, and ILF3 loci are not polymorphic or have very low allelic frequency in East Asian populations [6, 7]. In contrast, association with RA for the SNPs rs12026490 close to SLAMF6 was not reproduced in Europeans due to a < 1% frequency of the risk allele [7]. One may expect more population-specific association hits to be discovered in the future. Currently, 18% of known associations in European populations are not confirmed for Asian populations, while only 16% independently reach convincing genome-wide significance in both populations. On the other hand, comparing the effect size for the majority of associated SNPs from trans-ethnic meta-analyses reveals no differences, not only in the presence of significant associations in two different population, but also when one of the groups did not reach genome-wide significance for association [6, 7].
As mentioned previously, the data regarding non-HLA genetic associations with seronegative RA is less consistent and robust. This subgroup is usually a smaller fraction of large GWAS of RA and until recently, only three confirmed associations have demonstrated genome-wide significance (Table 1), close to loci ANKRD55, IRF4, and LINC01898 [34, 46, 55]. At least some of these associations match to hits for association with ACPA-positive RA and, therefore, may also occur due to misclassification within the seronegative group, as discussed above. Most recent trans-ancestry meta-analysis did not confirm genome-wide significant association for seronegative RA [8]. Therefore, GWAS of ACPA-negative RA will be an emerging area of research, although it requires an increase in the size of studies and better phenotypic data for this RA subpopulation.
Common genetic risk factors for different autoimmune diseases were found previously and investigated specifically with Immunochip genotyping [62]. Subsequently, several studies confirmed pleiotropic effects from these genes and by cross-investigation of autoimmune diseases discovered possible new associations for RA [23, 24].
Further integration of genetic association data is required, together with available omics data. Our PPI analysis using inBio Discover (INTOMICS, Denmark) of the gene list from Table 1 generated at least two comprehensive networks with only direct PPI and demonstrated possible involvement in the generation of RA risk for several signaling pathways in immune cells (Figs. 2 and 3).

PPI networks for the genes associated with rheumatoid arthritis

Hypothetical cell pathways based on PPI networks for some of the genes associated with rheumatoid arthritis
More analyses in serologically stratified RA cohorts and in populations with different ancestries are required to elucidate genetic risk of RA using data from genetic association studies.
Gene–gene and gene-environment interaction in risk of rheumatoid arthritis
An investigation of genetic risk factors for common complex diseases should consider the phenomena of gene-environment and gene–gene interaction. There are several statistical approaches for detecting interactions and when discussing interaction, it is important to clarify which definition of interaction is being used. Most often, interaction is part of a statistical model that tests for multiplicativity of contribution for selected parameters in the development of disease risk. The null hypothesis is that the coefficient for interaction mode in the regression model is null. In terms of odds ratio to develop disease, this transfers to the expectation that in a group of individuals with a combination of two parameters, the odds ratio will represent the product of odds for separate parameters. Deviation from the null hypothesis will be the indication for “multiplicative interaction.” In epidemiological studies following the Rothman hypothesis [63], the null hypothesis based on expectation for additivity of odds ratios for a group of individuals with a combination of two parameters was compared to groups with only one of these risk factors. Therefore, “additive interaction” arises in the case of significant deviation from the sum of contribution of each risk factor.
In previous studies, considerable interaction between cigarette smoking and SE alleles was detected in generation of risk, first for RF positive [10], and later for ACPA-positive RA [64]. Further analyses of other environmental factors for interaction with these alleles were performed and it was confirmed for independent Asian, African American, and European populations [65,66,67,68,69,70,71]. Several other studies of environmental effects, including exposure to textile dust and alcohol consumption, also found interaction with SE alleles [72, 73]. Additionally, two genetic loci, genes GSTT1 and HMOX1, at chromosome 22 were identified to increase risk of RA due to interaction with smoking. These genes are functionally related to protection from oxidative stress [74].
Several studies of gene–gene interaction in the development of risk of ACPA-positive RA were performed previously, with PTPN22 risk allele and variation close to MAP2K4 gene found to interact with SE alleles [75, 76]. In another study, gene–gene interaction between variations in the BANK1 and BLK genes from chromosomes 4 and 8, respectively, was shown to increase risk of RA [77]. Based on our dominion hypothesis [72], we performed a study showing the global effect of RA risk alleles in interaction with SE alleles on the development of additional risk for ACPA-positive RA [11]. Although this study was initially criticized for statistical biases [78], careful statistical modeling has confirmed that it does indeed describe true effects and could be applied to study gene–gene interactions in other binary phenotypes with a known dominant risk factor [79]. Therefore, the conclusion of this study is that significant interaction between the HLA-DRB1 SE alleles and the group of SNPs associated with ACPA-positive RA is a global feature that increases risk of ACPA-positive RA. It was also shown in this study that gradual decrease of the number of interacting risk alleles in a group of individuals with HLA-DRB1 SE alleles will decrease the risk for RA. No gene–gene or gene-environment interactions in development of the risk of ACPA-negative arthritis have so far been found. Further analysis of statistical interactions may help to integrate functionally relevant associations involved in the development of RA.
DNA methylation in association with rheumatoid arthritis
Development of tools for fast analysis of a massive number of methylation sites, like Illumina 450 and Illumina EPIC arrays, opened the possibility for genome-wide epigenetic study (GWES). To date, the largest GWES of RA was performed on whole blood genomic DNA of 354 ACPA-positive RA and 357 healthy controls [80]. Epigenetic changes at disease baseline are massive and obviously non-specific, reflecting basic inflammatory reactions. In an attempt to identify differentially methylated sites (DMS) that are more specifically related to RA development, genetic association signals for RA were integrated with data for DMS in genomic DNA from whole blood in ACPA-positive RA. This approach helped to focus only on a few DMS that are regulated by SNPs with association to the disease. Interestingly, almost all these DMS, 9 out of 10, were located within the HLA locus. Additionally, following known association of seropositive RA with cigarette smoking, one of these DMS, close to the HLA locus, was attributed as a major contributor to the risk of ACPA-positive RA due to gene-environment interaction [81]. However, DNA methylation profile reflects contribution of the multiple environmental factors and of the ongoing inflammation in RA patients and it is difficult to identify RA-specific casual relations between these factors and DMS changes. It was later shown that methylome of peripheral mononuclear cells can be used to anticipate the evolution of undifferentiated arthritis to RA, and these modifications could be annotated to a number of inflammatory pathways and transcription factors [82]. The significance of ACPAs during RA development was analyzed in serologically distinct RA subgroups in a study of twins discordant for ACPA status [83]. However, only marginal differences in the methylation profile of whole blood DNA of twins were found when data was corrected for cell composition; this highlights the importance of blood cell profiling in epigenetic studies of RA development. With the goal of determining RA-specific methylation events, several research groups conducted epigenetic studies with profiling of specific cell subpopulations. These studies demonstrated the following: significant changes in methylome of CD4 + memory cells compared to CD4 + naïve cells [84]; specific methylation-induced regulation of CTLA4 promoter [85] and FOXP3 enhancer in Tregs [86]; evidence for epigenetic regulation of inflammatory cytokines in monocytes [87]; and multiple immune-related pathways in fibroblast-like synoviocytes (latterly, in comparison with osteoarthritis) [88] in RA. Interestingly, a common pattern of epigenetic changes was found in a regulation of interferon-related genes for different autoimmune diseases, including RA, with hypomethylation of these genes in CD4 + cells being highly predictive to disease development [89].
While epigenetic studies most commonly focus on the available pool of peripheral blood cells, significant diversity was shown for the tissue from anatomically different joints. Synovial fibroblasts of different anatomical origins represent different transcriptomes that translate into unique joint-specific phenotypes of these cells, which is likely to influence methylation profile in RA with different localization [90].
Coming studies of methylome in RA should consider the important differences observed in genetic studies between the RA subgroups. It is desirable to build the experimental design either on stratification of seropositive and seronegative RA, or to adjust the differential methylation analysis for serological status. The major challenge for the analysis is in a study design in which healthy controls are not necessarily the optimal reference group. Epigenetic studies should be integrated with genetics, transcriptomics, and proteomics to reveal a true pattern of pathway modulation in the development of RA.
Conclusion
Multiple genetic polymorphisms contribute to predisposition to RA, and this is best investigated in seropositive disease. More detailed studies of seronegative RA are pending. As for many autoimmune diseases, the effect size for risk of seropositive RA is dominated by several HLA-DRB1 alleles. Although there are a few success stories with regard to translation of genetic studies of RA into the biological functions, most of the associations remain to be better interpreted and tested in the future. The major challenge will be the integration of genetic association studies with epigenetics, transcriptomics, and proteomics to produce an understanding that will enable personalized medical help and, ultimately, prevention of severely incapacitating chronic RA.
References
Almutairi KB et al (2021) The prevalence of rheumatoid arthritis: a systematic review of population-based studies. J Rheumatol 48(5):669–676
McInnes IB, Schett G (2011) The pathogenesis of rheumatoid arthritis. N Engl J Med 365(23):2205–2219
Catrina AI et al (2016) Mechanisms involved in triggering rheumatoid arthritis. Immunol Rev 269(1):162–174
Wellcome Trust Case Control C (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 447(7145):661–78.
Plenge RM et al (2007) TRAF1-C5 as a risk locus for rheumatoid arthritis–a genomewide study. N Engl J Med 357(12):1199–1209
Okada Y et al (2014) Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506(7488):376–381
Ha E, Bae SC, Kim K (2021) Large-scale meta-analysis across East Asian and European populations updated genetic architecture and variant-driven biology of rheumatoid arthritis, identifying 11 novel susceptibility loci. Ann Rheum Dis 80(5):558–565
Ishigaki K et al (2021) Trans-ancestry genome-wide association study identifies novel genetic mechanisms in rheumatoid arthritis. https://doi.org/10.1101/2021.12.01.21267132.
Aletaha D et al (2010) 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 62(9):2569–2581
Padyukov L et al (2004) A gene-environment interaction between smoking and shared epitope genes in HLA-DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheum 50(10):3085–3092
Diaz-Gallo LM et al (2018) Systematic approach demonstrates enrichment of multiple interactions between non-HLA risk variants and HLA-DRB1 risk alleles in rheumatoid arthritis. Ann Rheum Dis 77(10):1454–1462
Hannon E et al (2017) Pleiotropic effects of trait-associated genetic variation on DNA methylation: utility for refining GWAS loci. Am J Hum Genet 100(6):954–959
Houtman M et al (2018) T cells are influenced by a long non-coding RNA in the autoimmune associated PTPN2 locus. J Autoimmun 90:28–38
Gronwall C et al (2021) A comprehensive evaluation of the relationship between different IgG and IgA anti-modified protein autoantibodies in rheumatoid arthritis. Front Immunol 12:627986
Shi J et al (2011) Autoantibodies recognizing carbamylated proteins are present in sera of patients with rheumatoid arthritis and predict joint damage. Proc Natl Acad Sci USA 108(42):17372–17377
Sahlstrom P et al (2020) Different hierarchies of anti-modified protein autoantibody reactivities in rheumatoid arthritis. Arthritis Rheumatol 72(10):1643–1657
Nielen MM et al (2004) Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum 50(2):380–386
Rantapaa-Dahlqvist S et al (2003) Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum 48(10):2741–2749
Johansson L et al (2016) Antibodies directed against endogenous and exogenous citrullinated antigens pre-date the onset of rheumatoid arthritis. Arthritis Res Ther 18(1):127
Hansson M et al (2012) Validation of a multiplex chip-based assay for the detection of autoantibodies against citrullinated peptides. Arthritis Res Ther 14(5):R201
Laki J et al (2012) Very high levels of anti-citrullinated protein antibodies are associated with HLA-DRB1*15 non-shared epitope allele in patients with rheumatoid arthritis. Arthritis Rheum 64(7):2078–2084
International Genetics of Ankylosing Spondylitis C et al (2013) Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat Genet 45(7):730–8.
Marquez A et al (2018) Meta-analysis of Immunochip data of four autoimmune diseases reveals novel single-disease and cross-phenotype associations. Genome Med 10(1):97
Marquez A et al (2017) A combined large-scale meta-analysis identifies COG6 as a novel shared risk locus for rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis 76(1):286–294
Gregersen PK, Silver J, Winchester RJ (1987) The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum 30(11):1205–13
de Vries N et al (2002) Reshaping the shared epitope hypothesis: HLA-associated risk for rheumatoid arthritis is encoded by amino acid substitutions at positions 67–74 of the HLA-DRB1 molecule. Arthritis Rheum 46(4):921–928
van der Woude D et al (2010) Protection against anti-citrullinated protein antibody-positive rheumatoid arthritis is predominantly associated with HLA-DRB1*1301: a meta-analysis of HLA-DRB1 associations with anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis in four European populations. Arthritis Rheum 62(5):1236–1245
Irigoyen P et al (2005) Regulation of anti-cyclic citrullinated peptide antibodies in rheumatoid arthritis: contrasting effects of HLA-DR3 and the shared epitope alleles. Arthritis Rheum 52(12):3813–3818
Raychaudhuri S et al (2012) Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet 44(3):291–296
Okada Y et al (2016) Contribution of a non-classical HLA gene, HLA-DOA, to the risk of rheumatoid arthritis. Am J Hum Genet 99(2):366–374
Terao C et al (2019) Distinct HLA Associations with rheumatoid arthritis subsets defined by serological subphenotype. Am J Hum Genet 105(3):616–624
Regueiro C et al (2021) HLA-B*08 identified as the most prominently associated major histocompatibility complex locus for anti-carbamylated protein antibody-positive/anti-cyclic citrullinated peptide-negative rheumatoid arthritis. Arthritis Rheumatol 73(6):963–969
Han B et al (2014) Fine mapping seronegative and seropositive rheumatoid arthritis to shared and distinct HLA alleles by adjusting for the effects of heterogeneity. Am J Hum Genet 94(4):522–532
Bossini-Castillo L et al (2015) A genome-wide association study of rheumatoid arthritis without antibodies against citrullinated peptides. Ann Rheum Dis 74(3):e15
Verpoort KN et al (2005) Association of HLA-DR3 with anti-cyclic citrullinated peptide antibody-negative rheumatoid arthritis. Arthritis Rheum 52(10):3058–3062
Kim K et al (2016) Imputing variants in HLA-DR beta genes reveals that HLA-DRB1 is solely associated with rheumatoid arthritis and systemic lupus erythematosus. PLoS One 11(2):e0150283
Kochi Y et al (2004) Analysis of single-nucleotide polymorphisms in Japanese rheumatoid arthritis patients shows additional susceptibility markers besides the classic shared epitope susceptibility sequences. Arthritis Rheum 50(1):63–71
Guo J et al (2019) Sequencing of the MHC region defines HLA-DQA1 as the major genetic risk for seropositive rheumatoid arthritis in Han Chinese population. Ann Rheum Dis 78(6):773–780
Tan LK et al (2021) The spectrum of association in HLA region with rheumatoid arthritis in a diverse Asian population: evidence from the MyEIRA case-control study. Arthritis Res Ther 23(1):46
Danila MI et al (2017) Dense genotyping of immune-related regions identifies loci for rheumatoid arthritis risk and damage in African Americans. Mol Med 23:177–187
Govind N et al (2019) HLA-DRB1 amino acid positions and residues associated with antibody-positive rheumatoid arthritis in Black South Africans. J Rheumatol 46(2):138–144
Reynolds RJ et al (2014) HLA-DRB1-associated rheumatoid arthritis risk at multiple levels in African Americans: hierarchical classification systems, amino acid positions, and residues. Arthritis Rheumatol 66(12):3274–3282
Begovich AB et al (2004) A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet 75(2):330–337
Rodriguez MR et al (2002) Association of the CTLA4 3’ untranslated region polymorphism with the susceptibility to rheumatoid arthritis. Hum Immunol 63(1):76–81
Suzuki A et al (2003) Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet 34(4):395–402
Eyre S et al (2012) High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat Genet 44(12):1336–1340
Gregersen PK et al (2009) REL, encoding a member of the NF-kappaB family of transcription factors, is a newly defined risk locus for rheumatoid arthritis. Nat Genet 41(7):820–823
Kwon YC et al (2020) Genome-wide association study in a Korean population identifies six novel susceptibility loci for rheumatoid arthritis. Ann Rheum Dis 79(11):1438–1445
Myouzen K et al (2012) Functional variants in NFKBIE and RTKN2 involved in activation of the NF-kappaB pathway are associated with rheumatoid arthritis in Japanese. PLoS Genet 8(9):e1002949
Okada Y et al (2012) Meta-analysis identifies nine new loci associated with rheumatoid arthritis in the Japanese population. Nat Genet 44(5):511–516
Raychaudhuri S et al (2009) Genetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis risk. Nat Genet 41(12):1313–1318
Stahl EA et al (2010) Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet 42(6):508–514
Suzuki A et al (2008) Functional SNPs in CD244 increase the risk of rheumatoid arthritis in a Japanese population. Nat Genet 40(10):1224–1229
Terao C et al (2011) The human AIRE gene at chromosome 21q22 is a genetic determinant for the predisposition to rheumatoid arthritis in Japanese population. Hum Mol Genet 20(13):2680–2685
Wei WH et al (2017) Genotypic variability based association identifies novel non-additive loci DHCR7 and IRF4 in sero-negative rheumatoid arthritis. Sci Rep 7(1):5261
Zhao J et al (2017) A missense variant in NCF1 is associated with susceptibility to multiple autoimmune diseases. Nat Genet 49(3):433–437
Kim K et al (2015) High-density genotyping of immune loci in Koreans and Europeans identifies eight new rheumatoid arthritis risk loci. Ann Rheum Dis 74(3):e13
Saad MN et al (2019) Studying the effects of haplotype partitioning methods on the RA-associated genomic results from the North American Rheumatoid Arthritis Consortium (NARAC) dataset. J Adv Res 18:113–126
McAllister K et al (2013) Identification of BACH2 and RAD51B as rheumatoid arthritis susceptibility loci in a meta-analysis of genome-wide data. Arthritis Rheum 65(12):3058–3062
Thomson W et al (2007) Rheumatoid arthritis association at 6q23. Nat Genet 39(12):1431–1433
Kim K et al (2016) Association-heterogeneity mapping identifies an Asian-specific association of the GTF2I locus with rheumatoid arthritis. Sci Rep 6:27563
Cortes A, Brown MA (2011) Promise and pitfalls of the Immunochip. Arthritis Res Ther 13(1):101
Rothman KJ, Greenland S, Walker AM (1980) Concepts of interaction. Am J Epidemiol 112(4):467–470
Klareskog L et al (2006) A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum 54(1):38–46
Bang SY et al (2010) Smoking increases rheumatoid arthritis susceptibility in individuals carrying the HLA-DRB1 shared epitope, regardless of rheumatoid factor or anti-cyclic citrullinated peptide antibody status. Arthritis Rheum 62(2):369–377
Costenbader KH et al (2008) Genetic polymorphisms in PTPN22, PADI-4, and CTLA-4 and risk for rheumatoid arthritis in two longitudinal cohort studies: evidence of gene-environment interactions with heavy cigarette smoking. Arthritis Res Ther 10(3):R52
Criswell LA et al (2006) Smoking interacts with genetic risk factors in the development of rheumatoid arthritis among older Caucasian women. Ann Rheum Dis 65(9):1163–1167
Karlson EW et al (2010) Gene-environment interaction between HLA-DRB1 shared epitope and heavy cigarette smoking in predicting incident rheumatoid arthritis. Ann Rheum Dis 69(1):54–60
Mikuls TR et al (2010) Associations of cigarette smoking with rheumatoid arthritis in African Americans. Arthritis Rheum 62(12):3560–3568
Morgan AW et al (2009) Reevaluation of the interaction between HLA-DRB1 shared epitope alleles, PTPN22, and smoking in determining susceptibility to autoantibody-positive and autoantibody-negative rheumatoid arthritis in a large UK Caucasian population. Arthritis Rheum 60(9):2565–2576
Traylor M et al (2017) Genetic and environmental risk factors for rheumatoid arthritis in a UK African ancestry population: the GENRA case-control study. Rheumatol (Oxford) 56(8):1282–1292
Padyukov L (2014) Between the lines of genetic code : genetic interactions in understanding disease and complex phenotypes, vol. xxi. Academic Press, Amsterdam, p 203
Too CL et al (2016) Occupational exposure to textile dust increases the risk of rheumatoid arthritis: results from a Malaysian population-based case-control study. Ann Rheum Dis 75(6):997–1002
Keenan BT et al (2010) Effect of interactions of glutathione S-transferase T1, M1, and P1 and HMOX1 gene promoter polymorphisms with heavy smoking on the risk of rheumatoid arthritis. Arthritis Rheum 62(11):3196–3210
Kallberg H et al (2007) Gene-gene and gene-environment interactions involving HLA-DRB1, PTPN22, and smoking in two subsets of rheumatoid arthritis. Am J Hum Genet 80(5):867–875
Shchetynsky K et al (2015) Gene-gene interaction and RNA splicing profiles of MAP2K4 gene in rheumatoid arthritis. Clin Immunol 158(1):19–28
Genin E et al (2013) Epistatic interaction between BANK1 and BLK in rheumatoid arthritis: results from a large trans-ethnic meta-analysis. PLoS One 8(4):e61044
Kim K (2019) Massive false-positive gene-gene interactions by Rothman’s additive model. Ann Rheum Dis 78(3):437–439
Diaz-Gallo LM et al (2021) Understanding interactions between risk factors, and assessing the utility of the additive and multiplicative models through simulations. PLoS One 16(4):e0250282
Liu Y et al (2013) Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol 31(2):142–147
Meng W et al (2017) DNA methylation mediates genotype and smoking interaction in the development of anti-citrullinated peptide antibody-positive rheumatoid arthritis. Arthritis Res Ther 19(1):71
de la Calle-Fabregat C et al (2021) The DNA methylation profile of undifferentiated arthritis patients anticipates their subsequent differentiation to rheumatoid arthritis. Arthritis Rheumatol 73(12):2229–2239
Gomez-Cabrero D et al (2016) High-specificity bioinformatics framework for epigenomic profiling of discordant twins reveals specific and shared markers for ACPA and ACPA-positive rheumatoid arthritis. Genome Med 8(1):124
Guderud K et al (2020) Rheumatoid arthritis patients, both newly diagnosed and methotrexate treated, show more DNA methylation differences in CD4(+) memory than in CD4(+) naive T cells. Front Immunol 11:194
Cribbs AP et al (2014) Treg cell function in rheumatoid arthritis is compromised by ctla-4 promoter methylation resulting in a failure to activate the indoleamine 2,3-dioxygenase pathway. Arthritis Rheumatol 66(9):2344–2354
Kennedy A et al (2014) A novel upstream enhancer of FOXP3, sensitive to methylation-induced silencing, exhibits dysregulated methylation in rheumatoid arthritis Treg cells. Eur J Immunol 44(10):2968–2978
Rodriguez-Ubreva J et al (2019) Inflammatory cytokines shape a changing DNA methylome in monocytes mirroring disease activity in rheumatoid arthritis. Ann Rheum Dis 78(11):1505–1516
Ai R et al (2018) Comprehensive epigenetic landscape of rheumatoid arthritis fibroblast-like synoviocytes. Nat Commun 9(1):1921
Chen S et al (2019) Genome-wide DNA methylation profiles reveal common epigenetic patterns of interferon-related genes in multiple autoimmune diseases. Front Genet 10:223
Frank-Bertoncelj M et al (2017) Epigenetically-driven anatomical diversity of synovial fibroblasts guides joint-specific fibroblast functions. Nat Commun 8:14852
Acknowledgements
The author would like to thank his colleagues from Division of Rheumatology at Karolinska Institutet for the discussions and support during his work, Dr. L Bossini-Castillo for sharing unpublished data, Dr Marina Bobkova for help with figures, and Ms Janet Ahlberg for language editing.
Funding
Open access funding provided by Karolinska Institute. Partial financial support was received from the Swedish Research Council, grant 2018–02884.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is a contribution to the special issue on: Genetics and functional genetics of Autoimmune diseases - Guest Editors: Yukinori Okada & Kazuhiko Yamamoto
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Padyukov, L. Genetics of rheumatoid arthritis. Semin Immunopathol 44, 47–62 (2022). https://doi.org/10.1007/s00281-022-00912-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-022-00912-0