
Abstract
Introduction
Progression of joint damage despite appropriate therapy remains a significant problem for patients with rheumatoid arthritis (RA). This study was undertaken to identify profiles of immune response that correlate with radiographic joint damage as a first step toward the discovery of new pathogenic mechanisms of joint destruction in RA.
Methods
The study included 58 patients with RA and 15 healthy controls. The profiles of cytokine release from peripheral blood mononuclear cells (PBMC) in response to stimulation for 48 hours with one of six stimuli, or in media alone, were measured. Immune response profiles identified for each stimulus were correlated with radiographic joint damage as defined by the Sharp-van der Heijde score (SHS), before and after multivariable adjustment. For profiles correlated with the SHS, the distributions of individual cytokines were evaluated in patients according to the severity of joint damage and compared to healthy controls.
Results
The immune response profile for cytomegalovirus (CMV)/Epstein-Barr virus (EBV) stimulation was correlated with both the SHS total and erosion scores (r = 0.31, P = 0.018 and r = 0.33, P = 0.011, respectively). After adjusting for age, sex, disease duration, autoantibody status, CMV/EBV serological status, current disease activity, disability and treatments, the correlation of the CMV/EBV immune response and the SHS erosion score became stronger (r = 0.43, P < 0.003). The CMV/EBV immune response correlated with CMV IgG (r = 0.44, P < 0.001), but not with EBV IgG. The most important cytokines for the CMV/EBV immune response profile were IFN-γ, IL-2, IL-4, IL-5, IL-13 and IL-17A, all of which are associated with T-cell immunity. Both the summary immune response score and the individual responses of IFN-γ and IL-13 to CMV/EBV stimulation were associated with greater joint damage.
Conclusions
A profile of immune response to purified CMV/EBV lysates is associated with radiographic joint damage. The correlation of this immune response to CMV serology implies possible involvement of latent CMV infection. Therefore, the findings suggest that the immune response to latent CMV infection could play a fundamental role in the progression of inflammation and structural joint damage in patients with RA.
Similar content being viewed by others


Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune disease that causes chronic, persistent joint inflammation, leading to irreversible structural damage. The etiology remains unclear, but the prevailing disease model is that multiple genes, especially HLA-DRB1 alleles, interact with environmental risk factors, including tobacco smoking, culminating in an adaptive immune response to citrullinated autoantigens that perpetuates joint inflammation [1]. T lymphocytes are key "conductors" of the inflammatory process, providing help to B cells and leading to the production of autoantibodies and the recruitment/activation of other immune effectors [2, 3]. Ultimately, the activation of plasma cells, macrophages, fibroblasts, chondrocytes and osteoclasts, and downstream production of inflammatory cytokines (that is, TNF, IL-1, and IL-6), reactive oxygen species, matrix metalloproteinases and other toxic molecules, mediates destruction of articular cartilage and bone [4].
Advances in therapeutics have dramatically improved the outlook for joint structure and function in patients with RA. Current treatment strategies retard joint destruction in the majority. Nonetheless, between 5 to 30% of patients, depending on the treatment strategy used, still experience rapid progression of joint damage [5, 6]. Models describing the probability of rapid radiographic progression have limited accuracy and reliability and do not fully account for joint damage [7–9]. More patients suffer slow structural deterioration over time, which can occur despite intensive treatment [10–12]. The most likely explanation is that inflammation can persist despite treatment, even when the disease appears to be in clinical remission [13–16]. Therefore, there is an urgent need to identify new pathogenic mechanisms of persistent, treatment-refractory inflammation, in hopes of developing novel targeted therapies that prevent structural deterioration over time.
We have devised an approach to discover immunological correlates of disease phenotypes based on profiles of ex vivo cytokine production from peripheral blood mononuclear cells (PBMC) in response to a number of non-specific stimuli. This approach was based on the concept that we could sample a broad array of activation pathways and, thereby, develop a general profile of immune responsiveness rather than define the responsiveness of specific innate or adaptive immune pathways. Our previous studies demonstrated the success of this approach by characterizing differences in the profiles of immune response between patients with early versus late RA and healthy controls, and also between patients with and without myocardial dysfunction [17, 18]. The results informed our hypothesis that the broad responsiveness of one or more immune pathways is associated with the potential of the inflammatory process to mediate joint destruction. The objective of this study was to identify a profile of immune response based on ex vivo cytokine production that is associated with the severity of radiographic joint damage in patients with RA, after accounting for known predictors of severe disease.
Materials and methods
Study design and participants
We conducted a cross-sectional correlative study in the outpatient practice of the Division of Rheumatology at Mayo Clinic Rochester, MN, USA. A sample of patients referred by the 16 clinicians in the Division during the period of May 2008 through November 2009 was recruited. Adult patients of ≥ 18 years of age with a diagnosis of RA were eligible to participate if they met the following inclusion criteria: fulfillment of the 1987 American College of Rheumatology classification criteria for RA and seropositive status for rheumatoid factor (RF), anti-citrullinated protein antibodies (ACPA), or both. Patients with any of the following were excluded: clinically apparent acute infections, known chronic infections (that is, hepatitis C), recent malignancies in the past two years (excluding non-melanoma skin cancers), radiation or chemotherapy in the past two years, history of advanced chronic kidney disease or kidney transplantation, history of chronic liver disease, recent major surgery, or use of prednisone for indications other than RA. For controls, we included data on healthy blood donors from our initial study, which used identical methods as the present one [17]. The Mayo Foundation institutional review board approved this study, which was conducted according to the principles of the Declaration of Helsinki. All patients provided written informed consent.
Data collection
One consultant rheumatologist (JMD) evaluated tender and swollen joints using modified 28-joint counts [19] and the physician global assessment of disease activity using a 0 to 100 mm visual analog scale (VAS). Patient reported outcomes included levels of pain and fatigue (0 to 100 mm VAS), duration of morning stiffness (minutes), the Health Assessment Questionnaire (HAQ) disability index [20, 21], and the Medical Outcomes Study Short-Form 36 (SF-36) [22, 23]. Disease activity was defined by the Disease Activity Score in 28 joints, four-variable version, using C-reactive protein (DAS28) [19]. Data were collected on patient demographics, disease duration, RF and ACPA status, and current use of disease-modifying antirheumatic drugs (DMARDs), including biologic response modifiers. C-reactive protein (CRP) was measured by turbidometric assay (Roche, Indianapolis, IN, USA). Enzyme-linked immunosorbent assays were done to assess past CMV infection using the VIDAS® CMV IgG (bioMerieux, Inc., Durham, NC, USA), and multiplexed immunoassays were done to assess past EBV infection using the BioPlex™ 2200 System EBV IgG and EBV IgM (Bio-Rad Laboratories, Hercules, CA, USA).
Radiographic scoring
Radiographs, including single views of both hands and feet, were obtained using a standardized research protocol. A board-certified musculoskeletal radiologist (JAS) interpreted all radiographs blinded to clinical and experimental information, according to the modified Sharp-van der Heijde score (SHS) system [24, 25]. Separate evaluations were done as planned a priori for the total SHS as well as erosion and joint space narrowing (JSN) scores.
Immune response assays
Our approach to quantify the broad responsiveness of ex vivo cytokine production was previously described [17]. Functional peripheral blood mononuclear cells (PBMCs) were procured by Ficoll density gradient centrifugation. Within one to two hours, 4 × 105 PBMCs were cultured in 200 μL of medium (RPMI-1640 + 10% fetal bovine serum + 1× penicillin, streptomycin, and glutamine) in the presence of one of a panel of six stimuli, or in medium alone, in a 96-well culture plate. The rationale for the use of each stimulus was detailed in our previous publication [17]. The stimulation panel included immobilized anti-CD3 and anti-CD28 monoclonal antibodies (anti-CD3/anti-CD28; Dynabeads Human T-Activator, Invitrogen, Grand Island, NY, USA); bacterial CpG oligonucleotides (CpG); combined lysates of purified CMV and EBV, containing both viral peptides and DNA (CMV/EBV; Advanced Biotechnologies, Columbia, MD, USA); PMA with ionomycin (PMA/ionomycin; Sigma, St. Louis, MO, USA); phytohemagglutinin (PHA; Sigma); and staphylococcus enterotoxins A and B (SEA and SEB; Toxin Technology, Sarasota, FL, USA). The final concentrations of each stimulant were as follows: anti-CD3/anti-CD28, 0.5 × 106 beads per culture well (1:1 ratio of beads to PBMCs per manufacturer's instructions); PHA, 5 μg/mL; Staphylococcus enterotoxin A, 10 ng/mL, with Staphylococcus enterotoxin B, 10 ng/mL; CMV, 1 μg/mL, with EBV, 1 μg/mL; CpG, 10 μg/mL; PMA, 1 μg/mL, with ionomycin, 700 ng/mL. The PBMCs were incubated at 37°C in 5% CO2 for 48 hours; the supernatants were then harvested, transferred to a storage plate, and frozen at -80°C for subsequent analysis.
Multiplexed cytokine analysis was performed to measure cytokine release from the PBMCs into the culture supernatants in response to stimulation using the MSD® 96-Well MULTI-SPOT® Human Cytokine Assays tissue culture kit (Meso Scale Discovery (MSD), Gaithersburg, MD, USA). The cytokine panel included IL-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8 (CXCL8), IL-10, IL-12, IL-13, IL-17A, IFN-γ, TNF-α, MCP-1 (CCL2), MIP-1β (CCL4), G-CSF, and GM-CSF. Using the Sector 2400 instrument (MSD) and manufacturer-supplied reagents, the cytokine concentrations were determined based on a standard curve generated for each plate. The results were analyzed using the Discovery Workbench Software v2.0 (MSD). Previous studies have demonstrated acceptable reproducibility relative to the high degree of informative, biological variation [17].
Statistical analysis
The patient characteristics were summarized as median (interquartile range (IQR)) or number (%). Mixed effects models were used to normalize the cytokine data as previously described [17]. In brief, fixed effects were included for age, sex, cytokine-plate lot, and random effects were included for subject and individual cytokine plate used. This resulted in effective normalization of assay effects and adjustment of the cytokine data for age and sex.
The analytic approach was based on the premise that each stimulus activates a finite number of immune pathways. The foundational concept of principal components analysis (PCA) is that for a large number of independent variables, common pathways can be identified by variables that are highly correlated. We used PCA to identify immune response profiles associated with each stimulus based on the first and second principal components. To create stimulus-cytokine PCA scores for the first and second principal components of each stimulus, all cytokine concentrations were converted to Z-scores (the mean cytokine value for the patients was subtracted from each individual cytokine value, and this difference was divided by the standard deviation of the cytokine value), then the Z-scores were multiplied by the component loadings and summed to obtain a single continuous score. For ease of interpretation, these scores were rescaled to range from 0 to 100.
The study objective was to determine the associations between key immune profiles for each stimulus and radiographic joint damage. Spearman correlations were determined between each of the stimulus-cytokine PCA scores and the SHS total, erosion, and JSN scores. Partial Spearman correlations were also assessed after adjusting for age, sex, disease duration, RF status, ACPA status, CMV/EBV serological status, RA drug treatments (methotrexate, biologic response modifiers, and prednisone), the HAQ disability index, and DAS28.
For stimulus-cytokine scores significantly correlated with joint damage, individual cytokine values with component loadings > 0.5 were compared between RA patients and controls using Wilcoxon rank-sum tests. The patients were analyzed in two groups based on a dichotomous cutoff for the total SHS of 20 units. The rationale for this particular cutoff was that the progression of radiographic joint damage by 20 Sharp units corresponds to an irreversible deterioration in physical function as defined by the HAQ disability index of 0.2, approximately the minimum clinically important difference for this outcome [26]. The rescaled stimulus-cytokine PCA score was also analyzed among the groups. Statistical significance was set at the 0.05 level; all tests were two-sided.
Results
Subject characteristics
The study included 58 patients with RA (Table 1) and 15 controls. On average, the patients had relatively early disease, with median (IQR) disease duration of 18.8 months (11.3, 35.9). According to the selection criteria, all patients were positive for ACPA or RF; 44 (80%) were positive for ACPA, and 53 (93%) were positive for RF. Overall, the cohort had moderate disease activity, with a median (IQR) DAS28 of 4.4 (3.4, 5.4), and moderate disability, with a median (IQR) HAQ disability index of 0.5 (0.1, 1.4). Overall, 56 (97%) of the patients had erosive disease as defined by the SHS erosion score ≥ 1. The majority of patients were taking methotrexate, and nearly half were taking prednisone; only 14 were taking biologic response modifiers (12 patients were taking anti-TNF agents, and 2 patients had received rituximab). The control group included eight females and seven males with a mean (SD) age of 44.6 (16.8) years.
Discovery of a correlation between CMV/EBV immunity and radiographic joint damage using principal components analysis
The analysis of the first and second principal components for each of the seven stimulus-cytokine combinations resulted in identification of distinct cytokine profiles; based on the analytical premise, these were considered to reflect activation of canonical immune pathways (Table 2). For CMV/EBV stimulation, the stimulus-cytokine combinations with component loadings > 0.5 were IL-13, IL-2, IFN-γ, IL-5, IL-4, and IL-17A (in diminishing importance in the PCA score). This profile clearly reflected activation of T-cell cytokine production, across canonical T-cell subsets, in response to CMV/EBV lysate. The first and second principal components explained 30.2% and 20.7%, respectively, of the variation in cytokine production from PBMC in response to CMV/EBV stimulation.
The next step was to screen each of the immune response profiles identified using PCA for association with radiographic joint damage as defined by the SHS scores. Among all of the first and second principal components for the seven stimulation conditions, only one had a correlation coefficient of r > 0.3, a level which might be biologically meaningful: the first principal component for CMV/EBV stimulation (CMV/EBV-1) (Figure 1). Statistically significant correlations of the CMV/EBV-1 PCA score with the total SHS (r = 0.31; P = 0.018) and the SHS erosion score (r = 0.33; P = 0.011) were identified (Table 3). After adjusting for potential confounders of the relationship between viral immunity and joint damage (Table 3 and Figure 1), including markers of RA disease activity, severity and treatment status, the correlation of CMV/EBV-1 with both the SHS total and erosion scores became modestly stronger (r = 0.39; P = 0.006; see Table 3 for the list of covariates).

Results of the data reduction strategy identifying immune response profiles correlated with radiographic joint damage. Shown are stem-and-leaf schematics summarizing the hierarchy of correlation coefficients for each of the stimulus-cytokine principal component analysis scores. (A) Unadjusted Spearman correlation coefficients. (B) Partial Spearman correlation coefficients, adjusted for age, sex, disease duration, rheumatoid factor status, anti-citrullinated protein antibody status, the Health Assessment Questionnaire (HAQ) disability index, the Disease Activity Score in 28 joints (DAS28), and use of methotrexate, biologic response modifiers, and/or prednisone. The decimal point is one digit to the left of the vertical bar (for example, the bottom value in the left panel is r = 0.31). The highest unadjusted and adjusted correlation coefficients (r = 0.31 and r = 0.39, respectively) correspond to the CMV/EBV first principal component score. CMV, cytomegalovirus; EBV, Epstein Barr virus
Additionally, a significant correlation of the PMA/ionomycin second principal component (PMA/ionomycin-2) with the erosion score (r = 0.27; P = 0.042), but not the total SHS (r = 0.24; P = 0.07), was identified (Table 3). After adjustment for covariates, the association between PMA/ionomycin-2 and the erosion score became non-significant (r = 0.09, P = 0.53). This association was confounded by the use of prednisone and disease duration, both of which correlated with PMA/ionomycin-2 (r = -0.26, P = 0.046 and r = 0.29, P = 0.025, respectively).
Analysis of the CMV/EBV immune response profile in patients with RA and control subjects according to viral serological status
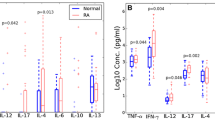
The next step was to analyze the individual cytokines comprising CMV/EBV-1 among the patient groups with 'high' or 'low' joint damage and healthy controls. Significant differences in the production of T-cell cytokines were observed among the groups (Figure 2A). The RA group with high joint damage (SHS ≥ 20) had significantly increased production of IFN-γ (Th1) and IL-13 (Th2) compared to the group with low joint damage (SHS < 20). IL-2, the other Th1 cytokine studied, had a slightly lower median in the overall RA group relative to controls (statistically non-significant), yet there was a subgroup of patients with high joint damage who had markedly increased levels compared to the low-damage group or controls. The production of IL-4 and IL-5, the other Th2 cytokines, in response to CMV/EBV stimulation was significantly increased among the patients overall compared to controls. Though not statistically significant, IL-17A production also appeared to be increased overall, with both higher median and 90th percentiles among the patients with RA compared to controls.

A profile of immune response mainly to CMV correlates with radiographic joint damage in RA. (A) Shown are normalized cytokine concentrations released from peripheral blood mononuclear cells into culture supernatants following 48 hours of stimulation with combined CMV/EBV lysates among healthy blood donors (Control), patients with 'low' joint damage (SHS < 20), and patients with 'high' joint damage (SHS > 20). Cytokines were selected based on the results of the principal components analysis (PCA) with factor loadings > 0.5 for the first factor. All data are adjusted for age and sex using mixed effects models. Statistical significance was tested using 2 degree of freedom tests in the mixed effects models. *P < 0.05. **P < 0.01. (B) Shown are the distributions of the CMV/EBV PCA score for the first principal component among healthy blood donors (Control), patients with negative CMV IgG (CMV IgG-), and patients with positive CMV IgG (CMV IgG+). *P < 0.05 for the Kruskal-Wallis test. (C) Shown are the distributions of radiographic joint damage defined by the total Sharp-van der Heijde score according to the tertiles of the CMV/EBV-1 PCA score. *P < 0.05 for the univariate Spearman correlation coefficient. In all panels, the horizontal lines are the medians. CMV, cytomegalovirus; EBV, Epstein-Barr virus; IFN-γ, interferon gamma; IgG, immunoglobulin G; IL, interleukin; PCA, principal components analysis; pg/ml, picograms per milliliter; RA, rheumatoid arthritis; SHS, Sharp-van der Heijde total score
After recognizing the association of CMV/EBV immunity and joint damage, we evaluated the serological status of the patients for past CMV and EBV infection. A total of 28 (48%) had past CMV infection whereas nearly all of the patients (95%) had prior EBV infection (Table 1). Positive CMV IgG correlated significantly with joint space narrowing (r = 0.29, P = 0.025), but not with the total SHS (r = 0.18, P = 0.19) or erosion score (r = 0.10, P = 0.44). The CMV/EBV-1 score correlated with positive CMV IgG (r = 0.44, P < 0.001), but not with EBV-IgG status (r = -0.11, P = 0.39). This suggests that the mechanism underpinning the association between CMV/EBV immunity and joint damage is primarily specific to CMV (Figure 2B). Further adjustment for CMV-IgG status beyond RA disease characteristics (Table 3) revealed an even higher correlation of the CMV/EBV-1 PCA score with the SHS erosion score (r = 0.43; P = 0.003). Finally, we observed linear variation of radiographic joint damage as defined by the total SHS according to the tertile of the CMV/EBV-1 immune response score (Figure 2C).
Correlation of CMV/EBV immune response with other clinical characteristics
To clarify the significance of the CMV/EBV immune response profile that correlated with radiographic joint damage, the next analysis considered the relationship of the CMV/EBV-1 PCA score to other clinical characteristics. The CMV/EBV-1 score was not significantly associated with the DAS28, suggesting that the correlation of CMV/EBV immunity with joint damage is not explained by clinical disease activity. Additionally, there were no statistically significant--nor biologically relevant--correlations of the CMV/EBV PCA score with age, sex, disease duration (from symptom onset), RF status, ACPA status, pain, fatigue, HAQ disability, the SF-36 physical or mental component summary scores, or treatment with methotrexate, biologic response modifiers, or prednisone (P > 0.2 for all; data not shown).
Discussion
The current study represents a translational effort to discover new immune pathways underpinning structural joint damage. Our approach was to induce cytokine response profiles using a panel of stimuli designed to activate both innate and adaptive immunity, then to screen these profiles for correlation with radiographic joint damage in patients with early RA. The singular finding of this study is the association between a profile of cytokine production in response to stimulation with purified CMV/EBV and radiographic joint damage. This finding assumes significance in view of the literature supporting a potential role of latent herpesvirus infection in the pathophysiology of RA.
Both CMV and EBV are in the family of human herpes viruses, which have large double-stranded DNA genomes enclosed in a protein nucleocapsid, surrounded by a protein tegument and also an outer glycoprotein-studded lipid membrane [27, 28]. Infection with these viruses is ubiquitous in people worldwide. In the United States, 50 to 60% of adults have ever been infected with CMV, whereas 90 to 95% of adults have ever been infected with EBV [27, 29]. Of crucial relevance to our findings is the knowledge that herpes viruses induce a robust, primary immune response capable of controlling viral replication, yet they are still able to establish a latent infection in cell reservoirs [30, 31]. CMV primarily infects monocytes and macrophages but also infects dendritic cells, epithelial cells, endothelial cells and fibroblasts [28]. EBV primarily infects B cells yet can also exist latently within nasopharyngeal epithelial cells [27]. Remarkably, 10% of the entire memory T-cell repertoire in humans, on average, is devoted to defense against latent CMV infection, underscoring the impact of this virus on the immune system [32]. Previous epidemiological studies report associations between infection with these herpes viruses (as defined by serological assays) and RA [33–38]. Indeed, latent infection with CMV and EBV is consistently detectable in the synovium [39–44], and CMV has been isolated from synovial cells of a patient with RA [45]. Studies also report the detection of viral replication in RA joints, suggesting active subclinical infection [44, 46, 47].
Considering herpes virus biology, our findings suggest that the immune response to latent infection with CMV or EBV may play a fundamental role in the pathophysiology of chronic inflammation and joint destruction in RA. Specifically, the key observation is that a profile of cytokine production in response to stimulation of PBMC with purified CMV and EBV lysates is associated with the severity of radiographic joint damage in patients with RA. Several pathways could give explanation for our findings. Innate immune receptors could recognize various non-self, pathogen-associated molecular patterns of the viruses [48]. Toll-like receptors (TLR) 2, 3, 7 and 9 likely have important roles in recognizing and activating responses to both CMV [49–54] and EBV [55–59]. Additionally, the protein DAI (ZBP1), and the AIM2 inflammasome are crucial sensors of viral double-stranded DNA in defense against CMV infection [48, 60–63]. However, our results do not clearly suggest activation of innate immune cells by such pathways. In this case, one would expect to see production of IL-1β, IL-6, IL-8, IL-12 or TNF in response to the viruses [50, 54–56], but this is not what we observed. The lack of correlation between the immune response to CpG oligonucleotide, a TLR-9 ligand and radiographic joint damage further suggests that recognition of CMV/EBV by pathogen recognition receptors and innate immune activation may not be the main explanation for our findings.
Another possible explanation for our findings is activation of cytokine production in memory T cells specific for CMV or EBV. The composition of the CMV/EBV immune profile suggests the main source of cytokine production is CD4+ or CD8+ T cells. These should be memory T cells, considering the high prevalence of CMV and EBV infection in our sample, in addition to the short time-period for the development of an immune response to the viruses. A key result is the significant correlation of the CMV/EBV immune response to CMV IgG+. The significance is that patients with RA and other immune-mediated disorders have large, clonally expanded populations of 'CD28null' T cells in the blood [64–67]. It happens that these CD28null T cells are actually highly differentiated, effector-memory T cells, which are largely antigen-specific for CMV [64–66, 68–70]. The down-regulation of the co-stimulatory molecule CD28 signifies chronic activation of the cells, which corresponds to progressive attenuation of proliferative capacity and increase in cytokine production [69, 71, 72]. In contrast, EBV-specific effector-memory T cells are typically CD28+ [69, 73]. Of further note, clonally expanded populations of CD8+ T cells specific for CMV are present in RA synovial fluid [74–76]. Their frequencies are higher in synovial fluid than peripheral blood, suggesting selective enrichment of CD8+ effector-memory T cells in joints [74]. Using HLA class I tetramers, CD8+ T cells have been shown to react specifically with CMV pp65 epitopes [74]. Further, CMV-specific CD28null T cells have a known association with the systemic complications of RA, including lung disease, vasculitis, cardiovascular disease and mortality, highlighting their potential role in the pathophysiology of RA [65, 67, 77, 78]. In view of the evidence, CMV appears to be the more compelling of the two viruses for further study.
CMV-specific effector-memory T cells could possibly mediate inflammation in RA in many, complementary ways. They could do so in an antigen-specific manner, by targeting cells that harbor latent virus, and thus amplify the autoimmune joint disease [79]. Alternatively, CMV-specific effectors could mediate inflammation non-specifically, by migrating into inflamed joints according to chemokine gradients and becoming activated through cell-cell contact, or, by binding other cytokines in the inflammatory milieu, leading to activation of macrophage TNF production [80]. This type of 'bystander activation' of effector-memory T cells is considered a critical mechanism in RA pathophysiology [79–81]. CMV-specific effector-memory CD4+ T cells are equipped to kill target cells rapidly and effectively by production of inflammatory cytokines (that is, IFN-γ or TNF) as well as perforin- and granzyme-dependent cytotoxic function [82, 83]. Additionally, the expansion of CMV-specific memory T cells can overwhelm immunological niches, leading to the loss of both naïve T cells and smaller populations of memory T cells [84]. Indeed, latent CMV infection causes telomere shortening in T cells, indicative of immune senescence [85, 86]. One intriguing possibility relevant to RA is that latent CMV infection is associated with increased differentiation of diverse T-cell specificities, suggesting that this could augment the function of autoreactive T cells in the joints of patients with RA [87]. Further research is necessary to elucidate the contribution of CMV-specific T-cell immunity to the progression of joint destruction in patients with RA.
This study has a number of limitations. The first is its discovery-oriented nature; our findings must be replicated in other patient populations. The study included relatively few patients currently receiving biologic therapies, limiting generalization of our findings to patients mainly on oral disease-modifying agents. Because we only collected data on current use, the results could underestimate the prevalence of ever having taken a biologic DMARD. Another limitation is the study of mixed populations of cells, precluding determination of the specific populations producing our results. We acknowledge that differences in the frequencies of cell types could contribute to our results, but in previous studies we could not explain the associations of ex vivo cytokine production with disease characteristics on the basis of such differences [17, 18]. The use of a mixture of both CMV and EBV lysates creates uncertainties. We cannot be sure that CMV is more important then EBV in regard to RA, but overall the serological findings point to CMV as the most promising candidate for further study. The specific viral moieties that mediate the observed cytokine production are unknown. It certainly remains possible that activation of TLR pathways in innate immune cells with downstream activation of T cells could contribute to our results. Finally, this is a correlative study, so future prospective longitudinal studies are imperative to determine the causal relationship between CMV immunity and joint destruction in RA.
Conclusion
We report the identification of a profile of ex vivo immune response to CMV/EBV stimulation that correlates with radiographic joint damage in patients with RA. The correlation of this immune response to CMV serology suggests the underlying drive is latent CMV infection. The nature of the response suggests the involvement of T-cell immunity. The findings assume significance in the knowledge that the magnitude of CMV-specific CD28null T cells predicts severe manifestations of RA and other immune-mediated diseases. Based on our findings and the literature, we hypothesize that the immune response to latent CMV infection contributes to the propagation of inflammation and progression of structural joint damage in patients with RA. Further research is necessary to elucidate the determinants of the immune response to CMV and/or EBV that aggravate joint destruction in this disease.
Abbreviations
- ACPA:
-
anti-citrullinated protein antibodies
- anti-CD3/anti-CD28:
-
anti-CD3 and anti-CD28 monoclonal antibodies
- CMV:
-
cytomegalovirus
- CMV/EBV-1:
-
CMV/EBV first principal component score
- CpG:
-
bacterial CpG oligonucleotides
- CRP:
-
C-reactive protein
- DAS28:
-
Disease Activity Score in 28 joints
- DMARDs:
-
disease-modifying antirheumatic drugs
- EBV:
-
Epstein-Barr virus
- HAQ:
-
Health Assessment Questionnaire
- HLA-DRB1:
-
human leukocyte antigen DRB1
- IgG:
-
immunoglobulin G
- IFN-γ:
-
interferon gamma
- IL:
-
interleukin
- IQR:
-
interquartile range
- JSN:
-
joint space narrowing
- MCP-1:
-
monocyte chemoattractant protein-1
- MHV:
-
murine gamma-herpesvirus
- MIP-1β:
-
monocyte inflammatory protein-1β
- MSD:
-
Meso Scale Discovery
- PBMC:
-
peripheral blood mononuclear cells
- PCA:
-
principal components analysis
- PMA:
-
phorbol myristate acetate
- PMA/ionomycin-2:
-
PMA/ionomycin stimulation second principal component score
- RA:
-
rheumatoid arthritis
- RF:
-
rheumatoid factor
- SEA/SEB:
-
Staphylococcal enterotoxins A and B
- SF-36:
-
Medical Outcomes Study short form 36
- SHS:
-
Sharp-van der Heijde score
- TLR:
-
Toll-like receptor
- TNF:
-
tumor necrosis factor
- VAS:
-
visual analog scale
References
Scott DL, Wolfe F, Huizinga TW: Rheumatoid arthritis. Lancet. 2010, 376: 1094-1108. 10.1016/S0140-6736(10)60826-4.
Cope AP: T cells in rheumatoid arthritis. Arthritis Res Ther. 2008, 10 (Suppl 1): S1-10.1186/ar2412.
Weyand CM: Immunopathologic aspects of rheumatoid arthritis: who is the conductor and who plays the immunologic instrument?. J Rheumatol Suppl. 2007, 79: 9-14.
van den Berg WB, van Lent PL, Joosten LA, Abdollahi-Roodsaz S, Koenders MI: Amplifying elements of arthritis and joint destruction. Ann Rheum Dis. 2007, 66 (Suppl 3): iii45-48.
Emery P, Breedveld F, van der Heijde D, Ferraccioli G, Dougados M, Robertson D, Pedersen R, Koenig AS, Freundlich B: Two-year clinical and radiographic results with combination etanercept-methotrexate therapy versus monotherapy in early rheumatoid arthritis: a two-year, double-blind, randomized study. Arthritis Rheum. 2010, 62: 674-682. 10.1002/art.27268.
van der Heijde D, Klareskog L, Rodriguez-Valverde V, Codreanu C, Bolosiu H, Melo-Gomes J, Tornero-Molina J, Wajdula J, Pedersen R, Fatenejad S: Comparison of etanercept and methotrexate, alone and combined, in the treatment of rheumatoid arthritis: two-year clinical and radiographic results from the TEMPO study, a double-blind, randomized trial. Arthritis Rheum. 2006, 54: 1063-1074. 10.1002/art.21655.
Durnez A, Vanderschueren G, Lateur L, Westhovens R, Verschueren P: Effectiveness of initial treatment allocation based on expert opinion for prevention of rapid radiographic progression in daily practice of an early RA cohort. Ann Rheum Dis. 2011, 70: 634-637. 10.1136/ard.2010.135319.
Vastesaeger N, Xu S, Aletaha D, St Clair EW, Smolen JS: A pilot risk model for the prediction of rapid radiographic progression in rheumatoid arthritis. Rheumatology (Oxford). 2009, 48: 1114-1121. 10.1093/rheumatology/kep155.
Visser K, Goekoop-Ruiterman YP, de Vries-Bouwstra JK, Ronday HK, Seys PE, Kerstens PJ, Huizinga TW, Dijkmans BA, Allaart CF: A matrix risk model for the prediction of rapid radiographic progression in patients with rheumatoid arthritis receiving different dynamic treatment strategies: post hoc analyses from the BeSt study. Ann Rheum Dis. 2010, 69: 1333-1337. 10.1136/ard.2009.121160.
Bakker MF, Jacobs JW, Welsing PM, Vreugdenhil SA, van Booma-Frankfort C, Linn-Rasker SP, Ton E, Lafeber FP, Bijlsma JW: Early clinical response to treatment predicts 5-year outcome in RA patients: follow-up results from the CAMERA study. Ann Rheum Dis. 2011, 70: 1099-1103. 10.1136/ard.2010.137943.
Markatseli TE, Voulgari PV, Alamanos Y, Drosos AA: Prognostic factors of radiological damage in rheumatoid arthritis: a 10-year retrospective study. J Rheumatol. 2011, 38: 44-52. 10.3899/jrheum.100514.
van Tuyl LH, Boers M, Lems WF, Landewe RB, Han H, van der Linden S, van de Laar M, Westhovens R, van Denderen JC, Westedt ML, Peeters AJ, Jacobs P, Huizinga TW, van de Brink H, Dijkmans BA, Voskuyl AE: Survival, comorbidities and joint damage 11 years after the COBRA combination therapy trial in early rheumatoid arthritis. Ann Rheum Dis. 2010, 69: 807-812. 10.1136/ard.2009.108027.
Brown AK, Conaghan PG, Karim Z, Quinn MA, Ikeda K, Peterfy CG, Hensor E, Wakefield RJ, O'Connor PJ, Emery P: An explanation for the apparent dissociation between clinical remission and continued structural deterioration in rheumatoid arthritis. Arthritis Rheum. 2008, 58: 2958-2967. 10.1002/art.23945.
Brown AK, Quinn MA, Karim Z, Conaghan PG, Peterfy CG, Hensor E, Wakefield RJ, O'Connor PJ, Emery P: Presence of significant synovitis in rheumatoid arthritis patients with disease-modifying antirheumatic drug-induced clinical remission: evidence from an imaging study may explain structural progression. Arthritis Rheum. 2006, 54: 3761-3773. 10.1002/art.22190.
Hetland ML, Ejbjerg BJ, Horslev-Petersen K, Jacobsen S, Vestergaard A, Jurik AG, Stengaard-Pedersen K, Junker P, Lottenburger T, Hansen I, Andersen LS, Tarp U, Skjodt H, Pedersen JK, Majgaard O, Svendsen AJ, Ellingsen T, Lindegaard HM, Christensen AF, Vallo J, Torfing T, Narvestad E, Thomsen HS, Ostergaard M, CIMESTRA study group: MRI bone oedema is the strongest predictor of subsequent radiographic progression in early rheumatoid arthritis. Results from a 2 year randomized controlled trial (CIMESTRA). Ann Rheum Dis. 2009, 68: 384-390. 10.1136/ard.2008.088245.
Hetland ML, Stengaard-Pedersen K, Junker P, Ostergaard M, Ejbjerg BJ, Jacobsen S, Lottenburger T, Hansen I, Tarp U, Andersen LS, Svendsen A, Pedersen JK, Lauridsen UB, Ellingsen T, Lindegaard H, Podenphant J, Vestergaard A, Jurik AG, Horslev-Petersen K: Radiographic progression and remission rates in early rheumatoid arthritis - MRI bone oedema and anti-CCP predicted radiographic progression in the 5-year extension of the double-blind randomised CIMESTRA trial. Ann Rheum Dis. 2010, 69: 1789-1795. 10.1136/ard.2009.125534.
Davis JM, Knutson KL, Strausbauch MA, Crowson CS, Therneau TM, Wettstein PJ, Matteson EL, Gabriel SE: Analysis of complex biomarkers for human immune-mediated disorders based on cytokine responsiveness of peripheral blood cells. J Immunol. 2010, 184: 7297-7304. 10.4049/jimmunol.0904180.
Davis JM, Knutson KL, Strausbauch MA, Crowson CS, Therneau TM, Wettstein PJ, Roger VL, Matteson EL, Gabriel SE: A signature of aberrant immune responsiveness identifies myocardial dysfunction in rheumatoid arthritis. Arthritis Rheum. 2011, 63: 1497-1506.
Prevoo ML, van 't Hof MA, Kuper HH, van Leeuwen MA, van der Putte LB, van Riel PL: Modified disease activity scores that include twenty-eight-joint counts: development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum. 1995, 38: 44-48. 10.1002/art.1780380107.
Fries JF, Spitz P, Kraines RG, Holman HR: Measurement of patient outcome in arthritis. Arthritis Rheum. 1980, 23: 137-145. 10.1002/art.1780230202.
Fries JF, Spitz PW, Young DY: The dimensions of health outcomes: the health assessment questionnaire, disability and pain scales. J Rheumatol. 1982, 9: 789-793.
McHorney CA, Ware JE, Lu JF, Sherbourne CD: The MOS 36-item Short-Form Health Survey (SF-36): III. Tests of data quality, scaling assumptions, and reliability across diverse patient groups. Med Care. 1994, 32: 40-66. 10.1097/00005650-199401000-00004.
McHorney CA, Ware JE, Raczek AE: The MOS 36-Item Short-Form Health Survey (SF-36): II. Psychometric and clinical tests of validity in measuring physical and mental health constructs. Med Care. 1993, 31: 247-263. 10.1097/00005650-199303000-00006.
van der Heijde D: How to read radiographs according to the Sharp/van der Heijde method. J Rheumatol. 2000, 27: 261-263.
van der Heijde DMA, van Riel PL, Koster AM, van 't Hof MA, van Rijswijk MH, van der Putte LB: Biannual radiographic assessment of hands and feet in a three-year prospective followup of patients with early rheumatoid arthritis. Arthritis Rheum. 1992, 35: 26-34. 10.1002/art.1780350105.
Smolen JS, Aletaha D, Grisar JC, Stamm TA, Sharp JT: Estimation of a numerical value for joint damage-related physical disability in rheumatoid arthritis clinical trials. Ann Rheum Dis. 2010, 69: 1058-1064. 10.1136/ard.2009.114652.
Johannsen E, Kaye KM: Epstein-Barr virus (infectious mononucleosis, Epstein-Barr virus-associated malignant diseases, and other diseases. Principles and Practice of Infectious Diseases. Edited by: Mandell GL, Bennett JE, Dolin R. 2009, Philadelphia, PA: Churchill Livingstone, 1989-2010. 7
Crumpacker CS, Zhang JL: Cytomegalovirus. Principles and Practice of Infectious Diseases. Edited by: Mandell GL, Bennett JE, Dolin R. 2009, Philadelphia, PA: Churchill Livingstone, 1971-1987. 7
Cannon MJ, Schmid DS, Hyde TB: Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev Med Virol. 2010, 20: 202-213. 10.1002/rmv.655.
Jackson SE, Mason GM, Wills MR: Human cytomegalovirus immunity and immune evasion. Virus Res. 2011, 157: 151-160. 10.1016/j.virusres.2010.10.031.
Vossen MT, Westerhout EM, Soderberg-Naucler C, Wiertz EJ: Viral immune evasion: a masterpiece of evolution. Immunogenetics. 2002, 54: 527-542. 10.1007/s00251-002-0493-1.
Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, Sleath PR, Grabstein KH, Hosken NA, Kern F, Nelson JA, Picker LJ: Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med. 2005, 202: 673-685. 10.1084/jem.20050882.
Alspaugh MA, Henle G, Lennette ET, Henle W: Elevated levels of antibodies to Epstein-Barr virus antigens in sera and synovial fluids of patients with rheumatoid arthritis. J Clin Invest. 1981, 67: 1134-1140. 10.1172/JCI110127.
Catalano MA, Carson DA, Slovin SF, Richman DD, Vaughan JH: Antibodies to Epstein-Barr virus-determined antigens in normal subjects and in patients with seropositive rheumatoid arthritis. Proc Natl Acad Sci USA. 1979, 76: 5825-5828. 10.1073/pnas.76.11.5825.
Elson CJ, Crawford DH, Bucknall RC, Allen C, Thompson JL, Epstein MA, Hall ND, Bacon PA: Infection with E.B. virus and rheumatoid arthritis. Lancet. 1979, 1: 105-
Ferrell PB, Aitcheson CT, Pearson GR, Tan EM: Seroepidemiological study of relationships between Epstein-Barr virus and rheumatoid arthritis. J Clin Invest. 1981, 67: 681-687. 10.1172/JCI110083.
Tsuchiya N, Murayama T, Yoshinoya S, Matsuta K, Shiota M, Furukawa T, Ito K: Antibodies to human cytomegalovirus 65-kilodalton Fc binding protein in rheumatoid arthritis: idiotypic mimicry hypothesis of rheumatoid factor production. Autoimmunity. 1993, 15: 39-48. 10.3109/08916939309004837.
Venables PJ, Ross MG, Charles PJ, Melsom RD, Griffiths PD, Maini RN: A seroepidemiological study of cytomegalovirus and Epstein-Barr virus in rheumatoid arthritis and sicca syndrome. Ann Rheum Dis. 1985, 44: 742-746. 10.1136/ard.44.11.742.
Einsele H, Steidle M, Muller CA, Fritz P, Zacher J, Schmidt H, Saal JG: Demonstration of cytomegalovirus (CMV) DNA and anti-CMV response in the synovial membrane and serum of patients with rheumatoid arthritis. J Rheumatol. 1992, 19: 677-681.
Mehraein Y, Lennerz C, Ehlhardt S, Remberger K, Ojak A, Zang KD: Latent Epstein-Barr virus (EBV) infection and cytomegalovirus (CMV) infection in synovial tissue of autoimmune chronic arthritis determined by RNA- and DNA-in situ hybridization. Mod Pathol. 2004, 17: 781-789. 10.1038/modpathol.3800119.
Mousavi-Jazi M, Bostrom L, Lovmark C, Linde A, Brytting M, Sundqvist VA: Infrequent detection of cytomegalovirus and Epstein-Barr virus DNA in synovial membrane of patients with rheumatoid arthritis. J Rheumatol. 1998, 25: 623-628.
Murayama T, Jisaki F, Ayata M, Sakamuro D, Hironaka T, Hirai K, Tsuchiya N, Ito K, Furukawa T: Cytomegalovirus genomes demonstrated by polymerase chain reaction in synovial fluid from rheumatoid arthritis patients. Clin Exp Rheumatol. 1992, 10: 161-164.
Stahl HD, Hubner B, Seidl B, Liebert UG, van der Heijden IM, Wilbrink B, Kraan MC, Emmrich F, Tak PP: Detection of multiple viral DNA species in synovial tissue and fluid of patients with early arthritis. Ann Rheum Dis. 2000, 59: 342-346. 10.1136/ard.59.5.342.
Takeda T, Mizugaki Y, Matsubara L, Imai S, Koike T, Takada K: Lytic Epstein-Barr virus infection in the synovial tissue of patients with rheumatoid arthritis. Arthritis Rheum. 2000, 43: 1218-1225. 10.1002/1529-0131(200006)43:6<1218::AID-ANR4>3.0.CO;2-2.
Hamerman D, Gresser I, Smith C: Isolation of cytomegalovirus from synovial cells of a patient with rheumatoid arthritis. J Rheumatol. 1982, 9: 658-664.
Saal JG, Krimmel M, Steidle M, Gerneth F, Wagner S, Fritz P, Koch S, Zacher J, Sell S, Einsele H, Muller CA: Synovial Epstein-Barr virus infection increases the risk of rheumatoid arthritis in individuals with the shared HLA-DR4 epitope. Arthritis Rheum. 1999, 42: 1485-1496. 10.1002/1529-0131(199907)42:7<1485::AID-ANR24>3.0.CO;2-7.
Yarilin DA, Valiando J, Posnett DN: A mouse herpesvirus induces relapse of experimental autoimmune arthritis by infection of the inflammatory target tissue. J Immunol. 2004, 173: 5238-5246.
Pyzik M, Gendron-Pontbriand EM, Fodil-Cornu N, Vidal SM: Self or nonself? That is the question: sensing of cytomegalovirus infection by innate immune receptors. Mamm Genome. 2011, 22: 6-18. 10.1007/s00335-010-9286-2.
Compton T, Kurt-Jones EA, Boehme KW, Belko J, Latz E, Golenbock DT, Finberg RW: Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J Virol. 2003, 77: 4588-4596. 10.1128/JVI.77.8.4588-4596.2003.
Delale T, Paquin A, Asselin-Paturel C, Dalod M, Brizard G, Bates EE, Kastner P, Chan S, Akira S, Vicari A, Biron CA, Trinchieri G, Briere F: MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-alpha release and initiation of immune responses in vivo. J Immunol. 2005, 175: 6723-6732.
Edelmann KH, Richardson-Burns S, Alexopoulou L, Tyler KL, Flavell RA, Oldstone MB: Does Toll-like receptor 3 play a biological role in virus infections?. Virology. 2004, 322: 231-238. 10.1016/j.virol.2004.01.033.
Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, Orihuela MM, Akira S, Yokoyama WM, Colonna M: TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. 2004, 21: 107-119. 10.1016/j.immuni.2004.06.007.
Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, Mudd S, Shamel L, Sovath S, Goode J, Alexopoulou L, Flavell RA, Beutler B: Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci USA. 2004, 101: 3516-3521. 10.1073/pnas.0400525101.
Zucchini N, Bessou G, Traub S, Robbins SH, Uematsu S, Akira S, Alexopoulou L, Dalod M: Cutting edge: overlapping functions of TLR7 and TLR9 for innate defense against a herpesvirus infection. J Immunol. 2008, 180: 5799-5803.
Michaud F, Coulombe F, Gaudreault E, Kriz J, Gosselin J: Involvement of TLR2 in recognition of acute gammaherpesvirus-68 infection. PLoS One. 2010, 5: e13742-10.1371/journal.pone.0013742.
Fiola S, Gosselin D, Takada K, Gosselin J: TLR9 contributes to the recognition of EBV by primary monocytes and plasmacytoid dendritic cells. J Immunol. 2010, 185: 3620-3631. 10.4049/jimmunol.0903736.
Iwakiri D, Zhou L, Samanta M, Matsumoto M, Ebihara T, Seya T, Imai S, Fujieda M, Kawa K, Takada K: Epstein-Barr virus (EBV)-encoded small RNA is released from EBV-infected cells and activates signaling from Toll-like receptor 3. J Exp Med. 2009, 206: 2091-2099. 10.1084/jem.20081761.
Ariza ME, Glaser R, Kaumaya PT, Jones C, Williams MV: The EBV-encoded dUTPase activates NF-kappa B through the TLR2 and MyD88-dependent signaling pathway. J Immunol. 2009, 182: 851-859.
Guggemoos S, Hangel D, Hamm S, Heit A, Bauer S, Adler H: TLR9 contributes to antiviral immunity during gammaherpesvirus infection. J Immunol. 2008, 180: 438-443.
Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E, Fitzgerald KA: The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010, 11: 395-402. 10.1038/ni.1864.
DeFilippis VR, Alvarado D, Sali T, Rothenburg S, Fruh K: Human cytomegalovirus induces the interferon response via the DNA sensor ZBP1. J Virol. 2010, 84: 585-598. 10.1128/JVI.01748-09.
Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, Taniguchi T: DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007, 448: 501-505. 10.1038/nature06013.
Wang Z, Choi MK, Ban T, Yanai H, Negishi H, Lu Y, Tamura T, Takaoka A, Nishikura K, Taniguchi T: Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc Natl Acad Sci USA. 2008, 105: 5477-5482. 10.1073/pnas.0801295105.
Fasth AE, Snir O, Johansson AA, Nordmark B, Rahbar A, Af Klint E, Bjorkstrom NK, Ulfgren AK, van Vollenhoven RF, Malmstrom V, Trollmo C: Skewed distribution of proinflammatory CD4+CD28null T cells in rheumatoid arthritis. Arthritis Res Ther. 2007, 9: R87-10.1186/ar2286.
Martens PB, Goronzy JJ, Schaid D, Weyand CM: Expansion of unusual CD4+ T cells in severe rheumatoid arthritis. Arthritis Rheum. 1997, 40: 1106-1114. 10.1002/art.1780400615.
Thewissen M, Somers V, Hellings N, Fraussen J, Damoiseaux J, Stinissen P: CD4+CD28null T cells in autoimmune disease: pathogenic features and decreased susceptibility to immunoregulation. J Immunol. 2007, 179: 6514-6523.
Morgan MD, Pachnio A, Begum J, Roberts D, Rasmussen N, Neil DA, Bajema I, Savage CO, Moss PA, Harper L: CD4+CD28- T-cell expansion in Wegener's granulomatosis is driven by latent CMV and is associated with an increased risk of infection and mortality. Arthritis Rheum. 2011
Hooper M, Kallas EG, Coffin D, Campbell D, Evans TG, Looney RJ: Cytomegalovirus seropositivity is associated with the expansion of CD4+CD28- and CD8+CD28- T cells in rheumatoid arthritis. J Rheumatol. 1999, 26: 1452-1457.
Amyes E, Hatton C, Montamat-Sicotte D, Gudgeon N, Rickinson AB, McMichael AJ, Callan MF: Characterization of the CD4+ T cell response to Epstein-Barr virus during primary and persistent infection. J Exp Med. 2003, 198: 903-911. 10.1084/jem.20022058.
van Bergen J, Kooy-Winkelaar EM, van Dongen H, van Gaalen FA, Thompson A, Huizinga TW, Feltkamp MC, Toes RE, Koning F: Functional killer Ig-like receptors on human memory CD4+ T cells specific for cytomegalovirus. J Immunol. 2009, 182: 4175-4182. 10.4049/jimmunol.0800455.
Fritsch RD, Shen X, Sims GP, Hathcock KS, Hodes RJ, Lipsky PE: Stepwise differentiation of CD4 memory T cells defined by expression of CCR7 and CD27. J Immunol. 2005, 175: 6489-6497.
Almanzar G, Schwaiger S, Jenewein B, Keller M, Herndler-Brandstetter D, Wurzner R, Schonitzer D, Grubeck-Loebenstein B: Long-term cytomegalovirus infection leads to significant changes in the composition of the CD8+ T-cell repertoire, which may be the basis for an imbalance in the cytokine production profile in elderly persons. J Virol. 2005, 79: 3675-3683. 10.1128/JVI.79.6.3675-3683.2005.
Lunemann JD, Frey O, Eidner T, Baier M, Roberts S, Sashihara J, Volkmer R, Cohen JI, Hein G, Kamradt T, Munz C: Increased frequency of EBV-specific effector memory CD8+ T cells correlates with higher viral load in rheumatoid arthritis. J Immunol. 2008, 181: 991-1000.
Fazou C, Yang H, McMichael AJ, Callan MF: Epitope specificity of clonally expanded populations of CD8+ T cells found within the joints of patients with inflammatory arthritis. Arthritis Rheum. 2001, 44: 2038-2045. 10.1002/1529-0131(200109)44:9<2038::AID-ART353>3.0.CO;2-1.
Tan LC, Mowat AG, Fazou C, Rostron T, Roskell H, Dunbar PR, Tournay C, Romagne F, Peyrat MA, Houssaint E, Bonneville M, Rickinson AB, McMichael AJ, Callan MF: Specificity of T cells in synovial fluid: high frequencies of CD8(+) T cells that are specific for certain viral epitopes. Arthritis Res. 2000, 2: 154-164. 10.1186/ar80.
David-Ameline J, Lim A, Davodeau F, Peyrat MA, Berthelot JM, Semana G, Pannetier C, Gaschet J, Vie H, Even J, Bonneville M: Selection of T cells reactive against autologous B lymphoblastoid cells during chronic rheumatoid arthritis. J Immunol. 1996, 157: 4697-4706.
Gerli R, Schillaci G, Giordano A, Bocci EB, Bistoni O, Vaudo G, Marchesi S, Pirro M, Ragni F, Shoenfeld Y, Mannarino E: CD4+CD28- T lymphocytes contribute to early atherosclerotic damage in rheumatoid arthritis patients. Circulation. 2004, 109: 2744-2748. 10.1161/01.CIR.0000131450.66017.B3.
Gilani SR, Vuga LJ, Lindell KO, Gibson KF, Xue J, Kaminski N, Valentine VG, Lindsay EK, George MP, Steele C, Duncan SR: CD28 down-regulation on circulating CD4 T-cells is associated with poor prognoses of patients with idiopathic pulmonary fibrosis. PLoS One. 2010, 5: e8959-10.1371/journal.pone.0008959.
Posnett DN: Herpesviruses and autoimmunity. Curr Opin Investig Drugs. 2008, 9: 505-514.
Brennan FM, Smith NM, Owen S, Li C, Amjadi P, Green P, Andersson A, Palfreeman AC, Hillyer P, Foey A, Beech JT, Feldmann M: Resting CD4+ effector memory T cells are precursors of bystander-activated effectors: a surrogate model of rheumatoid arthritis synovial T-cell function. Arthritis Res Ther. 2008, 10: R36-10.1186/ar2390.
Di Genova G, Savelyeva N, Suchacki A, Thirdborough SM, Stevenson FK: Bystander stimulation of activated CD4+ T cells of unrelated specificity following a booster vaccination with tetanus toxoid. Eur J Immunol. 2010, 40: 976-985. 10.1002/eji.200940017.
Heller KN, Gurer C, Munz C: Virus-specific CD4+ T cells: ready for direct attack. J Exp Med. 2006, 203: 805-808. 10.1084/jem.20060215.
Haigh TA, Lin X, Jia H, Hui EP, Chan AT, Rickinson AB, Taylor GS: EBV latent membrane proteins (LMPs) 1 and 2 as immunotherapeutic targets: LMP-specific CD4+ cytotoxic T cell recognition of EBV-transformed B cell lines. J Immunol. 2008, 180: 1643-1654.
Moss P: The emerging role of cytomegalovirus in driving immune senescence: a novel therapeutic opportunity for improving health in the elderly. Curr Opin Immunol. 2010, 22: 529-534. 10.1016/j.coi.2010.07.001.
van de Berg PJ, Griffiths SJ, Yong SL, Macaulay R, Bemelman FJ, Jackson S, Henson SM, ten Berge IJ, Akbar AN, van Lier RA: Cytomegalovirus infection reduces telomere length of the circulating T cell pool. J Immunol. 2010, 184: 3417-3423. 10.4049/jimmunol.0903442.
Spyridopoulos I, Hoffmann J, Aicher A, Brummendorf TH, Doerr HW, Zeiher AM, Dimmeler S: Accelerated telomere shortening in leukocyte subpopulations of patients with coronary heart disease: role of cytomegalovirus seropositivity. Circulation. 2009, 120: 1364-1372. 10.1161/CIRCULATIONAHA.109.854299.
Fletcher JM, Vukmanovic-Stejic M, Dunne PJ, Birch KE, Cook JE, Jackson SE, Salmon M, Rustin MH, Akbar AN: Cytomegalovirus-specific CD4+ T cells in healthy carriers are continuously driven to replicative exhaustion. J Immunol. 2005, 175: 8218-8225.
Acknowledgements
This project was supported by NIH/NCRR CTSA Grant Numbers KL2 RR024151 and UL1 RR024150. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. Dr. Davis was also supported by a New Investigator Award from the Arthritis Foundation and a career development award from Mayo Foundation.
The authors thank Jeaneen Alcorn for coordinating the study and Jennifer Gall and Sherry Kallies for administrative support. The authors also wish to thank Larry R. Pease, PhD, for scientific contributions and institutional support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
JMD, KLK and SEG are inventors of technology referenced in this article. That technology is entitled "Cytokine Response Profiling in Rheumatoid Arthritis." The Mayo Clinic has filed a provisional patent application for this technology.
Authors' contributions
JMD, KLK and SEG designed the study. JMD recruited and clinically evaluated the patients. MSS performed the PBMC assays and multiplexed cytokine analysis. JAS interpreted and scored all joint radiographs. JMD, CSC, KLK and TMT performed the statistical analysis. All authors contributed to analysis and critical interpretation of the results as well as manuscript preparation. All authors have read and approved the final manuscript for publication.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Davis, J.M., Knutson, K.L., Skinner, J.A. et al. A profile of immune response to herpesvirus is associated with radiographic joint damage in rheumatoid arthritis. Arthritis Res Ther 14, R24 (2012). https://doi.org/10.1186/ar3706
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar3706