
Abstract
Bone destruction is a frequent and clinically serious event in patients with rheumatoid arthritis (RA). Local joint destruction can cause joint instability and often necessitates reconstructive or replacement surgery. Moreover, inflammation-induced systemic bone loss is associated with an increased fracture risk. Bone resorption is a well-controlled process that is dependent on the differentiation of monocytes to bone-resorbing osteoclasts. Infiltrating as well as resident synovial cells, such as T cells, monocytes and synovial fibroblasts, have been identified as sources of osteoclast differentiation signals in RA patients. Pro-inflammatory cytokines are amongst the most important mechanisms driving this process. In particular, macrophage colony-stimulating factor, RANKL, TNF, IL-1 and IL-17 may play dominant roles in the pathogenesis of arthritis-associated bone loss. These cytokines activate different intracellular pathways to initiate osteoclast differentiation. Thus, over the past years several promising targets for the treatment of arthritic bone destruction have been defined.
Similar content being viewed by others
Introduction
Chronic inflammation is a risk factor for bone loss. Many chronic inflammatory disorders, such as rheumatoid arthritis (RA), ankylosing spondylitis, inflammatory bowel disease and even low-grade inflammation in otherwise healthy individuals, have been linked to an increased fracture risk [1–5]. RA is of particular interest as both locally affected bones and sites distant of joint inflammation are prone to bone loss.
Chronic inflammation is the key mediator for local and systemic bone loss in RA patients. In RA patients, cytokines are abundantly present in the arthritic synovium as well as secreted into the systemic circulation [6, 7]. The discovery of RANKL (Receptor activator of NF-kB ligand) in 1998 as a crucial regulator of osteoclastogenesis opened avenues for the research of arthritis-driven bone loss [8]. Since then, several pro-inflammatory cytokines have been identified as direct or indirect stimulators of osteoclast differentiation, survival and activity. This review comprises the knowledge on the most important cytokines, which are both involved in RA pathophysiology and documented drivers of osteoclast differentiation, survival or activation (Figure 1). In addition to their pro-resorbing role, other cytokines can also act anti-osteoclastogenically, which is reviewed in the accompanying article of Zhao and Ivashkiv. This is especially evident, as other arthritic disorders such as psoriatic arthritis are characterised by strong repair responses within affected joints [9]. The balance of osteoclastogenic and anti-osteoclastogenic mediators thus decides the fate of bone destruction.

Cytokines activating osteoclastogenesis in rheumatoid arthritis. TNF, IL-1, IL-6 and IL-17 upregulate expression of RANKL (receptor activator of NF-kB ligand) in osteoblasts and synovial fibroblasts. RANKL mediates differentiation, survival and activation of osteoclasts. TNF, produced by fibroblasts and macrophages, promotes differentiation and survival of osteoclasts. IL-1 supports differentiation, survival and activation of osteoclasts. IL-6 and IL-17 promote osteoclastogenesis indirectly. IL-6 is largely produced by fibroblasts and macrophages; it enhances the expression of RANKL and contributes to the induction of Th17 cells. Th17 cells secrete IL-17, but a main source of synovial IL-17 is probably mast cells. IL-17 induces the expression of RANKL in osteoblasts and fibroblasts and enhances secretion of pro-inflammatory cytokines by macrophages. Macrophage colony-stimulating factor (M-CSF) and IL-34 promote differentiation and activation of osteoclasts; IL-33 supports osteoclast differentiation. TGF, transforming growth factor.
RANKL
RANKL is a member of the TNF family of cytokines and plays a key role in bone resorption. Osteoclasts are the sole bone resorbing cell. They are formed by fusion of mononuclear cells of the monocyte/macrophage lineage, but dendritic cells could also serve as osteoclast pre-cursors [10–12]. RANKL is a necessary factor for the differentiation of osteoclasts. Mice deficient for RANKL develop severe osteopetrosis due to a complete lack of osteoclastogenesis [13]. RANKL also serves as survival factor and activates osteoclasts. The physiological inhibitor of RANKL is osteoprotegerin (OPG), a decoy receptor that binds RANKL. OPG-deficient mice exhibit severe osteoporosis [14]. The main sources of RANKL are osteoblasts but RANKL can also be expressed in synovial cells, activated T cells, mature B cells and natural killer cells [15–18]. Expression of RANKL is upregulated by parathyroid hormone, 1,25(OH)2D3 and several pro-inflammatory cytokines, including IL-1, IL-6, IL-17 and TNF-α [8, 19–21]. RANKL functions both as a membrane-anchored molecule and as a soluble molecule. Both forms bind to RANK, the receptor of RANKL. RANK is expressed on osteoclast precursors and mature osteoclasts [22]. In vivo, RANKL-deficient mice are protected from bone erosions in the serum transfer model of arthritis [23]. OPG treatment protects human TNF-α transgenic mice from bone destruction [24]. In a rat collagen-induced arthritis (CIA) model, OPG inhibited bone destruction as well [25]. In these models, inhibition of RANKL affects only bone destruction and not inflammation. Denosumab, a humanized antibody against RANKL, is currently being evaluated in clinical trials. Application of denosumab inhibits glucocorticoid-induced bone loss in mice [26]. In a phase II study, the addition of denosumab to methotrexate treatment inhibited structural bone damage in patients with RA [27].
Tumour necrosis factor α
Activated macrophages but also synovial fibroblasts, T cells, B cells, natural killer cells, osteoblasts and osteoclasts can produce the pro-inflammatory cytokine TNF [28, 29]. Both soluble and membrane-bound TNF bind to the TNF receptors TNFR1 (p55) and TNFR2 (p75). TNFR1 mediates most of the biological effects of TNF. Osteoclasts and its precursors express both TNFR1 and TNFR2 [30–32]. TNF upregulates RANK expression and can thus enhance osteoclastogenesis [33]. TNF may also directly act on osteoclast precursors, but whether this is truly independent of RANKL signalling is still the subject of debate (reviewed in [34]). TNF promotes the survival of mature osteoclasts, but does not efficiently activate osteoclasts [34, 35]. Kitaura and colleagues demonstrated TNF-dependent secretion of macrophage colony-stimulating factor (M-CSF) by bone marrow stromal cells that induces osteoclastogenesis more efficiently than the direct stimulation of osteoclast precursors by TNF. The relevance of this finding is underlined by inhibition of osteoclastogenesis despite persistence of inflammation in a serum-transfer arthritis model using an anti-M-CSF receptor (c-fms) antibody [36].
TNF further supports osteoclastogenesis by interacting with the wingless (Wnt) signalling pathway. TNF is a strong inducer of Dkk-1 expression, a Wnt antagonist. Dkk-1 inhibits Wnt signalling by binding to LRP-5 (low density lipoprotein-coupled receptor related protein-5) and LRP-6 and the coreceptor Kremen-1/2 [37]. Active Wnt signalling induces OPG expression and therefore decreases the RANKL/OPG ratio, thus acting anti-osteoclastogenically [38]. Consequently, Dkk-1 promotes osteoclastogenesis by increasing the RANKL/OPG ratio. In RA patients, elevated serum levels of Dkk-1 have been observed. After initiation of anti-TNF therapy, serum levels of Dkk-1 decrease. Expression of Dkk-1 is also enhanced in animal models of erosive arthritis, such as human TNF transgenic mice, CIA and glucose-6-phosphate isomerase-induced arthritis [39].
The relevance of TNF for arthritic bone destruction has been demonstrated in several experimental models and was finally confirmed by clinical trials. In vivo, human TNF transgenic mice develop severe arthritis with chronic synovial inflammation, cartilage destruction, and systemic and local bone loss [40]. The latter pathology is quite unique, as many other rodent arthritis models are characterized by strong repair responses, which is rarely seen in RA. In CIA, the application of TNF-specific neutralizing antibodies reduced disease activity and bone damage [41]. The results in TNF-deficient mice are not as clear. Using the serum transfer model of arthritis, most TNF-deficient mice develop no clinical or histological signs of arthritis, but one-third of mice showed clinical signs of arthritis [42]. The efficacy and safety of the TNF antagonists infliximab, etanercept, adalimumab, golimumab and certolizumab in RA patients were demonstrated in several clinical studies and these drugs are now frequently used in clinical practice [43]. Interestingly, RA patients clinically not responding to anti-TNF treatment are still protected from development of new bone erosions. This underlines the important role of TNF for arthritic bone destruction.
Interleukin-1
In RA joints, activated macrophages and synovial fibroblasts are sources of IL-1 production [44, 45]. IL-1α and IL-1β share only 24% amino acid sequence identity but have largely identical biological functions mediated through the receptor IL-1R1 [46, 47]. IL-1 receptor antagonist (IL1-Ra) is a soluble protein that competes with IL-1 for binding to IL-1R1 [48]. Thus, the IL-1/IL-1Ra ratio has to increase to induce IL-1R1 activation. IL-1R1 and the decoy receptor IL-1R2 are expressed in osteoclasts. There is higher expression of IL-1R1 in large osteoclasts than in small osteoclasts [49]. The mechanism has not yet been completely established, but several in vitro studies provide evidence that IL-1 plays a significant role in osteoclast physiology. IL-1 promotes the fusion of osteoclast precursors [50] and prolongs the survival of mature osteoclasts [51]. It is also important for osteoclast activation in vitro [31, 52].
In vivo, IL-1 is a key regulatory cytokine in mouse models of inflammatory arthritis. Overexpression of IL1-α or IL-1β as well as deletion of IL-1Ra leads to development of arthritis with destruction of cartilage and bone [45, 53–55]. Mice deficient of IL-1R1 develop no arthritis in a model of serum transfer arthritis [42]. In human TNF transgenic mice deficient for IL-1 signalling, cartilage destruction was completely blocked and bone destruction partly reduced despite the presence of synovial inflammation [56]. Recent data show no systemic inflammatory bone loss in these IL-1-deficient human TNF transgenic mice in spite of ongoing inflammatory arthritis [57]. These data indicate that TNF-induced local bone destruction and systemic inflammatory bone loss are largely dependent on IL-1. Thus, IL-1 is an important downstream mediator of TNF. These in vivo data are supported by in vitro evidence showing that TNF-induced synthesis of RANKL is inhibited by IL-1Ra [58]. In contrast to these in vivo and in vitro data, targeting IL-1 has not yet provided powerful therapeutics for the treatment of RA [59]. However, few data exist regarding the bone-protective properties of IL-1 neutralization in RA patients.
Interleukin-6
IL-6 expression in RA synovial tissue has been localized to synovial fibroblasts, macrophages and T cells [60, 61]. There are two forms of the IL-6 receptor, a transmembrane variant and a soluble variant. The transmembrane form consists of an 80-kDa chain specific for IL-6 and the intracellular signal transducer glycoprotein 130 (gp130). This transmembrane form of the IL-6 receptor is only expressed in hepatocytes, monocytes/macrophages, osteoblasts and other leukocytes, while gp130 is expressed on almost all cells [62, 63]. The soluble receptor binds IL-6 and activates gp130 on cells that do not express the transmembrane receptor [62]. This soluble receptor is found in many body fluids, including serum and synovial fluid [64]. IL-6 is elevated in serum and synovial fluid of RA patients [65]. IL-6-deficient mice are protected from ovariectomy-induced bone loss and show delayed bone fracture healing related to a decreased number of osteoclasts [66, 67]. IL-6 overexpression is associated with enhanced bone resorption and increased osteoclast numbers and activity [68]. These data suggest an activating effect of IL-6 signalling on bone resorption.
In vitro, IL-6 stimulates the release of RANKL by osteoblasts and together with transforming growth factor-β and IL-1 promotes the development of Th17 cells [69–71]. A recent study showed reduced in vitro osteoclast differentiation due to the blockade of the IL-6 receptor [72]. There is also recent in vitro evidence of an inhibitory effect of IL-6 on osteoclastogenesis [73, 74]. However, these models do not consider potential effects of other cells and cytokines and the in vivo relevance of these in vitro findings is unclear [75].
IL-6-deficient mice are protected from CIA and adjuvant-induced arthritis [76–78]. In contrast, the IL-6-deficient mice develop arthritis in the K/BxN serum transfer model [42]. While overexpression of human IL-6 does not induce polyarthritis, an activating mutation in the mouse gp130 gene causes autoimmune polyarthritis [79, 80]. The application of an IL-6 receptor (IL-6R) neutralizing antibody in CIA reduced disease activity [81]. In human TNF transgenic mice, an anti-IL-6R antibody did not inhibit joint inflammation but reduced osteoclast formation in the inflamed joints and bone erosion [72]. In contrast to the different experimental findings, the IL-6R-specific antibody tocilizumab efficiently reduces disease activity and radiographic progression in RA patients and is now used in clinical practice [59].
Interleukin-17
IL-17 is present in synovial fluid of RA patients and its expression has been detected within the inflamed synovium in Th17 and other cells [20, 82, 83]. Recent evidence suggests that cells other than Th17 cells, such as mast cells, are probably a major source of IL-17 production within human arthritic joints [84]. There is good evidence for an important role of IL-17 in osteoclastogenesis, but the detailed mechanism is not yet completely understood [22].
In mice, the severity of CIA is reduced in IL-17-deficient animals and the local overexpression of IL-17 in a joint enhances severity of arthritis [85, 86]. The treatment of arthritic mice with an anti-IL-17 antibody reduces joint inflammation, cartilage destruction and bone erosion in CIA and antigen-induced arthritis [87, 88]. In vitro, IL-17 stimulates osteoclastogenesis preferentially indirectly [20]. In vitro studies show that, on the one hand, IL-17 elevates RANKL expression in osteoblasts and fibroblasts, and on the other, it induces the secretion of pro-inflammatory cytokines such as IL-6 and IL-8 by fibroblasts and endothelial and epithelial cells and the secretion of TNF and IL-1 from monocytes [22, 89–91]. Recent data provide evidence for an additional direct effect of IL-17 on osteoclast differentiation. In vitro, IL-17 upregulates RANK on human osteoclast precursors to sensitize them to RANKL [92]. Another recent study demonstrated that IL-17 induces osteoclastogenesis in cultures of human CD11b-positive cells in the absence of osteoblasts or exogenous RANKL. This is blocked by the application of OPG or infliximab, suggesting a RANKL- and TNF-dependent mechanism [93]. Further investigation is necessary to identify the exact mechanism of IL-17-induced osteoclastogenesis. Ongoing clinical trials are analysing the efficacy of anti-IL-17 antibodies in RA patients.
Macrophage colony-stimulating factor
M-CSF is a key cytokine providing osteoclast differentiation signals [94, 95]. It is secreted by synovial fibroblasts, osteoblasts, macrophages and T cells in RA patients [96–98]. The importance of M-CSF-induced osteoclastogenesis is confirmed in mouse models: op/op mice, which fail to express functional M-CSF, and c-fms (the M-CSF receptor) deficient mice show an osteoclast-poor osteopetrotic phenotype [99, 100]. In vitro, M-CSF modulates multiple steps in human osteoclastogenesis, including proliferation, differentiation and fusion of pre-cursors and at later differentiation stages bone resorbing activity but not survival [101]. The binding of M-CSF to c-fms leads to the activation of the ERK-Akt signalling pathway [102–104].
Intracellular signalling
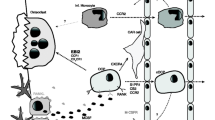
The stimulation of osteoclasts with RANKL leads to potent activation of NFATc1 (Nuclear factor of activated T cells, cytoplasmic 1), as demonstrated by gene expression profiling (Figure 2) [105]. NFATc1 is a key regulator of osteoclast differentiation in vitro and in vivo; it induces osteoclast-specific genes, including TNF-receptor associated protein (TRAP), calcitonin receptor and cathepsin K, and it positively regulates its own promoter. The activation of NFATc1 is regulated by RANKL in two ways: the NF-κB/AP-1/c-fos pathway and calcium signalling [105]. RANKL binds to its receptor RANK, which leads to the binding of RANK to its main adaptor molecule TNF receptor-associated factor (TRAF)6. TRAF5 is also involved in RANKL-mediated osteoclastogenesis [106]. This complex activates JNK, p38 and NF-κB [107]. In vitro, osteoclastogenesis is impaired in monocytes lacking p38α [108].

Intracellular signalling during inflammation-induced osteoclastogenesis. RANKL (receptor activator of NF-kB ligand) binds to its receptor RANK and induces the key regulator of osteoclast differentiation NFATc1 (Nuclear factor of activated T cells, cytoplasmic 1) through two different signalling pathways. On the one hand, RANK recruits TRAF6 (TNF receptor-associated factor 6) and activates NF-κB, JNK, p38, c-fos and AP-1. On the other hand, NFATc1 is activated by calcineurin that is in turn activated by elevated calcium in the cytoplasm. Phospholipase C (PLC)γ mediates the release of calcium in the cytoplasm; PLCγ is activated by RANK through Btk/Tec and by OSCAR (osteoclast-associated receptor) and TREM-2 (triggering receptor expressed by myeloid cells 2) through the Fc receptor gamma chain (FcRγ), DAP12 (DNAX-activating protein of 12 kDa) and Syk signalling. TNF binds to its receptor TNFR1, which recruits TRADD (TNFR-associated DD protein) and RIP-1 (Receptor interacting protein-1). TNF receptor-associated factor (TRAF)2, TRAF5 and TRAF6 mediate further signalling through NF-κB, JNK and p38. The IL-1 receptor IL-1R1 binds MYD88 (myeloid differentiation primary response gene 88) and RAK4, which activates TRAF6 through phosphorylation of IL-1 receptor-activating protein kinase (IRAK)2 and IRAK1. After binding IL-6, the IL-6 receptor recruits two gp130 molecules and activates the signal transducer and activator of transcription (STAT) pathway and the mitogen-activated protein kinase (MAPK) pathway (JNK, p38, ERK) through gp130. MMP, matrix metalloproteinase.
NF-κB is a family of dimeric transcription factors. In mammals there are five proteins: Rel (cRel), RelA (p65), RelB, NFκB1 (p50) and NFκB2 (p52) [109]. p50/p52-/-mice develop osteopetrosis while p50-/-mice show no bone phenotype [109–111]. There is a classical and an alternative NF-κB signalling pathway. The classical pathway includes activation of IκB kinase (IKK)β. Roucco and colleagues [112] showed impaired osteoclastogenesis in the absence of IKKβ in vitro and in vivo. The alternative pathway includes IKKα and NF-κB-inducing kinase (NIK). Osteoclastogenesis depends on IKKα and NIK only in vitro but not in vivo [112, 113]. Thus, the classical pathway seems to be of greater importance for osteo-clastogenesis. In addition, IKKβ prevents TNF-induced apoptosis of osteoclast precursors [112]. NF-κB induces c-fos, cyclic AMP-responsive element-binding protein (CREB) and calcium/calmodulin-dependent protein kinase type IV (CaMKIV) [114, 115].
The AP-1 transcription-factor is a dimeric complex composed of c-fos and Jun proteins. c-Fos-deficient mice develop severe osteopetrosis due to a complete block of osteoclastogenesis [116, 117]. Mice with conditional knockout of Jun proteins (c-Jun, JunB) show impaired osteoclastogenesis [118, 119], and mice expressing dominant negative c-Jun under the control of the TRAP promoter develop osteopetrosis [120]. AP-1 DNA binding activity is upregulated in the synovial tissue of RA patients and correlates with disease activity [121]. AP-1 cooperates with NFATc1, inducing osteoclast-specific genes [105].
On the other hand NFATc1 is dependent on calcium signalling. The phosphatase calcineurin specifically activates NFATc1 by dephosphorylating its amino-terminal regulatory domain. While the phosphorylated NFATc1 is localised in the cytoplasm, the dephosphorylated NFATc1 can enter the nucleus. The importance of this pathway is shown by the immunosuppressive drug cyclosporine, which inhibits calcineurin [122]. The activation of calcineurin is dependent on calcium and phospholipase C (PLC)γ, which mediates calcium release in the cytoplasm [105]. PLCγ2 is the isoform that regulates osteoclastogenesis, and PLCγ2-deficient mice develop osteopetrosis independent of PLCγ1 [123]. There are two links between calcium signalling and RANKL. The co-stimulatory receptors of RANK, OSCAR (osteoclast-associated receptor) and TREM-2 (triggering receptor expressed by myeloid cells 2), activate PLCγ through its adaptor proteins DAP12 (DNAX-activating protein of 12 kDa) and Fc receptor gamma chain (FcRγ) and the tyrosin kinase Syk [124, 125]. The second link is the Tec family tyrosine kinases Tec and Btk, which are activated by RANKL and are involved in the phosphorylation of PLCγ [126].
TNF signalling in osteoclasts and their precursors is primarily mediated by TNFR1. TNFR1 contains a cytoplasmic death domain and when unstimulated, this domain binds to the death domain of the protein SODD (silencer of death domain). TNF binding to TNFR1 leads to the release of SODD. This allows the binding of TRADD (TNFR-associated DD protein), which recruits RIP-1 (receptor interacting protein-1) and TRAF2. This TRADD-RIP-1-TRAF2 complex is released from TNFR1 and activates NFκB, JNK and p38 signalling [30]. TRAF2 is essential for osteoclastogenesis in vitro [127]. TRAF6 and TRAF5 also contribute to TNF-dependent osteoclastogenesis in vitro and activate NFκB, JNK and p38 signalling [106, 128, 129]. In line with this, TRAF6-deficient mice show severe osteopetrosis [130].
The binding of IL-1 to its receptor IL-1R1 induces a conformational change of the receptor. After recruitment of IL-1RacP, it binds to MYD88 (myeloid differentiation primary response gene 88) and IL-1 receptor-activating protein kinase (IRAK)4. This complex recruits TRAF6 through phosphorylation IRAK2 and IRAK1 [131].
The transmembrane or the soluble IL-6 receptor forms a complex with two gp130 molecules after binding IL-6. This leads to phosphorylation of Janus protein-tyrosine kinase, which causes the activation of intracellular signal transduction. gp130 can act though two intracellular signalling pathways: the signal transducer and activator of transcription (STAT) pathway and the mitogen-activated protein kinase (MAPK) pathway [75]. The mechanism of IL-6 signalling in bone turnover is not yet understood. Mice lacking the gp130 binding site for STAT show no alteration in osteoclast activity and one publication indicated that STAT3 downregulates NFATc1 [132, 133]. Mice lacking the gp130 binding site for MAPK signalling exhibit osteopenia. gp130-deficient mice develop osteopenia as well, although this mutation results in neonatal lethality [133]. A recent study shows that IL-6 suppresses NF-κB signalling [73]. Despite these findings, IL-6 seems to have potent osteoclast-activating functions in RA patients, as demonstrated by clinical trials using an antibody against the soluble IL-6 receptor.
Other cytokines
The recently discovered cytokine IL-34 binds to the M-CSF receptor c-fms. In functional studies it promotes monocyte viability and the formation of macrophage progenitor cells independent of M-CSF. Similar to M-CSF, IL-34 activates ERK signalling [134]. Baud'Huin and colleagues [135] demonstrated that IL-34 was able to support RANKL-induced osteoclastogenesis in the absence of M-CSF. However, higher concentrations of IL-34 than of M-CSF are required to exert an equivalent activity, probably due to a relatively lower binding affinity of IL-34 to c-fms. IL-34 activates the ERK-Akt signalling pathway in osteoclast progenitors and promotes osteoclastogenesis but has no effect on osteoclast survival.
There is recent evidence that the pro-inflammatory cytokine IL-33 participates in the pathogenesis of RA. It is expressed in the synovium of patients with RA and its expression appeared to correlate with the severity of inflammation [136]. IL-33 acts through the receptor ST2 [137], which is a member of the Toll-like/IL-1 receptor family and activates TRAF6 [138]. The ST2 transmembrane form is expressed predominantly on mast cells and Th2 cells. In murine antigen-induced arthritis, IL-33 exacerbates disease by activating mast cells [139]. Inhibition of IL-33 signalling reduced severity of bone erosion in an animal arthritis model [140]. Recent data show that IL-33 induces the formation of osteoclasts from human monocytes independent of RANKL [141]. IL-33 seems to activate MAPKs, NF-κB and the Syk/PLCγ signalling pathway in human monocytes. In contrast, IL-33 was found to inhibit murine osteoclastogenesis in vitro and in vivo [142].
There were previous reports that the culture medium of activated T cells directly stimulates osteoclastogenesis independent of RANKL [143, 144]. Rifas and colleagues [145] recently identified a new cytokine in the medium of activated T cells by chromatographic analysis. They called this new cytokine Secreted osteoclastogenic factor of activated T-cells (SOFAT). SOFAT induces the formation of human and mouse functional osteoclasts independent of RANKL and is secreted by T cells in a calcineurin-independent manner. It is derived from a mRNA splice variant encoded by the threonin synthase-like 2 gene homolog. RANKL-deficient mice have no osteoclasts and develop no bone erosions despite severe inflammation in the case of arthritis [13, 23]. These in vivo data show no relevant osteoclastogenesis independent of RANKL. Further investigation is needed to characterize the role of SOFAT in osteoclastogenesis.
In vitro data demonstrated that IL-15 directly promotes differentiation of rodent osteoclast progenitors into pre-osteoclasts [146] and neutralization of IL-15 prevented bone destruction in CIA [147]. IL-15 is elevated in synovial membrane and synovial fluid in RA patients [148]. In vitro, osteoclastogenesis and osteoclast function are reduced in IL-15R-deficient compared to wild-type spleen or bone marrow cells [149]. Bone mineral density was increased in IL-15R-deficient mice and was not reduced after ovariectomy. Serum levels of TRAP5b and osteocalcin were lower in IL-15R-deficient mice, consistent with a low bone turnover in the absence of IL-15 signalling.
Conclusion
Bone loss in RA patients is a frequent and clinically serious event. Considering bone remodelling in general, the balance between bone formation and bone resorption determines the net effect. In the past decade, significant gains in knowledge about the role of bone resorption during chronic erosive arthritis have been made. There is good evidence that inflammation itself triggers bone resorption by osteoclasts [5].
Pro-inflammatory cytokines are potent mediators of bone loss. These cytokines act both directly and indirectly to enhance osteoclastogenesis in the inflamed joint and systemic bone: first, many pro-inflammatory cytokines can alter the RANKL/OPG ratio in mesenchymal cells, such as osteoblasts and fibroblasts; second, some cytokines, such as M-CSF and RANKL, also directly affect osteoclast differentiation, survival and activity.
Bone erosions and osteoporosis significantly affect function and quality of life. Thus, anti-erosive therapies - besides anti-inflammatory therapy - for RA patients are of great interest. In the past, bisphosphonates had been used to inhibit structural damage in RA joints, but the effects were limited. Recently, the anti-RANKL antibody denosumab has been used in RA patients in a small study and promising results have been observed [150]. Denosumab-treated RA patients showed no radiographic progression compared to placebo-treated patients Furthermore, there is good evidence that TNF blockade also inhibits structural bone damage independent of its anti-inflammatory activity in RA patients [151]. Thus, our increased knowledge on the pathophysiology may lead to new therapeutic concepts in RA incorporating anti-erosive therapies. However, even nowadays up to 80% of RA patients experience structural bone damage during the course of disease [152]. Thus, further research is necessary to fully elucidate the pathophysiology of osteoclast-driven bone loss in RA patients.
Note
This article is part of the series Osteoimmunology, edited by Georg Schett. Other articles in this series can be found at http://arthritis-research.com/series/osteoimmunology
Abbreviations
- gp:
-
glycoprotein
- IKK:
-
IκB kinase
- IL:
-
interleukin
- IL1-Ra:
-
Interleukin 1 receptor antagonist
- MAPK:
-
mitogen-activated protein kinase
- M-CSF:
-
macrophage colony-stimulating factor
- NF:
-
nuclear factor
- NFATc1:
-
Nuclear factor of activated T cells, cytoplasmic 1
- NIK:
-
NF-κB-inducing kinase
- OPG:
-
osteoprotegerin
- PLC:
-
phospholipase C
- RA:
-
rheumatoid arthritis
- RANK:
-
receptor activator of NF-kB
- RANKL:
-
RANK ligand
- STAT:
-
signal transducer and activator of transcription
- TNF:
-
tumour necrosis factor
- TNFR:
-
tumour necrosis factor receptor
- TRAF:
-
TNF receptor-associated factor
- TRAP:
-
TNF-receptor associated protein.
References
Gough AK, Lilley J, Eyre S, Holder RL, Emery P: Generalised bone loss in patients with early rheumatoid arthritis. Lancet. 1994, 344: 23-27. 10.1016/S0140-6736(94)91049-9.
Spector TD, Hall GM, McCloskey EV, Kanis JA: Risk of vertebral fracture in women with rheumatoid arthritis. BMJ. 1993, 306: 558-10.1136/bmj.306.6877.558.
Magrey M, Khan MA: Osteoporosis in ankylosing spondylitis. Curr Rheumatol Rep. 2010, 12: 332-336. 10.1007/s11926-010-0122-1.
Loftus EV, Achenbach SJ, Sandborn WJ, Tremaine WJ, Oberg AL, Melton LJ: Risk of fracture in ulcerative colitis: a population-based study from Olmsted County, Minnesota. Clin Gastroenterol Hepatol. 2003, 1: 465-473. 10.1016/S1542-3565(03)00185-X.
Schett G, Kiechl S, Weger S, Pederiva A, Mayr A, Petrangeli M, Oberhollenzer F, Lorenzini R, Redlich K, Axmann R, Zwerina J, Willeit J: High-sensitivity C-reactive protein and risk of nontraumatic fractures in the Bruneck study. Arch Intern Med. 2006, 166: 2495-2501. 10.1001/archinte.166.22.2495.
Brennan FM, McInnes IB: Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008, 118: 3537-3545. 10.1172/JCI36389.
Matei I, Matei L: Cytokine patterns and pathogenicity in autoimmune diseases. Rom J Intern Med. 2002, 40: 27-41.
Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T: Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA. 1998, 95: 3597-3602. 10.1073/pnas.95.7.3597.
de Vlam K, Lories RJ, Luyten FP: Mechanisms of pathologic new bone formation. Curr Rheumatol Rep. 2006, 8: 332-337. 10.1007/s11926-006-0061-z.
Teitelbaum SL: Bone resorption by osteoclasts. Science. 2000, 289: 1504-1508. 10.1126/science.289.5484.1504.
Speziani C, Rivollier A, Gallois A, Coury F, Mazzorana M, Azocar O, Flacher M, Bella C, Tebib J, Jurdic P, Rabourdin-Combe C, Delprat C: Murine dendritic cell transdifferentiation into osteoclasts is differentially regulated by innate and adaptive cytokines. Eur J Immunol. 2007, 37: 747-757. 10.1002/eji.200636534.
Rivollier A, Mazzorana M, Tebib J, Piperno M, Aitsiselmi T, Rabourdin-Combe C, Jurdic P, Servet-Delprat C: Immature dendritic cell transdifferentiation into osteoclasts: a novel pathway sustained by the rheumatoid arthritis microenvironment. Blood. 2004, 104: 4029-4037. 10.1182/blood-2004-01-0041.
Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, Daro E, Smith J, Tometsko ME, Maliszewski CR, Armstrong A, Shen V, Bain S, Cosman D, Anderson D, Morrissey PJ, Peschon JJ, Schuh J: RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13: 2412-2424. 10.1101/gad.13.18.2412.
Mizuno A, Amizuka N, Irie K, Murakami A, Fujise N, Kanno T, Sato Y, Nakagawa N, Yasuda H, Mochizuki S, Gomibuchi T, Yano K, Shima N, Washida N, Tsuda E, Morinaga T, Higashio K, Ozawa H: Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem Biophys Res Commun. 1998, 247: 610-615. 10.1006/bbrc.1998.8697.
O'Brien CA: Control of RANKL gene expression. Bone. 2010, 46: 911-919. 10.1016/j.bone.2009.08.050.
Li Y, Toraldo G, Li A, Yang X, Zhang H, Qian WP, Weitzmann MN: B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood. 2007, 109: 3839-3848. 10.1182/blood-2006-07-037994.
Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, Khoo W, Wakeham A, Dunstan CR, Lacey DL, Mak TW, Boyle WJ, Penninger JM: OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999, 397: 315-323. 10.1038/16852.
Soderstrom K, Stein E, Colmenero P, Purath U, Muller-Ladner U, Teixeira de Matos C, Tarner IH, Robinson WH, Engleman EG: Natural killer cells trigger osteoclastogenesis and bone destruction in arthritis. Proc Natl Acad Sci USA. 2010, 107: 13028-13033. 10.1073/pnas.1000546107.
Nakashima T, Kobayashi Y, Yamasaki S, Kawakami A, Eguchi K, Sasaki H, Sakai H: Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-kappaB ligand: modulation of the expression by osteotropic factors and cytokines. Biochem Biophys Res Commun. 2000, 275: 768-775. 10.1006/bbrc.2000.3379.
Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, Saito S, Inoue K, Kamatani N, Gillespie MT, Martin TJ, Suda T: IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999, 103: 1345-1352. 10.1172/JCI5703.
Wittrant Y, Gorin Y, Mohan S, Wagner B, Abboud-Werner SL: Colony-stimulating factor-1 (CSF-1) directly inhibits receptor activator of nuclear factor-{kappa}B ligand (RANKL) expression by osteoblasts. Endocrinology. 2009, 150: 4977-4988. 10.1210/en.2009-0248.
Nakashima T, Takayanagi H: Osteoimmunology: crosstalk between the immune and bone systems. J Clin Immunol. 2009, 29: 555-567. 10.1007/s10875-009-9316-6.
Pettit AR, Ji H, von Stechow D, Muller R, Goldring SR, Choi Y, Benoist C, Gravallese EM: TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am J Pathol. 2001, 159: 1689-1699. 10.1016/S0002-9440(10)63016-7.
Redlich K, Hayer S, Maier A, Dunstan CR, Tohidast-Akrad M, Lang S, Türk B, Pietschmann P, Woloszczuk W, Haralambous S, Kollias G, Steiner G, Smolen JS, Schett G: Tumor necrosis factor alpha-mediated joint destruction is inhibited by targeting osteoclasts with osteoprotegerin. Arthritis Rheum. 2002, 46: 785-792. 10.1002/art.10097.
Stolina M, Schett G, Dwyer D, Vonderfecht S, Middleton S, Duryea D, Pacheco E, Van G, Bolon B, Feige U, Zack D, Kostenuik P: RANKL inhibition by osteoprotegerin prevents bone loss without affecting local or systemic inflammation parameters in two rat arthritis models: comparison with anti-TNFalpha or anti-IL-1 therapies. Arthritis Res Ther. 2009, 11: R187-10.1186/ar2879.
Hofbauer LC, Zeitz U, Schoppet M, Skalicky M, Schuler C, Stolina M, Kostenuik PJ, Erben RG: Prevention of glucocorticoid-induced bone loss in mice by inhibition of RANKL. Arthritis Rheum. 2009, 60: 1427-1437. 10.1002/art.24445.
Cohen SB, Dore RK, Lane NE, Ory PA, Peterfy CG, Sharp JT, van der Heijde D, Zhou L, Tsuji W, Newmark R: Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis Rheum. 2008, 58: 1299-1309. 10.1002/art.23417.
MacNaul KL, Hutchinson NI, Parsons JN, Bayne EK, Tocci MJ: Analysis of IL-1 and TNF-alpha gene expression in human rheumatoid synoviocytes and normal monocytes by in situ hybridization. J Immunol. 1990, 145: 4154-4166.
Danning CL, Illei GG, Hitchon C, Greer MR, Boumpas DT, McInnes IB: Macrophage-derived cytokine and nuclear factor kappaB p65 expression in synovial membrane and skin of patients with psoriatic arthritis. Arthritis Rheum. 2000, 43: 1244-1256. 10.1002/1529-0131(200006)43:6<1244::AID-ANR7>3.0.CO;2-2.
Bradley JR: TNF-mediated inflammatory disease. J Pathol. 2008, 214: 149-160. 10.1002/path.2287.
Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A: Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts. J Biol Chem. 2000, 275: 4858-4864. 10.1074/jbc.275.7.4858.
Nanes MS: Tumor necrosis factor-alpha: molecular and cellular mechanisms in skeletal pathology. Gene. 2003, 321: 1-15.
Zhang YH, Heulsmann A, Tondravi MM, Mukherjee A, Abu-Amer Y: Tumor necrosis factor-alpha (TNF) stimulates RANKL-induced osteoclastogenesis via coupling of TNF type 1 receptor and RANK signaling pathways. J Biol Chem. 2001, 276: 563-568.
David JP, Schett G: TNF and bone. Curr Dir Autoimmun. 2010, 11: 135-144.
Lee SE, Chung WJ, Kwak HB, Chung CH, Kwack KB, Lee ZH, Kim HH: Tumor necrosis factor-alpha supports the survival of osteoclasts through the activation of Akt and ERK. J Biol Chem. 2001, 276: 49343-49349. 10.1074/jbc.M103642200.
Kitaura H, Zhou P, Kim HJ, Novack DV, Ross FP, Teitelbaum SL: M-CSF mediates TNF-induced inflammatory osteolysis. J Clin Invest. 2005, 115: 3418-3427. 10.1172/JCI26132.
Mao B, Wu W, Li Y, Hoppe D, Stannek P, Glinka A, Niehrs C: LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature. 2001, 411: 321-325. 10.1038/35077108.
Glass DA 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, Karsenty G: Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005, 8: 751-764. 10.1016/j.devcel.2005.02.017.
Diarra D, Stolina M, Polzer K, Zwerina J, Ominsky MS, Dwyer D, Korb A, Smolen J, Hoffmann M, Scheinecker C, van der Heide D, Landewe R, Lacey D, Richards WG, Schett G: Dickkopf-1 is a master regulator of joint remodeling. Nat Med. 2007, 13: 156-163. 10.1038/nm1538.
Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G: Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991, 10: 4025-4031.
Williams RO, Feldmann M, Maini RN: Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc Natl Acad Sci USA. 1992, 89: 9784-9788. 10.1073/pnas.89.20.9784.
Ji H, Pettit A, Ohmura K, Ortiz-Lopez A, Duchatelle V, Degott C, Gravallese E, Mathis D, Benoist C: Critical roles for interleukin 1 and tumor necrosis factor alpha in antibody-induced arthritis. J Exp Med. 2002, 196: 77-85. 10.1084/jem.20020439.
Furst DE: Development of TNF inhibitor therapies for the treatment of rheumatoid arthritis. Clin Exp Rheumatol. 2010, 28 (3 Suppl 59): S5-12.
Ritchlin C: Fibroblast biology. Effector signals released by the synovial fibroblast in arthritis. Arthritis Res. 2000, 2: 356-360. 10.1186/ar112.
Niki Y, Yamada H, Seki S, Kikuchi T, Takaishi H, Toyama Y, Fujikawa K, Tada N: Macrophage- and neutrophil-dominant arthritis in human IL-1 alpha transgenic mice. J Clin Invest. 2001, 107: 1127-1135. 10.1172/JCI11530.
Dinarello CA: The interleukin-1 family: 10 years of discovery. Faseb J. 1994, 8: 1314-1325.
Dunn E, Sims JE, Nicklin MJ, O'Neill LA: Annotating genes with potential roles in the immune system: six new members of the IL-1 family. Trends Immunol. 2001, 22: 533-536. 10.1016/S1471-4906(01)02034-8.
Seckinger P, Klein-Nulend J, Alander C, Thompson RC, Dayer JM, Raisz LG: Natural and recombinant human IL-1 receptor antagonists block the effects of IL-1 on bone resorption and prostaglandin production. J Immunol. 1990, 145: 4181-4184.
Trebec DP, Chandra D, Gramoun A, Li K, Heersche JN, Manolson MF: Increased expression of activating factors in large osteoclasts could explain their excessive activity in osteolytic diseases. J Cell Biochem. 2007, 101: 205-220. 10.1002/jcb.21171.
Jimi E, Nakamura I, Duong LT, Ikebe T, Takahashi N, Rodan GA, Suda T: Interleukin 1 induces multinucleation and bone-resorbing activity of osteoclasts in the absence of osteoblasts/stromal cells. Exp Cell Res. 1999, 247: 84-93. 10.1006/excr.1998.4320.
Jimi E, Shuto T, Koga T: Macrophage colony-stimulating factor and interleukin-1 alpha maintain the survival of osteoclast-like cells. Endocrinology. 1995, 136: 808-811. 10.1210/en.136.2.808.
Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, Nakagawa N, Kinosaki M, Yamaguchi K, Shima N, Yasuda H, Morinaga T, Higashio K, Martin TJ, Suda T: Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med. 2000, 191: 275-286. 10.1084/jem.191.2.275.
Niki Y, Yamada H, Kikuchi T, Toyama Y, Matsumoto H, Fujikawa K, Tada N: Membrane-associated IL-1 contributes to chronic synovitis and cartilage destruction in human IL-1 alpha transgenic mice. J Immunol. 2004, 172: 577-584.
Ghivizzani SC, Kang R, Georgescu HI, Lechman ER, Jaffurs D, Engle JM, Watkins SC, Tindal MH, Suchanek MK, McKenzie LR, Evans CH, Robbins PD: Constitutive intra-articular expression of human IL-1 beta following gene transfer to rabbit synovium produces all major pathologies of human rheumatoid arthritis. J Immunol. 1997, 159: 3604-3612.
Horai R, Saijo S, Tanioka H, Nakae S, Sudo K, Okahara A, Ikuse T, Asano M, Iwakura Y: Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med. 2000, 191: 313-320. 10.1084/jem.191.2.313.
Zwerina J, Redlich K, Polzer K, Joosten L, Krönke G, Distler J, Hess A, Pundt N, Pap T, Hoffmann O, Gasser J, Scheinecker C, Smolen JS, van den Berg W, Schett G: TNF-induced structural joint damage is mediated by IL-1. Proc Natl Acad Sci USA. 2007, 104: 11742-11747. 10.1073/pnas.0610812104.
Polzer K, Joosten L, Gasser J, Distler JH, Ruiz G, Baum W, Redlich K, Bobacz K, Smolen JS, van den Berg W, Schett G, Zwerina J: Interleukin-1 is essential for systemic inflammatory bone loss. Ann Rheum Dis. 69: 284-290.
Wei S, Kitaura H, Zhou P, Ross FP, Teitelbaum SL: IL-1 mediates TNF-induced osteoclastogenesis. J Clin Invest. 2005, 115: 282-290.
Smolen JS, Landewé R, Breedveld FC, Dougados M, Emery P, Gaujoux-Viala C, Gorter S, Knevel R, Nam J, Schoels M, Aletaha D, Buch M, Gossec L, Huizinga T, Bijlsma JW, Burmester G, Combe B, Cutolo M, Gabay C, Gomez-Reino J, Kouloumas M, Kvien TK, Martin-Mola E, McInnes I, Pavelka K, van Riel P, Scholte M, Scott DL, Sokka T, Valesini G, et al: EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs. Ann Rheum Dis. 69: 964-975.
Okamoto H, Yamamura M, Morita Y, Harada S, Makino H, Ota Z: The synovial expression and serum levels of interleukin-6, interleukin-11, leukemia inhibitory factor, and oncostatin M in rheumatoid arthritis. Arthritis Rheum. 1997, 40: 1096-1105. 10.1002/art.1780400614.
Field M, Chu C, Feldmann M, Maini RN: Interleukin-6 localisation in the synovial membrane in rheumatoid arthritis. Rheumatol Int. 1991, 11: 45-50. 10.1007/BF00291144.
Rose-John S, Waetzig GH, Scheller J, Grotzinger J, Seegert D: The IL-6/sIL-6R complex as a novel target for therapeutic approaches. Expert Opin Ther Targets. 2007, 11: 613-624. 10.1517/14728222.11.5.613.
Taga T, Hibi M, Hirata Y, Yamasaki K, Yasukawa K, Matsuda T, Hirano T, Kishimoto T: Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell. 1989, 58: 573-581. 10.1016/0092-8674(89)90438-8.
Desgeorges A, Gabay C, Silacci P, Novick D, Roux-Lombard P, Grau G, Dayer JM, Vischer T, Guerne PA: Concentrations and origins of soluble interleukin 6 receptor-alpha in serum and synovial fluid. J Rheumatol. 1997, 24: 1510-1516.
Houssiau FA, Devogelaer JP, Van Damme J, de Deuxchaisnes CN, Van Snick J: Interleukin-6 in synovial fluid and serum of patients with rheumatoid arthritis and other inflammatory arthritides. Arthritis Rheum. 1988, 31: 784-788. 10.1002/art.1780310614.
Poli V, Balena R, Fattori E, Markatos A, Yamamoto M, Tanaka H, Ciliberto G, Rodan GA, Costantini F: Interleukin-6 deficient mice are protected from bone loss caused by estrogen depletion. EMBO J. 1994, 13: 1189-1196.
Yang X, Ricciardi BF, Hernandez-Soria A, Shi Y, Pleshko Camacho N, Bostrom MP: Callus mineralization and maturation are delayed during fracture healing in interleukin-6 knockout mice. Bone. 2007, 41: 928-936. 10.1016/j.bone.2007.07.022.
De Benedetti F, Rucci N, Del Fattore A, Peruzzi B, Paro R, Longo M, Vivarelli M, Muratori F, Berni S, Ballanti P, Ferrari S, Teti A: Impaired skeletal development in interleukin-6-transgenic mice: a model for the impact of chronic inflammation on the growing skeletal system. Arthritis Rheum. 2006, 54: 3551-3563. 10.1002/art.22175.
Palmqvist P, Persson E, Conaway HH, Lerner UH: IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF-kappa B ligand, osteoprotegerin, and receptor activator of NF-kappa B in mouse calvariae. J Immunol. 2002, 169: 3353-3362.
Wong PK, Quinn JM, Sims NA, van Nieuwenhuijze A, Campbell IK, Wicks IP: Interleukin-6 modulates production of T lymphocyte-derived cytokines in antigen-induced arthritis and drives inflammation-induced osteoclastogenesis. Arthritis Rheum. 2006, 54: 158-168. 10.1002/art.21537.
Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK: Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006, 441: 235-238. 10.1038/nature04753.
Axmann R, Bohm C, Kronke G, Zwerina J, Smolen J, Schett G: Inhibition of interleukin-6 receptor directly blocks osteoclast formation in vitro and in vivo. Arthritis Rheum. 2009, 60: 2747-2756. 10.1002/art.24781.
Yoshitake F, Itoh S, Narita H, Ishihara K, Ebisu S: Interleukin-6 directly inhibits osteoclast differentiation by suppressing receptor activator of NF-kappaB signaling pathways. J Biol Chem. 2008, 283: 11535-11540. 10.1074/jbc.M607999200.
Duplomb L, Baud'huin M, Charrier C, Berreur M, Trichet V, Blanchard F, Heymann D: Interleukin-6 inhibits receptor activator of nuclear factor kappaB ligand-induced osteoclastogenesis by diverting cells into the macrophage lineage: key role of Serine727 phosphorylation of signal transducer and activator of transcription 3. Endocrinology. 2008, 149: 3688-3697. 10.1210/en.2007-1719.
Le Goff B, Blanchard F, Berthelot JM, Heymann D, Maugars Y: Role for interleukin-6 in structural joint damage and systemic bone loss in rheumatoid arthritis. Joint Bone Spine. 2010, 77: 201-205. 10.1016/j.jbspin.2010.03.002.
Ohshima S, Saeki Y, Mima T, Sasai M, Nishioka K, Nomura S, Kopf M, Katada Y, Tanaka T, Suemura M, Kishimoto T: Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc Natl Acad Sci USA. 1998, 95: 8222-8226. 10.1073/pnas.95.14.8222.
Alonzi T, Fattori E, Lazzaro D, Costa P, Probert L, Kollias G, De Benedetti F, Poli V, Ciliberto G: Interleukin 6 is required for the development of collagen-induced arthritis. J Exp Med. 1998, 187: 461-468. 10.1084/jem.187.4.461.
Sasai M, Saeki Y, Ohshima S, Nishioka K, Mima T, Tanaka T, Katada Y, Yoshizaki K, Suemura M, Kishimoto T: Delayed onset and reduced severity of collagen-induced arthritis in interleukin-6-defi cient mice. Arthritis Rheum. 1999, 42: 1635-1643. 10.1002/1529-0131(199908)42:8<1635::AID-ANR11>3.0.CO;2-Q.
Suematsu S, Matsuda T, Aozasa K, Akira S, Nakano N, Ohno S, Miyazaki J, Yamamura K, Hirano T, Kishimoto T: IgG1 plasmacytosis in interleukin 6 transgenic mice. Proc Natl Acad Sci USA. 1989, 86: 7547-7551. 10.1073/pnas.86.19.7547.
Atsumi T, Ishihara K, Kamimura D, Ikushima H, Ohtani T, Hirota S, Kobayashi H, Park SJ, Saeki Y, Kitamura Y, Hirano T: A point mutation of Tyr-759 in interleukin 6 family cytokine receptor subunit gp130 causes autoimmune arthritis. J Exp Med. 2002, 196: 979-990. 10.1084/jem.20020619.
Takagi N, Mihara M, Moriya Y, Nishimoto N, Yoshizaki K, Kishimoto T, Takeda Y, Ohsugi Y: Blockage of interleukin-6 receptor ameliorates joint disease in murine collagen-induced arthritis. Arthritis Rheum. 1998, 41: 2117-2121. 10.1002/1529-0131(199812)41:12<2117::AID-ART6>3.0.CO;2-P.
Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, Miossec P: Human interleukin-17: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999, 42: 963-970. 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E.
Ziolkowska M, Koc A, Luszczykiewicz G, Ksiezopolska-Pietrzak K, Klimczak E, Chwalinska-Sadowska H, Maslinski W: High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol. 2000, 164: 2832-2838.
Hueber AJ, Asquith DL, Miller AM, Reilly J, Kerr S, Leipe J, Melendez AJ, McInnes IB: Mast cells express IL-17A in rheumatoid arthritis synovium. J Immunol. 2010, 184: 3336-3340. 10.4049/jimmunol.0903566.
Lubberts E, Joosten LA, van de Loo FA, Schwarzenberger P, Kolls J, van den Berg WB: Overexpression of IL-17 in the knee joint of collagen type II immunized mice promotes collagen arthritis and aggravates joint destruction. Inflamm Res. 2002, 51: 102-104. 10.1007/BF02684010.
Lubberts E, Koenders MI, van den Berg WB: The role of T-cell interleukin-17 in conducting destructive arthritis: lessons from animal models. Arthritis Res Ther. 2005, 7: 29-37. 10.1186/ar1478.
Lubberts E, Koenders MI, Oppers-Walgreen B, van den Bersselaar L, Coenende Roo CJ, Joosten LA, van den Berg WB: Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004, 50: 650-659. 10.1002/art.20001.
Koenders MI, Lubberts E, Oppers-Walgreen B, van den Bersselaar L, Helsen MM, Di Padova FE, Boots AM, Gram H, Joosten LA, van den Berg WB: Blocking of interleukin-17 during reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing RANKL and interleukin-1. Am J Pathol. 2005, 167: 141-149. 10.1016/S0002-9440(10)62961-6.
Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, Pin JJ, Garrone P, Garcia E, Saeland S, Blanchard D, Gaillard C, Das Mahapatra B, Rouvier E, Golstein P, Banchereau J, Lebecque S: T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. 1996, 183: 2593-2603. 10.1084/jem.183.6.2593.
Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, Zhang M, Mineau F, Pelletier JP: IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol. 1998, 160: 3513-3521.
Hwang SY, Kim JY, Kim KW, Park MK, Moon Y, Kim WU, Kim H Y: IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-kappaB- and PI3-kinase/Akt-dependent pathways. Arthritis Res Ther. 2004, 6: R120-128. 10.1186/ar1038.
Adamopoulos IE, Chao CC, Geissler R, Laface D, Blumenschein W, Iwakura Y, McClanahan T, Bowman EP: Interleukin-17A upregulates receptor activator of NF-kappaB on osteoclast precursors. Arthritis Res Ther. 2010, 12: R29-
Yago T, Nanke Y, Ichikawa N, Kobashigawa T, Mogi M, Kamatani N, Kotake S: IL-17 induces osteoclastogenesis from human monocytes alone in the absence of osteoblasts, which is potently inhibited by anti-TNF-alpha antibody: a novel mechanism of osteoclastogenesis by IL-17. J Cell Biochem. 2009, 108: 947-955. 10.1002/jcb.22326.
Teitelbaum SL, Ross FP: Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003, 4: 638-649. 10.1038/nrg1122.
Tanaka S, Takahashi N, Udagawa N, Tamura T, Akatsu T, Stanley ER, Kurokawa T, Suda T: Macrophage colony-stimulating factor is indispensable for both proliferation and differentiation of osteoclast progenitors. J Clin Invest. 1993, 91: 257-263. 10.1172/JCI116179.
Inoue H, Takamori M, Shimoyama Y, Ishibashi H, Yamamoto S, Koshihara Y: Regulation by PGE2 of the production of interleukin-6, macrophage colony stimulating factor, and vascular endothelial growth factor in human synovial fibroblasts. Br J Pharmacol. 2002, 136: 287-295. 10.1038/sj.bjp.0704705.
Atkins GJ, Haynes DR, Geary SM, Loric M, Crotti TN, Findlay DM: Coordinated cytokine expression by stromal and hematopoietic cells during human osteoclast formation. Bone. 2000, 26: 653-661. 10.1016/S8756-3282(00)00280-5.
Weitzmann MN, Cenci S, Rifas L, Brown C, Pacifici R: Interleukin-7 stimulates osteoclast formation by up-regulating the T-cell production of soluble osteoclastogenic cytokines. Blood. 2000, 96: 1873-1878.
Wiktor-Jedrzejczak W, Bartocci A, Ferrante AW, Ahmed-Ansari A, Sell KW, Pollard JW, Stanley ER: Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc Natl Acad Sci USA. 1990, 87: 4828-4832. 10.1073/pnas.87.12.4828.
Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, Sylvestre V, Stanley ER: Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood. 2002, 99: 111-120. 10.1182/blood.V99.1.111.
Hodge JM, Kirkland MA, Nicholson GC: Multiple roles of M-CSF in human osteoclastogenesis. J Cell Biochem. 2007, 102: 759-768. 10.1002/jcb.21331.
Jack GD, Zhang L, Friedman AD: M-CSF elevates c-Fos and phospho-C/EBPalpha(S21) via ERK whereas G-CSF stimulates SHP2 phosphorylation in marrow progenitors to contribute to myeloid lineage specification. Blood. 2009, 114: 2172-2180. 10.1182/blood-2008-11-191536.
Kelley TW, Graham MM, Doseff AI, Pomerantz RW, Lau SM, Ostrowski MC, Franke TF, Marsh CB: Macrophage colony-stimulating factor promotes cell survival through Akt/protein kinase B. J Biol Chem. 1999, 274: 26393-26398. 10.1074/jbc.274.37.26393.
Gingery A, Bradley E, Shaw A, Oursler MJ: Phosphatidylinositol 3-kinase coordinately activates the MEK/ERK and AKT/NFkappaB pathways to maintain osteoclast survival. J Cell Biochem. 2003, 89: 165-179. 10.1002/jcb.10503.
Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, Wagner EF, Mak TW, Kodama T, Taniguchi T: Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002, 3: 889-901. 10.1016/S1534-5807(02)00369-6.
Kanazawa K, Azuma Y, Nakano H, Kudo A: TRAF5 functions in both RANKL-and TNFalpha-induced osteoclastogenesis. J Bone Miner Res. 2003, 18: 443-450. 10.1359/jbmr.2003.18.3.443.
Takayanagi H: Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007, 7: 292-304. 10.1038/nri2062.
Böhm C, Hayer S, Kilian A, Zaiss MM, Finger S, Hess A, Engelke K, Kollias G, Krönke G, Zwerina J, Schett G, David JP: The alpha-isoform of p38 MAPK specifically regulates arthritic bone loss. J Immunol. 2009, 183: 5938-5947. 10.4049/jimmunol.0901026.
Asagiri M, Takayanagi H: The molecular understanding of osteoclast differentiation. Bone. 2007, 40: 251-264. 10.1016/j.bone.2006.09.023.
Iotsova V, Caamano J, Loy J, Yang Y, Lewin A, Bravo R: Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat Med. 1997, 3: 1285-1289. 10.1038/nm1197-1285.
Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD, Leonardi A, Tran T, Boyce BF, Siebenlist U: Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. 1997, 11: 3482-3496. 10.1101/gad.11.24.3482.
Ruocco MG, Maeda S, Park JM, Lawrence T, Hsu LC, Cao Y, Schett G, Wagner EF, Karin M: I{kappa}B kinase (IKK){beta}, but not IKK{alpha}, is a critical mediator of osteoclast survival and is required for inflammation-induced bone loss. J Exp Med. 2005, 201: 1677-1687. 10.1084/jem.20042081.
Novack DV, Yin L, Hagen-Stapleton A, Schreiber RD, Goeddel DV, Ross FP, Teitelbaum SL: The IkappaB function of NF-kappaB2 p100 controls stimulated osteoclastogenesis. J Exp Med. 2003, 198: 771-781. 10.1084/jem.20030116.
Yamashita T, Yao Z, Li F, Zhang Q, Badell IR, Schwarz EM, Takeshita S, Wagner EF, Noda M, Matsuo K, Xing L, Boyce BF: NF-kappaB p50 and p52 regulate receptor activator of NF-kappaB ligand (RANKL) and tumor necrosis factor-induced osteoclast precursor differentiation by activating c-Fos and NFATc1. J Biol Chem. 2007, 282: 18245-18253. 10.1074/jbc.M610701200.
Sato K, Suematsu A, Nakashima T, Takemoto-Kimura S, Aoki K, Morishita Y, Asahara H, Ohya K, Yamaguchi A, Takai T, Kodama T, Chatila TA, Bito H, Takayanagi H: Regulation of osteoclast differentiation and function by the CaMK-CREB pathway. Nat Med. 2006, 12: 1410-1416.
Johnson RS, Spiegelman BM, Papaioannou V: Pleiotropic effects of a null mutation in the c-fos proto-oncogene. Cell. 1992, 71: 577-586. 10.1016/0092-8674(92)90592-Z.
Wang ZQ, Ovitt C, Grigoriadis AE, Mohle-Steinlein U, Ruther U, Wagner EF: Bone and haematopoietic defects in mice lacking c-fos. Nature. 1992, 360: 741-745. 10.1038/360741a0.
David JP, Sabapathy K, Hoffmann O, Idarraga MH, Wagner EF: JNK1 modulates osteoclastogenesis through both c-Jun phosphorylation-dependent and-independent mechanisms. J Cell Sci. 2002, 115: 4317-4325. 10.1242/jcs.00082.
Kenner L, Hoebertz A, Beil T, Keon N, Karreth F, Eferl R, Scheuch H, Szremska A, Amling M, Schorpp-Kistner M, Angel P, Wagner EF: Mice lacking JunB are osteopenic due to cell-autonomous osteoblast and osteoclast defects. J Cell Biol. 2004, 164: 613-623. 10.1083/jcb.200308155.
Ikeda F, Nishimura R, Matsubara T, Tanaka S, Inoue J, Reddy SV, Hata K, Yamashita K, Hiraga T, Watanabe T, Kukita T, Yoshioka K, Rao A, Yoneda T: Critical roles of c-Jun signaling in regulation of NFAT family and RANKL-regulated osteoclast differentiation. J Clin Invest. 2004, 114: 475-484.
Asahara H, Fujisawa K, Kobata T, Hasunuma T, Maeda T, Asanuma M, Ogawa N, Inoue H, Sumida T, Nishioka K: Direct evidence of high DNA binding activity of transcription factor AP-1 in rheumatoid arthritis synovium. Arthritis Rheum. 1997, 40: 912-918. 10.1002/art.1780400520.
Sitara D, Aliprantis AO: Transcriptional regulation of bone and joint remodeling by NFAT. Immunol Rev. 2010, 233: 286-300. 10.1111/j.0105-2896.2009.00849.x.
Mao D, Epple H, Uthgenannt B, Novack DV, Faccio R: PLCgamma2 regulates osteoclastogenesis via its interaction with ITAM proteins and GAB2. J Clin Invest. 2006, 116: 2869-2879. 10.1172/JCI28775.
Koga T, Inui M, Inoue K, Kim S, Suematsu A, Kobayashi E, Iwata T, Ohnishi H, Matozaki T, Kodama T, Taniguchi T, Takayanagi H, Takai T: Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature. 2004, 428: 758-763. 10.1038/nature02444.
Mocsai A, Humphrey MB, Van Ziffle JA, Hu Y, Burghardt A, Spusta SC, Majumdar S, Lanier LL, Lowell CA, Nakamura MC: The immunomodulatory adapter proteins DAP12 and Fc receptor gamma-chain (FcRgamma) regulate development of functional osteoclasts through the Syk tyrosine kinase. Proc Natl Acad Sci USA. 2004, 101: 6158-6163. 10.1073/pnas.0401602101.
Shinohara M, Koga T, Okamoto K, Sakaguchi S, Arai K, Yasuda H, Takai T, Kodama T, Morio T, Geha RS, Kitamura D, Kurosaki T, Ellmeier W, Takayanagi H: Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell. 2008, 132: 794-806. 10.1016/j.cell.2007.12.037.
Kanazawa K, Kudo A: TRAF2 is essential for TNF-alpha-induced osteoclastogenesis. J Bone Miner Res. 2005, 20: 840-847.
Kaji K, Katogi R, Azuma Y, Naito A, Inoue JI, Kudo A: Tumor necrosis factor alpha-induced osteoclastogenesis requires tumor necrosis factor receptor-associated factor 6. J Bone Miner Res. 2001, 16: 1593-1599. 10.1359/jbmr.2001.16.9.1593.
Kadono Y, Okada F, Perchonock C, Jang HD, Lee SY, Kim N, Choi Y: Strength of TRAF6 signalling determines osteoclastogenesis. EMBO Rep. 2005, 6: 171-176. 10.1038/sj.embor.7400345.
Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, Morony S, Capparelli C, Van G, Kaufman S, van der Heiden A, Itie A, Wakeham A, Khoo W, Sasaki T, Cao Z, Penninger JM, Paige CJ, Lacey DL, Dunstan CR, Boyle WJ, Goeddel DV, Mak TW: TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999, 13: 1015-1024. 10.1101/gad.13.8.1015.
Weber A, Wasiliew P, Kracht M: Interleukin-1 (IL-1) pathway. Sci Signal. 2010, 3: cm1-10.1126/scisignal.3105cm1.
Kim K, Lee J, Kim JH, Jin HM, Zhou B, Lee SY, Kim N: Protein inhibitor of activated STAT 3 modulates osteoclastogenesis by down-regulation of NFATc1 and osteoclast-associated receptor. J Immunol. 2007, 178: 5588-5594.
Sims NA, Jenkins BJ, Quinn JM, Nakamura A, Glatt M, Gillespie MT, Ernst M, Martin TJ: Glycoprotein 130 regulates bone turnover and bone size by distinct downstream signaling pathways. J Clin Invest. 2004, 113: 379-389.
Lin H, Lee E, Hestir K, Leo C, Huang M, Bosch E, Halenbeck R, Wu G, Zhou A, Behrens D, Hollenbaugh D, Linnemann T, Qin M, Wong J, Chu K, Doberstein SK, Williams LT: Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science. 2008, 320: 807-811. 10.1126/science.1154370.
Baud'huin M, Renault R, Charrier C, Riet A, Moreau A, Brion R, Gouin F, Duplomb L, Heymann D: Interleukin-34 is expressed by giant cell tumours of bone and plays a key role in RANKL-induced osteoclastogenesis. J Pathol. 2010, 221: 77-86. 10.1002/path.2684.
Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, Bouche G, Girard JP: IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci USA. 2007, 104: 282-287. 10.1073/pnas.0606854104.
Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, Gorman DM, Bazan JF, Kastelein RA: IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005, 23: 479-490. 10.1016/j.immuni.2005.09.015.
Kakkar R, Lee RT: The IL-33/ST2 pathway: therapeutic target and novel biomarker. Nat Rev Drug Discov. 2008, 7: 827-840. 10.1038/nrd2660.
Xu D, Jiang HR, Kewin P, Li Y, Mu R, Fraser AR, Pitman N, Kurowska-Stolarska M, McKenzie AN, McInnes IB, Liew FY: IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc Natl Acad Sci USA. 2008, 105: 10913-10918. 10.1073/pnas.0801898105.
Palmer G, Talabot-Ayer D, Lamacchia C, Toy D, Seemayer CA, Viatte S, Finckh A, Smith DE, Gabay C: Inhibition of interleukin-33 signaling attenuates the severity of experimental arthritis. Arthritis Rheum. 2009, 60: 738-749. 10.1002/art.24305.
Mun SH, Ko NY, Kim HS, Kim JW, Kim do K, Kim AR, Lee SH, Kim YG, Lee CK, Lee SH, Kim BK, Beaven MA, Kim YM, Choi WS: Interleukin-33 stimulates formation of functional osteoclasts from human CD14(+) monocytes. Cell Mol Life Sci. 2010, 67: 3883-3892. 10.1007/s00018-010-0410-y.
Schulze J, Bickert T, Beil FT, Zaiss MM, Albers J, Wintges K, Streichert T, Klaetschke K, Keller J, Hissnauer TN, Spiro AS, Gessner A, Schett G, Amling M, McKenzie AN, Horst AK, Schinke T: Interleukin-33 is expressed in differentiated osteoblasts and blocks osteoclast formation from bone marrow precursor cells. J Bone Miner Res. 2011, 26: 704-17. 10.1002/jbmr.269.
Rifas L, Avioli LV: A novel T cell cytokine stimulates interleukin-6 in human osteoblastic cells. J Bone Miner Res. 1999, 14: 1096-1103. 10.1359/jbmr.1999.14.7.1096.
Weitzmann MN, Cenci S, Rifas L, Haug J, Dipersio J, Pacifici R: T cell activation induces human osteoclast formation via receptor activator of nuclear factor kappaB ligand-dependent and -independent mechanisms. J Bone Miner Res. 2001, 16: 328-337. 10.1359/jbmr.2001.16.2.328.
Rifas L, Weitzmann MN: A novel T cell cytokine, secreted osteoclastogenic factor of activated T cells, induces osteoclast formation in a RANKL-independent manner. Arthritis Rheum. 2009, 60: 3324-3335. 10.1002/art.24877.
Ogata Y, Kukita A, Kukita T, Komine M, Miyahara A, Miyazaki S, Kohashi O: A novel role of IL-15 in the development of osteoclasts: inability to replace its activity with IL-2. J Immunol. 1999, 162: 2754-2760.
Ferrari-Lacraz S, Zanelli E, Neuberg M, Donskoy E, Kim YS, Zheng XX, Hancock WW, Maslinski W, Li XC, Strom TB, Moll T: Targeting IL-15 receptor-bearing cells with an antagonist mutant IL-15/Fc protein prevents disease development and progression in murine collagen-induced arthritis. J Immunol. 2004, 173: 5818-5826.
McInnes IB, Liew FY: Interleukin 15: a proinflammatory role in rheumatoid arthritis synovitis. Immunol Today. 1998, 19: 75-79. 10.1016/S0167-5699(97)01205-X.
Djaafar S, Pierroz DD, Chicheportiche R, Zheng XX, Ferrari SL, Ferrari-Lacraz S: Inhibition of T cell-dependent and rankl-dependent osteoclastogenic processes associated with high bone mass in IL-15 receptor-deficient mice. Arthritis Rheum. 2010, 62: 3300-3310. 10.1002/art.27645.
Deodhar A, Dore RK, Mandel D, Schechtman J, Shergy W, Trapp R, Ory PA, Peterfy CG, Fuerst T, Wang H, Zhou L, Tsuji W, Newmark R: Denosumab-mediated increase in hand bone mineral density associated with decreased progression of bone erosion in rheumatoid arthritis patients. Arthritis Care Res (Hoboken). 2010, 62: 569-574. 10.1002/acr.20004.
Güler-Yüksel M, Allaart CF, Watt I, Goekoop-Ruiterman YP, de Vries-Bouwstra JK, van Schaardenburg D, van Krugten MV, Dijkmans BA, Huizinga TW, Lems WF, Kloppenburg M: Treatment with TNF-alpha inhibitor infliximab might reduce hand osteoarthritis in patients with rheumatoid arthritis. Osteoarthritis Cartilage. 2010, 18: 1256-1262. 10.1016/j.joca.2010.07.011.
Kitamura T, Murase T, Hashimoto J, Tomita T, Arimitsu S, Yoshikawa H, Sugamoto K: Radiographic study on the pattern of wrist joint destruction in rheumatoid arthritis. Clin Rheumatol. 2011, 30: 353-359. 10.1007/s10067-010-1575-3.
Acknowledgements
The present work was supported by grants from the German Research Society (DFG).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
JZ has received speaker's honoraria and travel grants from Amgen, Abbot, Wyeth, UCB and Roche and a research grant from Wyeth.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Braun, T., Zwerina, J. Positive regulators of osteoclastogenesis and bone resorption in rheumatoid arthritis. Arthritis Res Ther 13, 235 (2011). https://doi.org/10.1186/ar3380
Published:
DOI: https://doi.org/10.1186/ar3380