
Abstract
Studies on the immune regulation of osteoclasts in rheumatoid arthritis have promoted the new research field of 'osteoimmunology', which investigates the interplay between the skeletal and immune systems at the molecular level. Accumulating evidence lends support to the theory that bone destruction associated with rheumatoid arthritis is caused by the enhanced activity of osteoclasts, resulting from the activation of a unique helper T cell subset, 'Th17 cells'. Understanding the interaction between osteoclasts and the adaptive immune system in rheumatoid arthritis and the molecular mechanisms of Th17 development will lead to the development of potentially effective therapeutic strategies.
Similar content being viewed by others


Introduction
The bony skeleton enables locomotive activity, the storage of calcium, and the harboring of hematopoietic stem cells (HSCs) . This multifunctional organ is characterized by calcified hard tissue composed of type I collagen and highly organized deposits of calcium phosphate [1]. Although bone superficially seems to be metabolically inert, it is, in fact, restructured at such a high speed that approximately 10% of the total bone content is replaced each year in adult vertebrates. This process, called bone remodeling, is dependent on the dynamic balance of bone formation and resorption, which are mediated by osteoblasts and osteoclasts, respectively. A delicate regulation of this process is requisite for normal bone homeostasis, and an imbalance is often related to metabolic bone diseases in humans [2].
Accumulating evidence has indicated that the immune and skeletal systems share a number of regulatory molecules, including cytokines, receptors, signaling molecules, and transcription factors. Furthermore, immune cells are formed and HSCs are maintained in the bone marrow, where they interact with bone cells. Therefore, the evidence that the physiology and pathology of one system might affect those of the other is compelling and the term osteoimmunology was coined to cover these overlapping scientific fields. The most typical example of the interaction between the skeletal and immune systems is seen in the abnormal or prolonged activation of the immune system (or both) in autoimmune diseases such as rheumatoid arthritis (RA), which is characterized by progressive multiple joint destruction. Since autoreactive T lymphocytes are considered to play a key role in the pathogenesis of RA, attention must be paid to the relationship between osteoclast-mediated bone destruction and aberrant adoptive immune responses in order to develop effective therapeutic strategies against RA. Here, we summarize recent progress in the understanding of the relationship between bone and the adaptive immune system in arthritis by focusing mainly on osteoclasts and osteoclastogenic helper T cells, Th17 cells.
Osteoclasts and bone destruction
The role of RANK/RANKL in osteoclastogenesis
Osteoclasts are large, multinucleated cells formed by the fusion of precursor cells of monocyte/macrophage lineage [2]. Mature osteoclasts degrade bone matrix proteins by secreting proteolytic enzymes, such as cathepsin K and matrix metalloproteinase, and decalcify the inorganic components of bone by releasing hydrochloric acid. In the late 1980s, an in vitro osteoclast formation system that uses a system of culturing bone marrow-derived cells of monocyte/macrophage lineage together with osteoclastogenesis-supporting cells such as osteoblasts was established [3, 4]. These supporting mesenchymal cells provide certain factors that are necessary for osteoclast differentiation [5]. Analysis of op/op mice with osteopetrosis revealed one of these essential factors to be macrophage colony-stimulating factor (M-CSF) [6]. M-CSF stimulation alone, however, does not induce the differentiation of osteoclasts. Forced expression of anti-apoptotic molecule Bcl-2 partially rescues the osteopetrotic phenotype of the op/op mice [7], suggesting that M-CSF is a survival factor for osteoclast precursor cells. Ultimately, in 1998, Yasuda and colleagues [8] and Lacey and colleagues [9] did clone the long-sought ligand mediating the essential signal for osteoclast differentiation; this ligand was called ODF and osteoprotegerin ligand, respectively. Interestingly, this cytokine, which belongs to the tumor necrosis factor (TNF) family, was shown to be identical to receptor activator of nuclear factor-κB ligand (RANKL) and TNF-related activation-induced cytokine (TRANCE), both of which had been cloned in the immune system [10, 11]. The cloning of ODF (RANKL, hereafter) enabled investigation of the differentiation process in a sophisticated culture system employing recombinant RANKL and M-CSF [12].
The receptor for RANKL is RANK, a type I trans-membrane protein that possesses a high homology with CD40. RANK is expressed on osteoclast precursor cells and mature osteoclasts, and the binding of RANKL to RANK is inhibited by the decoy receptor osteoprotegerin (OPG) [13, 14]. In bone, RANKL is expressed by osteoclastogenesis-supporting cells, including osteoblasts, in response to osteoclastogenic factors, such as 1,25-dihydroxyvitamin D3, prostaglandin E2, and parathyroid hormone, and is a crucial determinant of the level of bone resorption in vivo [5, 12]. Mice with a disruption of either Rank or Rankl exhibit severe osteopetrosis accompanied by a tooth eruption defect resulting from a complete lack of osteoclasts [15–17]. In contrast, mice lacking Opg exhibit a severe form of osteoporosis caused by both an increased number and enhanced activity of osteoclasts [18, 19]. These genetic findings clearly demonstrate that RANK/RANKL signaling is essential for osteoclastogenesis in vivo. Furthermore, mutations in RANK, RANKL, and OPG have been identified in human patients with bone disorders such as familial expansile osteolysis, autosomal recessive osteopetrosis, and juvenile Paget's disease of bone, respectively [20–23].
RANKL signaling
The ligation of RANK with RANKL results in trimerization of RANK and recruitment of adaptor molecules such as the TNF receptor-associated factor (TRAF) family of proteins, among which TRAF6 has been shown to be the major adaptor molecule [24, 25]. TRAF6 trimerizes upon RANK stimulation and activates nuclear factor-κB (NF-κB) and mitogen-activated protein kinases, including Jun N-terminal kinase (JNK) and p38. RANK also activates the transcription-factor complex, activator protein 1 (AP-1), through the induction of its component c-Fos [26]. The induction mechanism of c-Fos is dependent on the activation of Ca2+/calmodulin-dependent protein kinase IV (CaMKIV) and cyclic adenosine monophosphate responsive-element-binding protein (CREB) [27] as well as the activation of NF-κB [28]. Importantly, RANKL specifically and potently induces nuclear factor of activated T cells cytoplasmic 1 (NFATc1), the master regulator of osteoclast differentiation, and this induction is dependent on both the TRAF6 and c-Fos pathways [29]. The activation of NFAT is mediated by a specific phosphatase, calcineurin, which is activated by calcium-calmodulin signaling. The NFATc1 promoter contains NFAT-binding sites, and NFATc1 specifically autoregulates its own promoter during osteoclastogenesis, thus enabling the robust induction of NFATc1 [30]. The essential role of NFATc1 has been conclusively demonstrated by genetic experiments [30–32]. NFATc1 regulates a number of osteoclast-specific genes, such as cathepsin K, tartrate-resistant acid phosphatase (TRAP), calcitonin receptor, osteoclast-associated receptor (OSCAR), and β3 integrin, in cooperation with other transcription factors such as AP-1, PU.1, microphthalmia-associated transcription factor (MITF), and CREB (Figure 1).

Signaling cascades during osteoclastogenesis. Receptor activator of nuclear factor-κB ligand (RANKL)-RANK binding results in the recruitment of tumor necrosis factor receptor-associated factor 6 (TRAF 6), which activates nuclear factor-κB (NF-κB) and mitogen-activated protein kinases. RANKL also stimulates the induction of c-Fos through NF-κB and Ca2+/calmodulin-dependent protein kinase IV (CaMKIV). NF-κB and c-Fos are important for the robust induction of nuclear factor of activated T cells cytoplasmic 1 (NFATc1). Several costimulatory receptors associate with the immunoreceptor tyrosine-based activation motif (ITAM)-harboring adaptors, Fc receptor common γ subunit (FcRγ), and DNAX-activating protein 12 (DAP12): osteoclast-associated receptor (OSCAR) and triggering receptor expressed in myeloid cells 2 (TREM2) associate with FcRγ, and signal-regulatory protein β1 (SIRPβ1) and paired immunoglobulin-like receptor-A (PIR-A) associate with DAP12. RANK signaling and ITAM signaling cooperate to phosphorylate phospholipase Cγ (PLCγ) and activate calcium signaling, the latter of which is critical for the activation and autoamplification of NFATc1. Tec family tyrosine kinases (Tec and Btk) activated by RANK are important for the formation of the osteoclastogenic signaling complex composed of Tec kinases, B-cell linker (BLNK)/SH2 domain-containing leukocyte protein of 76 kDa (SLP76) (activated by ITAM-spleen tyrosine kinase, or Syk), and PLCγ, all of which are essential for the efficient phosphorylation of PLCγ. AP-1, activator protein 1; CREB, cyclic adenosine monophosphate responsive-element-binding protein; MITF, microphthalmia-associated transcription factor; TRAP, tartrate-resistant acid phosphatase.
During osteoclastogenesis, activation of calcium signaling is dependent on costimulatory receptors for RANK, which are immunoglobulin-like receptors, such as OSCAR and triggering receptor expressed in myeloid cells-2 (TREM-2). These receptors associate with the adaptor molecules Fc receptor common γ subunit (FcRγ) and DNAX-activating protein 12 (DAP12), transducing signals by the phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) within the adaptor proteins, which, in turn, recruit the spleen tyrosine kinase (Syk) [33, 34] (Figure 1). As shown recently, Tec family tyrosine kinases (Tec and Btk) activated by RANK cooperate with Syk to induce efficient phosphorylation of phospholipase Cγ (PLCγ), which induces the release of calcium from the endoplasmic reticulum through the generation of inositol trisphosphate [35]. Although a series of genetically modified mice has clearly shown that ITAM-mediated signals are essential for osteoclasto-genesis, the ligands for the costimulatory receptors remain to be identified [33–35].
Mechanism of bone destruction in rheumatoid arthritis
The essential role of osteoclasts in bone destruction in rheumatoid arthritis
The bone destruction observed in the joints of patients with RA presents a challenging clinical problem. In the early 1980s, researchers observed osteoclast-like cells at the bone destruction sites [36], but it was not until RANKL was cloned that the importance of osteoclasts became generally accepted. We previously demonstrated efficient osteoclast formation in synovial cell cultures obtained from patients with RA [37]. Moreover, the expression of RANKL was detected specifically in the synovium of patients with RA [38, 39]. Recent studies have provided further direct genetic evidence: RANKL-deficient mice, which lack osteoclasts, were protected from bone destruction in an arthritis model induced by serum transfer [40]. Bone erosion was not observed in osteopetrotic Fos-/- mice, even when they were crossed with TNF-α transgenic mice, which develop erosive arthritis spontaneously [41]. In the two cases, a similar level of inflammation was observed, indicating that RANKL and osteoclasts are indispensable for the bone loss but not the inflammation. Consistent with this, anti-RANKL and anti-osteoclast therapies have been shown to be beneficial in the treatment of bone damage in animal models of arthritis [42, 43]. Inflammatory cytokines such as TNF-α, interleukin-1 (IL-1), and IL-6 have a potent capacity to induce RANKL expression on synovial fibroblasts/osteoblasts and to facilitate RANKL signaling, thus contributing directly to the bone destruction process. In particular, TNF-α is considered of special importance since anti-TNF therapy reduces bone erosion as well as inflammation [44].
Effect of T cells on osteoclastogenesis
As infiltration of T cells into the synovium is a pathological hallmark of RA, it is vital to address how T-cell immunity is linked to the enhanced expression of RANKL and eventual osteoclastic bone resorption. More specifically, as RANKL is known to be expressed in activated T cells, it is important to determine whether this source of RANKL can directly induce osteoclast differentiation. In 1999, Kong and colleagues [42] showed that the RANKL expressed on activated T cells acts directly on osteoclast precursor cells and induces osteoclastogenesis in vitro. Horwood and colleagues [45] reported that osteoclastogenesis could be induced in vitro by activated T cells. However, it is important to note that T cells produce various cytokines, including interferon-γ (IFN-γ), IL-4, and IL-10, which exert potent inhibitory effects on osteoclast differentiation [2]. In the former study, the T cells were fixed by formaldehyde and thus were unable to release any humoral factors [42]. In the latter study, the T cells and osteoclast precursor cells were derived from different species, suggesting that the effect of cytokines would, in all likelihood, be much lower than that on cells of the same species [45]. The question then arises as to how T-cell cytokines other than RANKL affect osteoclast differentiation.
Upon activation, naïve CD4+ T cells differentiate into different lineages of helper T (Th) cells, depending on the cytokine milieu [46]. Th1 and Th2 cells are traditionally thought to be the major subsets generated upon antigenic stimulation. Th1 cells, which are induced by IL-12, produce mainly IFN-γ and are involved in cellular immunity; Th2 cells produce mainly IL-4, IL-5, and IL-10 and contribute to humoral immunity. RA was previously considered to be a disease in which the Th1-Th2 balance is skewed toward Th1. However, IFN-γ is not highly expressed in the joints of patients with RA [47]. Notably, IFN-γ strongly inhibits osteoclastogenesis, even at minute concentrations, through ubiquitin-proteasome-mediated degradation of TRAF6 [48]. Moreover, the severity of collagen-induced arthritis was reported to be exaggerated in the absence of IFN-γ signaling [49, 50], suggesting that Th1 cells are not linked to bone damage in arthritis.
Th17 cells function as osteoclastogenic Th cells
It is worthwhile to define what is believed to be a very rare but pathologically important Th cell subset that is responsible for abnormal bone resorption as osteoclastogenic Th cells. Previous investigations in our laboratory together with other studies on synovial T cell in RA have clarified the characteristics of osteoclastogenic Th cells in autoimmune arthritis [51]. First, osteoclastogenic Th cells do not produce a large amount of IFN-γ. Second, they trigger both local inflammation and the production of inflammatory cytokines that induce RANKL expression on synovial fibroblasts. Third, osteoclastogenic Th cells express RANKL and might thereby participate directly in accelerated osteoclastogenesis. Because these Th cells have such osteoclastogenic characteristics, they can tip the balance in favor of osteoclastogenesis synergistically.
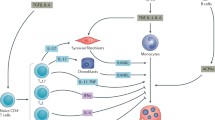
Th17 cells have recently been identified as a new effector Th cell subset characterized by the production of proinflammatory cytokines, including IL-17, IL-17F, IL-21, and IL-22. Th17 cell differentiation is induced by the combination of IL-6 and transforming growth factor-β (TGF-β). IL-23 is dispensable for the lineage commitment of Th17 cells but is required for the growth, survival, and effector functions of Th17 cells [52, 53]. Importantly, this unique subset plays a critical role in host defense against certain extracellular pathogens and also contributes to the pathogenesis of various auto-immune diseases [53]. Recent data from our laboratory indicate that Th17 cells represent the long sought-after osteoclastogenic Th-cell subset, fulfilling all of the criteria mentioned above [54]. IL-17 induces RANKL on osteoclastogenesis-supporting mesenchymal cells, such as osteoblasts and synovial fibroblasts [55]. IL-17 also enhances local inflammation and increases the production of inflammatory cytokines, which further promote RANKL expression and activity. Therefore, the infiltration of Th17 cells into the inflammatory lesion is the link between the abnormal T-cell response and bone damage (Figure 2).

Regulation of osteoclast differentiation by T cells in rheumatoid arthritis. Interleukin (IL)-17-producing helper T (Th17) cells have stimulatory effects on osteoclastogenesis and play an important role in the pathogenesis of rheumatoid arthritis through IL-17, whereas Th1 and Th2 cells have inhibitory effects on osteoclastogenesis through interferon-γ (IFN-γ) and IL-4, respectively. IL-17 not only induces receptor activator of nuclear factor-κB ligand (RANKL) on synovial fibroblasts of mesenchymal origin but also activates local inflammation, leading to the upregulation of proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α), IL-1, and IL-6. These cytokines activate osteoclastogenesis by either acting directly on osteoclast precursor cells or inducing RANKL on synovial fibroblasts. Th17 cells also express RANKL on their cellular membrane, and this partly contributes to the enhanced osteoclastogenesis. RANK, receptor activator of nuclear factor-κB.
Effects of regulatory T cells on osteoclastogenesis
CD4+ CD25+ regulatory T (Treg) cells are a specialized T-cell subset that engages in the maintenance of immunological self-tolerance and immune homeostasis, as evidenced by the development of severe autoimmune disease, allergy, and immunopathology in humans and mice with a mutation of forkhead box P3 (Foxp3), a master regulator for the Treg cell lineage [56]. Treg cells can be classified into two main populations: FoxP3+ naturally occurring Treg cells generated in the thymus and FoxP3+ Treg cells induced by antigen stimulation in a milieu rich in TGF-β in the periphery. Although the differences and similarities between these two populations are yet to be fully elucidated, both have been considered to be essential for immune homeostasis. Notably, Th 17 cells and Treg cells are reciprocally regulated during differentiation but exert the opposite effects on auto-immunity, and the balance between these populations is associated with inflammation and autoimmune diseases [53, 56]. In many studies, Treg cells were found in high numbers within joint fluid from patients with RA [57–59]. However, Treg cells in joint fluid from patients with RA failed to suppress effector T-cell proliferation or cytokine production. This is because inflammatory cytokines, including IL-6 and TNF-α, attenuate Treg function. Effector T cells in joint fluid were also reported to be resistant to suppression by Treg cells. In addition, serum markers of bone resorption such as C-terminal telopeptide of type I collagen inversely correlated with the number of CD4+CD25+ Treg cells in peripheral blood of healthy control and RA patients [60]. Thus, it is of key interest whether Treg cells affect inflammatory-associated bone destruction. Several groups have reported the inhibitory effect of Treg cells on osteoclastogenesis and bone resorption, but no consensus regarding their inhibitory mechanisms has been established. Kim and colleagues [61] reported that the human CD4+CD25+ Treg cells isolated from peripheral blood mononuclear cells (PBMCs) suppress osteoclast differentiation in a cytokine-dependent manner and proposed that TGF-β and IL-4 are required for the suppressive function of Treg cells. Zaiss and colleagues [62] demonstrated the inhibitory effect of CD4+CD25+ Treg cells purified from mouse spleen on osteoclast differentiation. However, the authors showed that CD4+CD25+ Treg cells inhibit osteoclastogenesis partially via IL-4 and IL-10 production but mainly through cell-to-cell contact via cytotoxic T lymphocyte antigen 4. It is notable that wild-type Treg cells failed to inhibit the differentiation of osteoclasts from CD80/86-/- monocytes [63]. A decrease in osteoclast number and bone resorption was observed after transfer of CD4+CD25+ Treg cells into Rag1-deficient mice, indicating that Treg cells could directly block osteoclastogenesis without engaging effector T cells [63]. Furthermore, Luo and colleagues [64] recently reported that human PBMC-derived CD4+CD25+ Treg cells suppress osteoclastogenesis and bone resorption in a TGF-β1 and IL-10 cytokine-dependent manner. Since TGF-β, IL-10, and IL-4 are cytokines that are well known to inhibit osteoclastogenesis, these cytokines produced by Treg cells may be involved, at least partially, in the suppressive function of Treg cells on osteoclastogenesis. In all studies by these three groups, Treg cells were activated before coculture experiments, but their culture conditions varied, and this may cause the difference among their results. Zaiss and colleagues [63] also reported increased bone mass and partial protection from bone loss after ovariectomy in Foxp3 transgenic mice. Foxp3+ Treg cells have been shown to protect against local and systemic bone destruction in the mouse model of TNF-α-induced arthritis [60]. It is likely that, taken as a whole, Foxp3+ Treg cells exert inhibitory effects on inflammatory-associated bone destruction, but it is important to consider the possibility that the characteristics of Treg cells are affected by the specific microenvironment such as autoimmune inflammation, as described above. Additional studies would be needed to determine how Treg cells affect osteoclast-mediated bone destruction under inflammatory conditions.
The involvement of B cells in bone destruction
B cells and antibodies make up the body's humoral immune response. B cells develop within bone marrow with the support of the stromal cells and the osteoblast lineage cells via various growth factors and cytokines, and are released into the blood and lymphatic systems. In the sera of most patients with RA, a variety of auto-antibodies such as rheumatoid factor and anti-cyclic citrullinated peptide antibodies can be detected [65]. The clinical benefit of the treatment of anti-CD20 antibody, rituximab, supports the notion that B cell-mediated immune responses contribute to the pathogenesis of RA [65, 66]. However, there are conflicting data on the role of B cells on bone remodeling: whereas some reported that activated B cells have the potential to promote osteoclastogenesis via RANKL expression [67, 68], others insisted that B cells have an inhibitory effect on osteoclastogenesis through TGF-β or IFN-γ production [69, 70]. Weitzmann and colleagues [71] reported that μMT heavy chain-deficient mice, which lack mature B cells, are osteoporotic. The authors proposed that B cells are critical regulators of physiological bone turnover by secreting OPG and that T cells promote enhanced OPG secretion by activated B cells via CD40/CD40L costimulation. Interestingly, T cell-deficient nude mice, CD40-deficient mice, and CD40L-deficient mice displayed osteoporosis and diminished bone marrow OPG production [71]. However, the other group reported that neither μMT-deficient mice nor Rag1-deficient mice have an obvious bone phenotype [72]. Thus, the role of B-cell lineages in physiological bone remodeling has not been firmly established.
IL-7, a major growth factor for B cells, has been reported to be upregulated under inflammatory conditions and during estrogen deficiency [73, 74]. Suda and colleagues [75] reported that systemic administration of IL-7 induced bone loss, which was similar to that of ovariectomized mice, and that IL-7Ra-deficient mice had increased bone mass. The authors proposed that increased B lymphopoiesis due to induction of IL-7 by estrogen deficiency may be involved in the elevated osteoclastogenesis. On the other hand, Weitzmann and colleagues [76] reported the other effect of IL-7 on bone metabolism; IL-7 promotes osteoclastogenesis by up-regulating T cell-derived osteoclastogenic cytokines, including RANKL. Indeed, IL-7 administration did not induce bone loss in T cell-deficient nude mice [77]. In contrast, Lorenzo and colleagues [78] reported that IL-7 inhibited osteoclast formation in bone marrow culture and that IL-7 deficiency caused increased osteoclastogenesis and decreased trabecular bone mass in vivo [79]. Wild-type and IL-7-deficient mice lose similar amounts of trabecular bone mass after ovariectomy. Consideration of the various effects of IL-7 on different target cells will be required to define the precise role of IL-7-mediated B lymphopoiesis on bone remodeling.
Kawai and colleagues [80] reported that, in case of bone destruction in periodontal disease, RANKL was highly expressed by activated B cells isolated from gingival tissues of patients. Furthermore, it has been recently reported that, after injection of lipopolysaccharide (LPS) into mouse gingival, alveolar bone destruction was more highly induced in B cell-reconstituted severe combined immunodeficiency (SCID) mice than in SCID mice and that LPS-stimulated B cells enhanced osteoclast differentiation by producing TNF-α in vitro [81]. These reports suggested that activated B cells have stimulatory effects on bone destruction under inflammatory conditions such as periodontitis, but further studies are needed to determine how B cell-mediated immune responses are directly involved in the osteoclast activation in RA.
Mechanisms involved in Th17 cell differentiation
The Th17 cell subset has emerged as an attractive therapeutic target for both inflammation and bone destruction. It is therefore important to understand the molecular mechanism underlying Th17 development in order to develop novel therapeutic strategies.
ROR nuclear receptors in Th17 development
Th cell differentiation is initiated by the T-cell receptor signal in combination with other cytokine receptor signals. These signals induce the activation of specific transcription factors to promote lineage-specific cytokine production [46]. For example, the T-box-containing protein expressed in T cells, which is activated by IL-12 and IFN-γ, is required for Th1 cell differentiation. Th2 cell differentiation requires the function of the GATA-binding protein 3, which is induced by the IL-4-activated signal transducer and activator of transcription (Stat) 6.
Soon after the discovery of Th17 cells, Littman and colleagues [82] reported that retinoid-related orphan receptor (ROR) γt is selectively expressed in Th17 cells and is required for Th17 cell differentiation. RORγt expression is induced by the combination of IL-6 and TGF-β through Stat3. Furthermore, RORγt deficiency was shown to lead to an impairment of Th17 cell differentiation both in vitro and in vivo. A subsequent study by Dong and colleagues [83] showed that another ROR family member, RORα, is highly induced during Th17 cell differentiation in a Stat3-dependent manner. Although RORα deletion in mice had only a minimal effect on IL-17 production, the deficiency of both RORα and RORγt completely abolished IL-17 production and protected mice from experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis. Thus, RORγt and RORα have redundant functions, but RORγt seems to be the major player in Th17 cell differentiation. Although the mechanisms by which the ROR nuclear receptors drive Th17 development and production of Th17-related cytokines such as IL-17 have not yet been fully elucidated, they are considered to be essential factors for Th17 development.
A role of cathepsin K in autoimmunity
Cathepsin K is a lysosomal cysteine protease that plays a pivotal role in osteoclast-mediated degradation of the bone matrices [84]. Thus, cathepsin K has been considered a potential therapeutic target for the treatment of bone diseases such as osteoporosis. We developed a new orally active cathepsin K inhibitor, NC-2300, and examined the effect of the inhibitor in osteoporosis as well as arthritis models [85]. We observed unexpected results that cathepsin K suppression leads to the reduction of inflammation in the latter model. Cathepsin K, despite a low expression level in dendritic cells, plays an important role in the activation of Toll-like receptor (TLR) 9 signaling. CpG (cytosine followed by guanine) DNA (a TLR9 ligand)-induced production of cytokines such as IL-6 and IL-23 was found to be impaired in cathepsin K inhibitor-treated or cathepsin K-deficient dendritic cells. The immune function of cathepsin K was further analyzed in EAE, and the severity of the disease was markedly suppressed in cathepsin K-deficient mice. The suppression of inflammation was associated with the reduced induction of Th17 cells, indicating that cathepsin K contributes to autoimmune inflammation by inducing Th17 cells, possibly through cytokines such as IL-6 and IL-23 in dendritic cells.
The detailed mechanism by which cathepsin K regulates TLR9 signaling remains elusive, but it has been reported that functional maturation of TLR9 requires its proteolytic cleavage [86, 87], to which cathepsin K might contribute. As cathepsin K is now known to be expressed by other cell types, including synovial cells [88], we cannot exclude the possibility that NC-2300 exerted an anti-arthritic effect through other cells. However, cathepsin K is an interesting example of a molecule that was originally found in bone and subsequently shown to regulate the immune system. Our study identified cathepsin K as a novel dendritic cell-specific regulator of TLR9 signaling and as a potential target of therapeutic intervention into inflammation-associated bone loss.
Regulation of Th17 development by IκBζ
We found that a nuclear IκB family member, IκBζ, was most highly expressed in Th 17 cells among the Th cell subsets [89]. IκBζ is a nuclear protein highly homologous to Bcl-3, which interacts with the NF-κB subunit via the ankyrin repeat domain [90]. Its expression is rapidly induced by TLR ligands or IL-1 stimulation in peritoneal macrophages. Yamamoto and colleagues [91], using IκBζ-deficient mice, demonstrated that IκBζ is essential for the LPS induction of a subset of secondary response genes, including IL-6 and the IL-12 p40 subunit, in macro-phages. However, no attempt to determine the function of IκBζ in T cells was reported in their study.
IκBζ expression was shown to be upregulated by the combination of IL-6 and TGF-β. IκBζ induction was mediated by Stat3, but not by RORγt, in Th17 cells. Importantly, not only IκBζ-deficient mice but also Rag2-deficient mice transferred with IκBζ-deficient CD4+ T cells were shown to be highly resistant to EAE. When naïve CD4+ T cells were activated in vitro under Th1- and Th2-polarizing conditions, IκBζ-deficient naïve CD4+ T cells normally produced IFN-γ and IL-4, respectively. On the other hand, when activated under Th17-polarizing conditions, IL-17 production in IκBζ-deficient T cells was markedly reduced compared with wild-type T cells. Since the expression of RORγt and RORα was shown to be normal in IκBζ-deficient T cells, it is unlikely that ROR nuclear receptors function downstream of IκBζ or vice versa.
Although ROR nuclear receptors have been proposed as essential regulators for Th17 development as described above, several groups have reported that the ectopic expression of RORγt or RORα leads to only modest IL-17 production in the absence of IL-6 and TGF-β [83, 92]. The ectopic expression of IκBζ in naïve CD4+ T cells did not induce IL-17 production in the absence of IL-6 and TGF-β. Interestingly, however, even in the absence of IL-6 and TGF-β, the ectopic expression of IκBζ, together with RORγt or RORα, potently induced IL-17 production. A reporter assay system showed that IκBζ moderately activated the promoter of the mouse Il17 gene as well as RORγt and RORα. When the ROR nuclear receptor was expressed, IκBζ highly activated the Il17 promoter. Previous studies showed that an evolutionarily conserved noncoding sequence 2 (CNS2) region in the Il17 locus is associated with histone H3 acetylation in a Th17 lineage-specific manner and that the ROR nuclear receptor is recruited to the CNS2 region during Th17 development [83, 93, 94]. In combination with RORγt and RORα, IκBζ potently induced the CNS2 enhancer activity. IκBζ was recruited to the CNS2 region in Th17 cells, and recruitment of IκBζ to the CNS2 region was dependent on RORγt function (Figure 3). Moreover, the expression of IL-17F, IL-21, and IL-23 receptor was decreased in IκBζ-deficient T cells. IκBζ also bound to the promoter or the enhancer region of these genes in Th17 cells. Collectively, these findings indicate that IκBζ is critical for the transcriptional program in Th17 cell lineage commitment [89].

IκBζ and ROR nuclear receptors synergistically promote Th17 development. Interleukin (IL)-6 and transforming growth factor-β (TGF-β) induce Th17 cell differentiation, in which the ROR nuclear receptors, RORγt and RORα, have an indispensable role. The expression of IκBζ is induced by the combination of IL-6 and TGF-β. IκBζ induction is mediated by signal transducer and activator of transcription 3 (Stat3), but not RORγt. IκBζ and ROR nuclear receptor bind directly to the CNS2 region of the Il17 promoter and cooperatively activate the Il17 promoter. Notably, recruitment of IκBζ to the CNS2 region was dependent on RORγt, suggesting that the binding of both IκBζ and ROR nuclear receptors to the Il17 promoter leads to an efficient recruitment of transcriptional coactivators having histone acetylase activity. CNS2, conserved noncoding sequence 2; MHC II, major histocompatibility complex class II; ROR, retinoid-related orphan receptor; TCR, T-cell receptor; Th, helper T.
Conclusions
The new field of osteoimmunology originated from studies on bone destruction in RA. Increasing evidence has made it evident that the skeletal and immune systems are connected in complex ways; in fact, it would be difficult to understand either system in depth without the insights afforded by studying their interaction in an osteoimmunological context [44]. The findings in RA might be applicable to numerous inflammatory or neoplastic diseases, such as periodontitis, infectious diseases, and primary or metastatic bone tumors.
Clearly, the Th17 cell subset is an auspicious target for future therapeutic investigation, and cytokines related to Th17 cell differentiation and function will be of great clinical importance. Antibodies against IL-17 or IL-23 would be expected to exert beneficial effects in auto-immune diseases, and antibodies targeting the IL-6 receptor might not only inhibit Th 17 development in RA but also effect a direct inhibition of local inflammation and osteoclastogenesis [95, 96]. The mechanism of Th17 development is currently one of the most important subjects in immunology. In recent years, several transcriptional regulators of Th17 development, including IRF4, BATF, Ahr, and Runx1, have been reported [92, 93, 97–99]. Although further studies will be required to determine whether or how IκBζ synergizes with other transcriptional regulators of Th17 cells, our results raise the possibility that the targeting of IκBζ may prove effective in the treatment of autoimmune diseases.
Importantly, Th17 cells are also implicated in host defense against a number of microorganisms. Inhibition of Th17 cells might thus carry a risk of increasing the susceptibility to infection. Therefore, great care will be required to effectively treat autoimmune diseases without compromising the host defense system. Understanding the precise role of Th17 cells in human autoimmune disorders therefore will be required for the development of effective therapeutic applications.
Note
This article is part of the series Osteoimmunology, edited by Georg Schett. Other articles in this series can be found at http://arthritis-research.com/series/osteoimmunology
Abbreviations
- AP-1:
-
activator protein 1
- CNS2:
-
conserved noncoding sequence 2
- CREB:
-
cyclic adenosine monophosphate responsive-element-binding protein
- EAE:
-
experimental autoimmune encephalomyelitis
- Foxp3:
-
forkhead box P3
- HSC:
-
hematopoietic stem cell
- IFN-γ:
-
interferon-γ
- IL:
-
interleukin
- ITAM:
-
immunoreceptor tyrosine-based activation motif
- LPS:
-
lipopolysaccharide
- M-CSF:
-
macrophage colony-stimulating factor
- NF-κB:
-
nuclear factor-κB
- NFAT:
-
nuclear factor of activated T cells
- NFATc1:
-
nuclear factor of activated T cells cytoplasmic 1
- OPG:
-
osteoprotegerin
- OSCAR:
-
osteoclast-associated receptor
- PBMC:
-
peripheral blood mononuclear cell
- RA:
-
rheumatoid arthritis
- RANK:
-
receptor activator of nuclear factor-κB
- RANKL:
-
receptor activator of nuclear factor-kappa-B ligand
- ROR:
-
retinoid-related orphan receptor
- SCID:
-
severe combined immunodeficiency
- Stat:
-
signal transducer and activator of transcription
- Syk:
-
spleen tyrosine kinase
- TGF-β:
-
transforming growth factor-β
- Th:
-
helper T
- TLR:
-
Toll-like receptor
- TNF:
-
tumor necrosis factor
- TRAF:
-
tumor necrosis factor receptor-associated factor
- Treg:
-
regulatory T.
References
Seeman E, Delmas PD: Bone quality--the material and structural basis of bone strength and fragility. N Engl J Med. 2006, 354: 2250-2261. 10.1056/NEJMra053077.
Takayanagi H: Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007, 7: 292-304. 10.1038/nri2062.
Takahashi N, Akatsu T, Udagawa N, Sasaki T, Yamaguchi A, Moseley JM, Martin TJ, Suda T: Osteoblastic cells are involved in osteoclast formation. Endocrinology. 1988, 123: 2600-2602. 10.1210/endo-123-5-2600.
Takahashi N, Yamana H, Yoshiki S, Roodman GD, Mundy GR, Jones SJ, Boyde A, Suda T: Osteoclast-like cell formation and its regulation by osteotropic hormones in mouse bone marrow cultures. Endocrinology. 1988, 122: 1373-1382. 10.1210/endo-122-4-1373.
Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ: Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev. 1999, 20: 345-357. 10.1210/er.20.3.345.
Yoshida H, Hayashi S, Kunisada T, Ogawa M, Nishikawa S, Okamura H, Sudo T, Shultz LD: The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990, 345: 442-444. 10.1038/345442a0.
Lagasse E, Weissman IL: Enforced expression of Bcl-2 in monocytes rescues macrophages and partially reverses osteopetrosis in op/op mice. Cell. 1997, 89: 1021-1031. 10.1016/S0092-8674(00)80290-1.
Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T: Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA. 1998, 95: 3597-3602. 10.1073/pnas.95.7.3597.
Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ: Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998, 93: 165-176. 10.1016/S0092-8674(00)81569-X.
Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, Teepe MC, DuBose RF, Cosman D, Galibert L: A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997, 390: 175-179. 10.1038/36593.
Wong BR, Rho J, Arron J, Robinson E, Orlinick J, Chao M, Kalachikov S, Cayani E, Bartlett FS, Frankel WN, Lee SY, Choi Y: TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J Biol Chem. 1997, 272: 25190-25194. 10.1074/jbc.272.40.25190.
Theill LE, Boyle WJ, Penninger JM: RANK-L and RANK: T cells, bone loss, and mammalian evolution. Annu Rev Immunol. 2002, 20: 795-823. 10.1146/annurev.immunol.20.100301.064753.
Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ: Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997, 89: 309-319. 10.1016/S0092-8674(00)80209-3.
Tsuda E, Goto M, Mochizuki S, Yano K, Kobayashi F, Morinaga T, Higashio K: Isolation of a novel cytokine from human fibroblasts that specifically inhibits osteoclastogenesis. Biochem Biophys Res Commun. 1997, 234: 137-142. 10.1006/bbrc.1997.6603.
Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveirados-Santos AJ, Van G, Itie A, Khoo W, Wakeham A, Dunstan CR, Lacey DL, Mak TW, Boyle WJ, Penninger JM: OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999, 397: 315-323. 10.1038/16852.
Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, Daro E, Smith J, Tometsko ME, Maliszewski CR, Armstrong A, Shen V, Bain S, Cosman D, Anderson D, Morrissey PJ, Peschon JJ, Schuh J: RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13: 2412-2424. 10.1101/gad.13.18.2412.
Li J, Sarosi I, Yan XQ, Morony S, Capparelli C, Tan HL, McCabe S, Elliott R, Scully S, Van G, Kaufman S, Juan SC, Sun Y, Tarpley J, Martin L, Christensen K, McCabe J, Kostenuik P, Hsu H, Fletcher F, Dunstan CR, Lacey DL, Boyle WJ: RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci USA. 2000, 97: 1566-1571. 10.1073/pnas.97.4.1566.
Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, Boyle WJ, Simonet WS: Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12: 1260-1268. 10.1101/gad.12.9.1260.
Mizuno A, Amizuka N, Irie K, Murakami A, Fujise N, Kanno T, Sato Y, Nakagawa N, Yasuda H, Mochizuki S, Gomibuchi T, Yano K, Shima N, Washida N, Tsuda E, Morinaga T, Higashio K, Ozawa H: Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem Biophys Res Commun. 1998, 247: 610-615. 10.1006/bbrc.1998.8697.
Hughes AE, Ralston SH, Marken J, Bell C, MacPherson H, Wallace RG, van Hul W, Whyte MP, Nakatsuka K, Hovy L, Anderson DM: Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nat Genet. 2000, 24: 45-48. 10.1038/71667.
Whyte MP, Obrecht SE, Finnegan PM, Jones JL, Podgornik MN, McAlister WH, Mumm S: Osteoprotegerin deficiency and juvenile Paget's disease. N Engl J Med. 2002, 347: 175-184. 10.1056/NEJMoa013096.
Sobacchi C, Frattini A, Guerrini MM, Abinun M, Pangrazio A, Susani L, Bredius R, Mancini G, Cant A, Bishop N, Grabowski P, Del Fattore A, Messina C, Errigo G, Coxon FP, Scott DI, Teti A, Rogers MJ, Vezzoni P, Villa A, Helfrich MH: Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL. Nat Genet. 2007, 39: 960-962. 10.1038/ng2076.
Guerrini MM, Sobacchi C, Cassani B, Abinun M, Kilic SS, Pangrazio A, Moratto D, Mazzolari E, Clayton-Smith J, Orchard P, Coxon FP, Helfrich MH, Crockett JC, Mellis D, Vellodi A, Tezcan I, Notarangelo LD, Rogers MJ, Vezzoni P, Villa A, Frattini A: Human osteoclast-poor osteopetrosis with hypogammaglobulinemia due to TNFRSF11A (RANK) mutations. Am J Hum Genet. 2008, 83: 64-76. 10.1016/j.ajhg.2008.06.015.
Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, Morony S, Capparelli C, Van G, Kaufman S, van der Heiden A, Itie A, Wakeham A, Khoo W, Sasaki T, Cao Z, Penninger JM, Paige CJ, Lacey DL, Dunstan CR, Boyle WJ, Goeddel DV, Mak TW: TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999, 13: 1015-1024. 10.1101/gad.13.8.1015.
Naito A, Azuma S, Tanaka S, Miyazaki T, Takaki S, Takatsu K, Nakao K, Nakamura K, Katsuki M, Yamamoto T, Inoue J: Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells. 1999, 4: 353-362. 10.1046/j.1365-2443.1999.00265.x.
Wagner EF, Eferl R: Fos/AP-1 proteins in bone and the immune system. Immunol Rev. 2005, 208: 126-140. 10.1111/j.0105-2896.2005.00332.x.
Sato K, Suematsu A, Nakashima T, Takemoto-Kimura S, Aoki K, Morishita Y, Asahara H, Ohya K, Yamaguchi A, Takai T, Kodama T, Chatila TA, Bito H, Takayanagi H: Regulation of osteoclast differentiation and function by the CaMK-CREB pathway. Nat Med. 2006, 12: 1410-1416.
Yamashita T, Yao Z, Li F, Zhang Q, Badell IR, Schwarz EM, Takeshita S, Wagner EF, Noda M, Matsuo K, Xing L, Boyce BF: NF-κB p50 and p52 regulate receptor activator of NF-κB ligand (RANKL) and tumor necrosis factorinduced osteoclast precursor differentiation by activating c-Fos and NFATc1. J Biol Chem. 2007, 282: 18245-18253. 10.1074/jbc.M610701200.
Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, Wagner EF, Mak TW, Kodama T, Taniguchi T: Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002, 3: 889-901. 10.1016/S1534-5807(02)00369-6.
Asagiri M, Sato K, Usami T, Ochi S, Nishina H, Yoshida H, Morita I, Wagner EF, Mak TW, Serfling E, Takayanagi H: Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med. 2005, 202: 1261-1269. 10.1084/jem.20051150.
Winslow MM, Pan M, Starbuck M, Gallo EM, Deng L, Karsenty G, Crabtree GR: Calcineurin/NFAT signaling in osteoblasts regulates bone mass. Dev Cell. 2006, 10: 771-782. 10.1016/j.devcel.2006.04.006.
Aliprantis AO, Ueki Y, Sulyanto R, Park A, Sigrist KS, Sharma SM, Ostrowski MC, Olsen BR, Glimcher LH: NFATc1 in mice represses osteoprotegerin during osteoclastogenesis and dissociates systemic osteopenia from inflammation in cherubism. J Clin Invest. 2008, 118: 3775-3789. 10.1172/JCI35711.
Koga T, Inui M, Inoue K, Kim S, Suematsu A, Kobayashi E, Iwata T, Ohnishi H, Matozaki T, Kodama T, Taniguchi T, Takayanagi H, Takai T: Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature. 2004, 428: 758-763. 10.1038/nature02444.
Mocsai A, Humphrey MB, Van Ziffle JA, Hu Y, Burghardt A, Spusta SC, Majumdar S, Lanier LL, Lowell CA, Nakamura MC: The immunomodulatory adapter proteins DAP12 and Fc receptor γ-chain (FcRγ) regulate development of functional osteoclasts through the Syk tyrosine kinase. Proc Natl Acad Sci USA. 2004, 101: 6158-6163. 10.1073/pnas.0401602101.
Shinohara M, Koga T, Okamoto K, Sakaguchi S, Arai K, Yasuda H, Takai T, Kodama T, Morio T, Geha RS, Kitamura D, Kurosaki T, Ellmeier W, Takayanagi H: Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell. 2008, 132: 794-806. 10.1016/j.cell.2007.12.037.
Bromley M, Woolley DE: Chondroclasts and osteoclasts at subchondral sites of erosion in the rheumatoid joint. Arthritis Rheum. 1984, 27: 968-975. 10.1002/art.1780270902.
Takayanagi H, Oda H, Yamamoto S, Kawaguchi H, Tanaka S, Nishikawa T, Koshihara Y: A new mechanism of bone destruction in rheumatoid arthritis: synovial fibroblasts induce osteoclastogenesis. Biochem Biophys Res Commun. 1997, 240: 279-286. 10.1006/bbrc.1997.7404.
Gravallese EM, Manning C, Tsay A, Naito A, Pan C, Amento E, Goldring SR: Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum. 2000, 43: 250-258. 10.1002/1529-0131(200002)43:2<250::AID-ANR3>3.0.CO;2-P.
Takayanagi H, Iizuka H, Juji T, Nakagawa T, Yamamoto A, Miyazaki T, Koshihara Y, Oda H, Nakamura K, Tanaka S: Involvement of receptor activator of nuclear factor κB ligand/osteoclast differentiation factor in osteoclastogenesis from synoviocytes in rheumatoid arthritis. Arthritis Rheum. 2000, 43: 259-269. 10.1002/1529-0131(200002)43:2<259::AID-ANR4>3.0.CO;2-W.
Pettit AR, Ji H, von Stechow D, Muller R, Goldring SR, Choi Y, Benoist C, Gravallese EM: TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am J Pathol. 2001, 159: 1689-1699. 10.1016/S0002-9440(10)63016-7.
Redlich K, Hayer S, Ricci R, David JP, Tohidast-Akrad M, Kollias G, Steiner G, Smolen JS, Wagner EF, Schett G: Osteoclasts are essential for TNF-α-mediated joint destruction. J Clin Invest. 2002, 110: 1419-1427.
Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe S, Wong T, Campagnuolo G, Moran E, Bogoch ER, Van G, Nguyen LT, Ohashi PS, Lacey DL, Fish E, Boyle WJ, Penninger JM: Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999, 402: 304-309. 10.1038/46303.
Takayanagi H, Juji T, Miyazaki T, Iizuka H, Takahashi T, Isshiki M, Okada M, Tanaka Y, Koshihara Y, Oda H, Kurokawa T, Nakamura K, Tanaka S: Suppression of arthritic bone destruction by adenovirus-mediated csk gene transfer to synoviocytes and osteoclasts. J Clin Invest. 1999, 104: 137-146. 10.1172/JCI6093.
Takayanagi H: Osteoimmunology and the effects of the immune system on bone. Nat Rev Rheumatol. 2009, 5: 667-676. 10.1038/nrrheum.2009.217.
Horwood NJ, Kartsogiannis V, Quinn JM, Romas E, Martin TJ, Gillespie MT: Activated T lymphocytes support osteoclast formation in vitro. Biochem Biophys Res Commun. 1999, 265: 144-150. 10.1006/bbrc.1999.1623.
Zhou L, Chong MM, Littman DR: Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009, 30: 646-655. 10.1016/j.immuni.2009.05.001.
Firestein GS, Zvaifler NJ: How important are T cells in chronic rheumatoid synovitis?. Arthritis Rheum. 1990, 33: 768-773. 10.1002/art.1780330602.
Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, Takaoka A, Yokochi T, Oda H, Tanaka K, Nakamura K, Taniguchi T: T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-γ. Nature. 2000, 408: 600-605. 10.1038/35046102.
Manoury-Schwartz B, Chiocchia G, Bessis N, Abehsira-Amar O, Batteux F, Muller S, Huang S, Boissier MC, Fournier C: High susceptibility to collagen-induced arthritis in mice lacking IFN-γ receptors. J Immunol. 1997, 158: 5501-5506.
Vermeire K, Heremans H, Vandeputte M, Huang S, Billiau A, Matthys P: Accelerated collagen-induced arthritis in IFN-γ receptor-deficient mice. J Immunol. 1997, 158: 5507-5513.
Sato K, Takayanagi H: Osteoclasts, rheumatoid arthritis, and osteoimmunology. Curr Opin Rheumatol. 2006, 18: 419-426. 10.1097/01.bor.0000231912.24740.a5.
Kastelein RA, Hunter CA, Cua DJ: Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu Rev Immunol. 2007, 25: 221-242. 10.1146/annurev.immunol.22.012703.104758.
Korn T, Bettelli E, Oukka M, Kuchroo VK: IL-17 and Th17 cells. Annu Rev Immunol. 2009, 27: 485-517. 10.1146/annurev.immunol.021908.132710.
Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y, Cua DJ, Takayanagi H: Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006, 203: 2673-2682. 10.1084/jem.20061775.
Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, Saito S, Inoue K, Kamatani N, Gillespie MT, Martin TJ, Suda T: IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999, 103: 1345-1352. 10.1172/JCI5703.
Sakaguchi S, Yamaguchi T, Nomura T, Ono M: Regulatory T cells and immune tolerance. Cell. 2008, 133: 775-787. 10.1016/j.cell.2008.05.009.
Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, Mauri C: Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFα therapy. J Exp Med. 2004, 200: 277-285. 10.1084/jem.20040165.
Boissier MC, Assier E, Falgarone G, Bessis N: Shifting the imbalance from Th1/Th2 to Th17/treg: the changing rheumatoid arthritis paradigm. Joint Bone Spine. 2008, 75: 373-375. 10.1016/j.jbspin.2008.04.005.
Nistala K, Wedderburn LR: Th17 and regulatory T cells: rebalancing pro- and anti-inflammatory forces in autoimmune arthritis. Rheumatology (Oxford). 2009, 48: 602-606. 10.1093/rheumatology/kep028.
Zaiss MM, Frey B, Hess A, Zwerina J, Luther J, Nimmerjahn F, Engelke K, Kollias G, Hunig T, Schett G, David JP: Regulatory T cells protect from local and systemic bone destruction in arthritis. J Immunol. 2010, 184: 7238-7246. 10.4049/jimmunol.0903841.
Kim YG, Lee CK, Nah SS, Mun SH, Yoo B, Moon HB: Human CD4+CD25+ regulatory T cells inhibit the differentiation of osteoclasts from peripheral blood mononuclear cells. Biochem Biophys Res Commun. 2007, 357: 1046-1052. 10.1016/j.bbrc.2007.04.042.
Zaiss MM, Axmann R, Zwerina J, Polzer K, Guckel E, Skapenko A, Schulze-Koops H, Horwood N, Cope A, Schett G: Treg cells suppress osteoclast formation: a new link between the immune system and bone. Arthritis Rheum. 2007, 56: 4104-4112. 10.1002/art.23138.
Zaiss MM, Sarter K, Hess A, Engelke K, Bohm C, Nimmerjahn F, Voll R, Schett G, David JP: Increased bone density and resistance to ovariectomy-induced bone loss in FoxP3-transgenic mice based on impaired osteoclast differentiation. Arthritis Rheum. 2010, 62: 2328-2338. 10.1002/art.27535.
Luo CY, Wang L, Sun C, Li DJ: Estrogen enhances the functions of CD4+CD25+Foxp3+ regulatory T cells that suppress osteoclast differentiation and bone resorption in vitro. Cell Mol Immunol. 2010, 8: 50-58.
Townsend MJ, Monroe JG, Chan AC: B-cell targeted therapies in human autoimmune diseases: an updated perspective. Immunol Rev. 2010, 237: 264-283. 10.1111/j.1600-065X.2010.00945.x.
Browning JL: B cells move to centre stage: novel opportunities for autoimmune disease treatment. Nat Rev Drug Discov. 2006, 5: 564-576. 10.1038/nrd2085.
Manabe N, Kawaguchi H, Chikuda H, Miyaura C, Inada M, Nagai R, Nabeshima Y, Nakamura K, Sinclair AM, Scheuermann RH, Kuro-o M: Connection between B lymphocyte and osteoclast differentiation pathways. J Immunol. 2001, 167: 2625-2631.
Choi Y, Woo KM, Ko SH, Lee YJ, Park SJ, Kim HM, Kwon BS: Osteoclastogenesis is enhanced by activated B cells but suppressed by activated CD8(+) T cells. Eur J Immunol. 2001, 31: 2179-2188. 10.1002/1521-4141(200107)31:7<2179::AID-IMMU2179>3.0.CO;2-X.
Weitzmann MN, Cenci S, Haug J, Brown C, DiPersio J, Pacifici R: B lymphocytes inhibit human osteoclastogenesis by secretion of TGFβ. J Cell Biochem. 2000, 78: 318-324. 10.1002/(SICI)1097-4644(20000801)78:2<318::AID-JCB13>3.0.CO;2-N.
Choi Y, Kim JJ: B cells activated in the presence of Th1 cytokines inhibit osteoclastogenesis. Exp Mol Med. 2003, 35: 385-392.
Li Y, Toraldo G, Li A, Yang X, Zhang H, Qian WP, Weitzmann MN: B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood. 2007, 109: 3839-3848. 10.1182/blood-2006-07-037994.
Horowitz MC, Xi Y, Pfl ugh DL, Hesslein DG, Schatz DG, Lorenzo JA, Bothwell AL: Pax5-deficient mice exhibit early onset osteopenia with increased osteoclast progenitors. J Immunol. 2004, 173: 6583-6591.
Weitzmann MN, Roggia C, Toraldo G, Weitzmann L, Pacifici R: Increased production of IL-7 uncouples bone formation from bone resorption during estrogen deficiency. J Clin Invest. 2002, 110: 1643-1650.
Churchman SM, Ponchel F: Interleukin-7 in rheumatoid arthritis. Rheumatology (Oxford). 2008, 47: 753-759. 10.1093/rheumatology/ken053.
Miyaura C, Onoe Y, Inada M, Maki K, Ikuta K, Ito M, Suda T: Increased B-lymphopoiesis by interleukin 7 induces bone loss in mice with intact ovarian function: similarity to estrogen deficiency. Proc Natl Acad Sci USA. 1997, 94: 9360-9365. 10.1073/pnas.94.17.9360.
Weitzmann MN, Cenci S, Rifas L, Brown C, Pacifici R: Interleukin-7 stimulates osteoclast formation by up-regulating the T-cell production of soluble osteoclastogenic cytokines. Blood. 2000, 96: 1873-1878.
Toraldo G, Roggia C, Qian WP, Pacifici R, Weitzmann MN: IL-7 induces bone loss in vivo by induction of receptor activator of nuclear factor κB ligand and tumor necrosis factor alpha from T cells. Proc Natl Acad Sci USA. 2003, 100: 125-130. 10.1073/pnas.0136772100.
Lee SK, Kalinowski JF, Jastrzebski SL, Puddington L, Lorenzo JA: Interleukin-7 is a direct inhibitor of in vitro osteoclastogenesis. Endocrinology. 2003, 144: 3524-3531. 10.1210/en.2002-221057.
Lee SK, Kalinowski JF, Jacquin C, Adams DJ, Gronowicz G, Lorenzo JA: Interleukin-7 influences osteoclast function in vivo but is not a critical factor in ovariectomy-induced bone loss. J Bone Miner Res. 2006, 21: 695-702. 10.1359/jbmr.060117.
Kawai T, Matsuyama T, Hosokawa Y, Makihira S, Seki M, Karimbux NY, Goncalves RB, Valverde P, Dibart S, Li YP, Miranda LA, Ernst CW, Izumi Y, Taubman MA: B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. Am J Pathol. 2006, 169: 987-998. 10.2353/ajpath.2006.060180.
Kozuka Y, Ozaki Y, Ukai T, Kaneko T, Hara Y: B cells play an important role in lipopolysaccharide-induced bone resorption. Calcif Tissue Int. 2006, 78: 125-132. 10.1007/s00223-005-0149-x.
Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR: The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006, 126: 1121-1133. 10.1016/j.cell.2006.07.035.
Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, Watowich SS, Tian Q, Jetten AM, Dong C: T helper 17 lineage differentiation is programmed by orphan nuclear receptors RORα and RORγ. Immunity. 2008, 28: 29-39. 10.1016/j.immuni.2007.11.016.
Gelb BD, Shi GP, Chapman HA, Desnick RJ: Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science. 1996, 273: 1236-1238. 10.1126/science.273.5279.1236.
Asagiri M, Hirai T, Kunigami T, Kamano S, Gober HJ, Okamoto K, Nishikawa K, Latz E, Golenbock DT, Aoki K, Ohya K, Imai Y, Morishita Y, Miyazono K, Kato S, Saftig P, Takayanagi H: Cathepsin K-dependent toll-like receptor 9 signaling revealed in experimental arthritis. Science. 2008, 319: 624-627. 10.1126/science.1150110.
Ewald SE, Lee BL, Lau L, Wickliff e KE, Shi GP, Chapman HA, Barton GM: The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature. 2008, 456: 658-662. 10.1038/nature07405.
Park B, Brinkmann MM, Spooner E, Lee CC, Kim YM, Ploegh HL: Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nat Immunol. 2008, 9: 1407-1414. 10.1038/ni.1669.
Hou WS, Li Z, Gordon RE, Chan K, Klein MJ, Levy R, Keysser M, Keyszer G, Bromme D: Cathepsin k is a critical protease in synovial fibroblastmediated collagen degradation. Am J Pathol. 2001, 159: 2167-2177. 10.1016/S0002-9440(10)63068-4.
Okamoto K, Iwai Y, Oh-Hora M, Yamamoto M, Morio T, Aoki K, Ohya K, Jetten AM, Akira S, Muta T, Takayanagi H: IκBζ regulates TH17 development by cooperating with ROR nuclear receptors. Nature. 2010, 464: 1381-1385. 10.1038/nature08922.
Muta T: IκB-ζ: an inducible regulator of nuclear factor-κB. Vitam Horm. 2006, 74: 301-316.
Yamamoto M, Yamazaki S, Uematsu S, Sato S, Hemmi H, Hoshino K, Kaisho T, Kuwata H, Takeuchi O, Takeshige K, Saitoh T, Yamaoka S, Yamamoto N, Yamamoto S, Muta T, Takeda K, Akira S: Regulation of Toll/IL-1-receptormediated gene expression by the inducible nuclear protein IκBζ. Nature. 2004, 430: 218-222. 10.1038/nature02738.
Brustle A, Heink S, Huber M, Rosenplanter C, Stadelmann C, Yu P, Arpaia E, Mak TW, Kamradt T, Lohoff M: The development of inflammatory TH-17 cells requires interferon-regulatory factor 4. Nat Immunol. 2007, 8: 958-966. 10.1038/ni1500.
Zhang F, Meng G, Strober W: Interactions among the transcription factors Runx1, RORγt and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat Immunol. 2008, 9: 1297-1306. 10.1038/ni.1663.
Akimzhanov AM, Yang XO, Dong C: Chromatin remodeling of interleukin-17 (IL-17)-IL-17F cytokine gene locus during inflammatory helper T cell differentiation. J Biol Chem. 2007, 282: 5969-5972.
Takatori H, Kanno Y, Chen Z, O'Shea JJ: New complexities in helper T cell fate determination and the implications for autoimmune diseases. Mod Rheumatol. 2008, 18: 533-541. 10.1007/s10165-008-0099-z.
Mihara M, Ohsugi Y, Kishimoto T: Evidence for the role of Th17 cell inhibition in the prevention of autoimmune diseases by anti-interluekin-6 receptor antibody. Biofactors. 2009, 35: 47-51. 10.1002/biof.9.
Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B: The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008, 453: 106-109. 10.1038/nature06881.
Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL: Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008, 453: 65-71. 10.1038/nature06880.
Schraml BU, Hildner K, Ise W, Lee WL, Smith WA, Solomon B, Sahota G, Sim J, Mukasa R, Cemerski S, Hatton RD, Stormo GD, Weaver CT, Russell JH, Murphy TL, Murphy KM: The AP-1 transcription factor Batf controls TH17 differentiation. Nature. 2009, 460: 405-409.
Acknowledgements
This work was supported in part by Grants-in-Aid for the Global Center of Excellence Program from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT), and ERATO, Takayanagi Osteonetwork Project from the Japan Science and Technology Agency (Saitama, Japan).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Okamoto, K., Takayanagi, H. Regulation of bone by the adaptive immune system in arthritis. Arthritis Res Ther 13, 219 (2011). https://doi.org/10.1186/ar3323
Published:
DOI: https://doi.org/10.1186/ar3323