
Abstract
Type I interferons are implicated in the pathogenesis of systemic lupus erythematosus (SLE). Type I interferon-inducible mRNAs are widely and concordantly overexpressed in the periphery and involved tissues of a subset of SLE patients, and provide utility as pharmacodynamic biomarkers to aid dose selection, as well as potential indicators of patients who might respond favorably to anti-IFNα therapy in SLE. We implemented a three-tiered approach to identify a panel of type I interferon-inducible mRNAs to be used as potential pharmacodynamic biomarkers to aid dose selection in clinical trials of sifalimumab, an anti-IFNα monoclonal antibody under development for the treatment of SLE. In a single-dose escalation phase 1 trial, we observed a sifalimumab-specific and dose-dependent inhibition of the overexpression of type I interferon-inducible mRNAs in the blood of treated subjects. Inhibition of expression of type I interferon-inducible mRNAs and proteins was also observed in skin lesions of SLE subjects from the same trial. Inhibiting IFNα resulted in a profound downstream effect in these SLE subjects that included suppression of mRNAs of B-cell activating factor belonging to the TNF family and the signaling pathways of TNFα, IL-10, IL-1β, and granulocyte-macrophage colony-stimulating factor in both the periphery and skin lesions. A scoring method based on the expression of type I interferon-inducible mRNAs partitioned SLE patients into two distinct subpopulations, which suggests the possibility of using these type I interferon-inducible genes as predictive biomarkers to identify SLE patients who might respond more favorably to anti-type I interferon therapy.
Similar content being viewed by others


Role of IFNα in the pathogenesis of systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by multiple immune system abnormalities, including production of autoantibodies that can lead to inflammation and tissue damage [1]. SLE symptoms can range from a mild rash to life-threatening nephritis and central nervous system disease. These disease manifestations cause a significant burden of illness and can cause reduced physical function, loss of employment, lower health-related quality of life, and a lifespan shortened by about 10 years [2]. Increased hospitalizations and side effects of medications including chronic corticosteroid and other immunosuppressive treatments add to the disease burden in SLE [2]. No new treatment for SLE has been approved by the US Food and Drug Administration in about 50 years since hydroxychloroquine was approved for use in discoid lupus and SLE; otherwise, the existing standard of care for SLE consists of off -label medications.
The disease pathogenesis of SLE includes activation of innate immunity, with increased production of type I interferons, including IFNα, and increased plasmacytoid and myeloid dendritic cells in involved tissue [3–8]. Specific immunity, including both humoral and cellular immune systems, is activated. Autoantibodies are universally present and may precede development of clinically apparent disease [9]. SLE-associated autoantibodies include anti-dsDNA, anti-nucleosomes, anti-RNP (ribonucleoprotein complex), and anti-Sm antibodies. Immune complexes containing anti-dsDNA or anti-RNP antibodies can activate type I interferon production [3, 4]. After internalization through Fc receptors, autoantibody-containing immune complexes bind endosomal Toll-like receptors 7 and 9, and stimulate production of type I interferon. Type I interferon stimulates myeloid dendritic cell maturation, which promotes loss of tolerance and generation of autoreactive T cells and B cells, autoantibody production, immune complex formation and further production of type I interferon, creating a self-perpetuating cycle of autoimmunity [5, 10, 11].
Type I interferons include 14 IFNα family members, IFNβ, IFNτ, IFNκ and IFNω [12]. This cytokine family regulates immune functions of cellular components of both innate and adaptive immune systems, including dendritic cells, T cells, B cells, and natural killer cells. For example, type I interferons promote dendritic cell maturation, memory CD8+ T-cell proliferation, natural killer-cell activation, and B-cell differentiation [5, 13]. Type I interferons also enhance the expression of immunologically important molecules such as MHC class I, CD38, interleukins such as BLyS, IL-6, IL-10 and IL-15, and multiple chemokines [14–17].
Emerging data indicate a role for type I interferons in disease pathogenesis in SLE. Genetic polymorphisms associated with type I interferon pathways are associated with susceptibility to SLE [18, 19]. Treatment with IFNα has been associated with the development of autoantibodies and new or worsening clinical features of the SLE [20, 21]. Higher IFNα levels and type I interferon activity are associated with greater disease activity in SLE [3, 7]. Patients with high anti-dsDNA antibody titers, lupus nephritis, and progressive skin rashes have high serum levels of IFNα [3]. In addition, patients with acute skin involvement tend to have elevated IFNα in blood and skin [7].
These clinical observations in humans are supported by data that show a key role for type I interferon in animal models of SLE. IFNα can induce glomerulonephritis in normal mice and accelerates the onset of the spontaneous autoimmune disease of NZB/W mice [22]. Autoimmune-predisposed mice deficient in the IFNα/β receptor exhibit significantly reduced anti-erythrocyte autoantibodies, hemolytic anemia, anti-DNA autoantibodies, kidney disease, and mortality [23]. Furthermore, IFNα kinoid vaccine has been shown to induce neutralizing antibodies to prevent clinical manifestations in a lupus flare murine model [24].
Together, these human and animal data support the hypothesis that inhibiting type I interferon may reduce disease activity in SLE.
Increased expression of the type I interferon signature in the blood and involved tissues of SLE patients
Molecular signatures or mRNA expression patterns in the periphery and disease tissues have been used to identify novel therapeutic targets and to predict patients that might respond favorably to therapeutic interventions [25–28]. One distinct observation in SLE is the robust and prevalent overexpression of type I interferon-inducible mRNAs (type I interferon signature) in the blood and involved tissues of patients. Increased expression of mRNAs induced by type I interferon is prominent in peripheral blood mononuclear cells and in whole blood (WB) in approximately 60% of SLE patients, and is associated with greater disease activity [29–34]. Type I interferon is the most activated signaling pathway and type I interferon-inducible mRNAs are the most overexpressed mRNAs in WB of patients with SLE [35]. Skin biopsies from a subset of patients with SLE also show increased expression of a type I interferon signature [36–38]. Expression of type I interferon-inducible IP-10/CXCL10 mRNAs is increased in SLE patients with active central nervous system symptoms [39]. Selected type I interferon-inducible mRNAs (IFI44, IFI27, and IFI44L) and type I interferon-inducible proteins (IFI27, STAT1) are overexpressed in the synovium of SLE patients with arthritis [40].
Detection of type I interferon proteins by ELISA has yielded positive results in only a small population of SLE patients [41], whereas detection of overexpression of the type I interferon signature is positive in about 60% of patients overall. This discrepancy suggests the type I interferon signature is the more sensitive measurement to detect increased type I interferon activity in SLE. The type I interferon-signature thus provides a potential pharmacodynamic (PD) marker to evaluate inhibition of the molecular target and to help guide dose selection for anti-type I interferon therapies in SLE.
Increased expression of the type I interferon signature in other rheumatic diseases
In addition to SLE, overexpression of the type I interferon signature has been reported in the peripheral blood of patients with a variety of inflammatory and autoimmune diseases that include dermatomyositis, polymyositis, multiple sclerosis, rheumatoid arthritis, scleroderma and Sjögren's syndrome [42–46]. The presence of increased numbers of plasmacytoid dendritic cells and overexpression of type I interferon-inducible mRNAs and/or proteins has also been reported at the disease tissues in some of the diseases [47, 48].
Figure 1 shows a heatmap of the relative expression of 807 type I interferon-inducible mRNAs (identified by ex vivo stimulation of healthy donor WB with type I interferon proteins, as described in [35]) in WB of 106 SLE patients, 22 dermatomyositis patients, 20 polymyositis patients, and 66 rheumatoid arthritis subjects compared with 24 normal healthy control individuals. For each autoimmune disease (blocks of columns), the subjects (individual columns) are sorted, from left to right, from those showing the greatest overexpression of the 807 type I interferon-inducible mRNAs (defined as the median fold change for each of the 807 mRNAs versus healthy controls; rows) to those with the least expression. When compared with the 24 normal healthy control individuals, a subpopulation of patients within each of these autoimmune diseases demonstrated overexpression of the type I interferon signature in WB.

Relative expression of type I interferon-inducible mRNAs in the whole blood of rheumatic disease patients. Representative heatmap visualizing the relative expression of 807 type I interferon-inducible mRNAs in whole blood from 106 systemic lupus erythematosus (SLE) subjects (red), 20 polymyositis (PM) subjects (green), 22 dermatomyositis (DM) subjects (blue), and 66 rheumatoid arthritis (RA) subjects (pink) compared with whole blood from 24 healthy subjects (black). Fold change values are graphed for each subject and are grouped by disease.
The evidence that the type I interferon signature is highly overexpressed in peripheral blood and tissue in multiple autoimmune diseases raises an important question as to whether type I interferon is involved in the pathogenesis of these diseases, and whether the activation of this signaling pathway is shared among the diseases. One key issue that needs to be addressed is whether activation of the type I interferon signaling pathway is concordant in WB and disease tissues of patients with these autoimmune diseases. Further more, it remains to be seen whether targeting type I interferon could benefit a subgroup of patients with distinct rheumatic diseases where the different disease pathogenesis and manifestations are different.
Development of the type I interferon signature as a pharmacodynamic marker for sifalimumab
Sifalimumab is a fully human IgG1κ monoclonal antibody that was generated by Medarex in mice transgenic for human immunoglobulin genes. The type I interferon signature was chosen as a potential PD marker to test whether sifalimumab inhibited its intended target, IFNα, in early clinical trials in SLE. This choice was made because increased expression of type I interferon-inducible mRNAs is a direct molecular consequence of increased expression of IFNα proteins, and because type I interferon-induced mRNAs are easy to measure by either RT-PCR or microarray-based technologies and demonstrate concordant overexpression in blood and involved tissue in SLE [38].
A three-tiered approach was taken to develop a panel of type I interferon-inducible mRNAs as potential PD markers for sifalimumab in SLE. First, the prevalence and magnitude of the overexpression of type I interferon mRNAs was determined in WB of SLE patients. WB samples from healthy donors were stimulated ex vivo with each of 10 IFNα subtypes or IFNβ, and then mRNAs (n = 807) were identified that were uniformly upregulated.
Secondly, mRNA expression was measured in the WB from 41 SLE patients and compared with mRNA expression in the WB of 24 normal healthy controls, using significance analysis of microarrays and the false discovery rate. Overall, 110 of the overexpressed mRNAs in the WB of these SLE patients were type I interferon-inducible, with type I interferon-inducible mRNAs among the most overexpressed mRNAs in the WB of SLE patients. The type I interferon signaling pathway was the most activated pathway in WB of these SLE patients. These results were confirmed in an independent set of 56 SLE WB samples.
Finally, mRNAs were identified whose expression was both stimulated when healthy donor peripheral blood mononuclear cells were exposed ex vivo to SLE patient sera and inhibited by sifalimumab in a dose-dependent manner in the same culture system. Overall, 77 type I interferon-inducible mRNAs were identified as overexpressed in each of the three different experiments. A subset of 21 of these 77 mRNAs was selected to be used as candidate PD biomarkers for sifalimumab in SLE, based on the magnitude and prevalence of overexpression in SLE [35]. In the end, this three-tiered approach yielded a small, robust panel of type I interferon-inducible mRNAs that could be used in a high-throughput PD biomarker assay to evaluate target inhibition by sifalimumab in SLE trials.
The 21-mRNA panel demonstrated many desirable characteristics for a PD biomarker. Overexpression of the mRNAs within this panel provides a sensitive and specific marker for overexpression of type I interferons within WB of SLE patients [35, 38]. Elevated expression versus normal expression of this mRNA panel was a consistent characteristic of an individual SLE patient, in the absence of therapeutic intervention with sifalimumab [29–35], which allows normalization of expression of this PD biomarker to be interpreted as treatment effect. Expression of the mRNAs within this panel can be measured quantitatively by multiple assays [35], allowing flexibility of the approach during clinical testing.
The level of expression of this mRNA panel calculated as a type I interferon signature score (Figure 2) in an individual subject provides a continuous measurement that can be used to classify subpopulations of SLE patients. The distribution of this type I interferon signature score in the WB of SLE patients exhibited a bimodal distribution, with relatively few SLE patient scores falling between subpopulations with normal levels and increased levels of expression (moderate and high over expression) of the type I interferon signature (Figure 2). This ability to differentiate between SLE patients with increased and normal expression of the type I interferon signature suggests potential utility of the type I interferon signature as a possible diagnostic marker to identify a subpopulation of SLE patients with increased expression of type I interferon. This hypothesis remains to be validated in clinical trials.

Distribution and density of type I interferon signature scores in systemic lupus erythematosus patients. (a) Distribution of the type I interferon signature scores in 202 systemic lupus erythematosus (SLE) patients measured at baseline. The type I interferon signature score is calculated as the median fold change (relative to a pooled normal healthy donor sample) of the 21-member type I interferon-inducible transcript pharmacodynamic (PD) marker panel for each patient. The cut-off point for an SLE patient scored as type I interferon signature positive was identified at a value of at least 2 (1 on a log2 scale), as represented by both the red points in (a) and the rightmost density in (b). Patients scored as type I interferon signature negative are represented by the blue points in and the leftmost density in (b). y axis, type I interferon signature score calculated for the 21-member mRNA PD marker panel on a log2 scale. x axis, type I interferon signature score calculated for the 21-member transcript PD marker panel on a log2 scale. (b) Density plot of the type I interferon signature scores for these 202 SLE patients. Two distributional modes are illustrated, indicating a clear partitioning between the SLE populations of type I interferon signature positive and negative.
Sifalimumab reduces the type I interferon signature in whole blood of SLE patients, in a dose-dependent manner
The safety and tolerability of sifalimumab were evaluated in a phase 1 trial in 62 mild-to-moderately active adult SLE subjects who were receiving standard-of-care therapy [38, 49]. The trial evaluated intravenously administered sifalimumab over a dose range of 0.3 to 30.0 mg/kg, compared with placebo. The phase 1 trial integrated translational research to evaluate PD effects of the drug and downstream effects of inhibiting IFNα in SLE.
Sifalimumab caused specific and dose-dependent inhibition of overexpression of the type I interferon signature in WB of treated SLE subjects (Figure 3). Concordant overexpression of the type I interferon signature was observed in the majority of subjects who had matched WB and lesional skin biopsies [38]. Seven out of eight patients who exhibited positive overexpression of the type I interferon signature in WB and skin lesions showed a similar trend of target neutralization in both sites at day 14 post sifalimumab or placebo treatment [38]. Inhibition of expression of both type I interferon-inducible mRNAs and protein occurred in skin lesions of SLE subjects, as measured by TaqMan quantitative RT-PCR and immunohistochemistry in subjects treated with sifalimumab. Reductions of plasmacytoid dendritic cells, myeloid dendritic cells, CD4+ T cells and dendritic cells were detected in the lesional skin of SLE patients, along with reduced levels of type I interferon-inducible mRNAs and proteins in patients who had skin biopsies that responded to sifalimumab treatment (Figure 4). These changes were not observed in the majority of the placebo-treated patients or in some subjects who did not respond to sifalimumab treatment, as evaluated by immunohistochemistry [38]. In a small number of SLE subjects with overexpression of mRNAs of B-cell activating factor belonging to the TNF family and activation of TNFα, IL-10, IL-1β and granulocyte-macrophage colony-stimulating factor signaling pathways in WB or skin lesions - as assessed by both TaqMan quantitative RT-PCR and microarray - a trend toward inhibition of the expression of these mRNAs was observed in response to sifalimumab treatment.

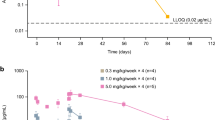
Sifalimumab inhibition of overexpression of the type I interferon signature in systemic lupus erythematosus patients. Dose responses of sifalimumab therapy in whole blood of systemic lupus erythematosus (SLE) patients with an overexpressed type I interferon-inducible gene signature as measured by the type I interferon-inducible gene signature score. Neutralization of the 21 type I interferon-inducible mRNAs by treatment averaged for each dose cohort observed from day 0 (pretreatment) to day 7 (posttreatment). y axis, values calculated as the fraction of neutralization of certain days post-treatment/pretreatment (day 0 predosing), subtracted from 1 for each patient separately. Those values that exceed 1 from this formula are a result of an increase in transcript levels of type I interferon-inducible mRNAs in whole blood following treatment (mostly in placebo-treated SLE patients). Significance for the difference between dose levels and placebo treatment using Hotelling's T2 test: 0.3 mg/kg, P = 0.09; 1 mg/kg, P = 0.04; 3 mg/kg, P = 0.01; 10 mg/kg, P = 0.007; 30 mg/kg, P = 0.004. From top to bottom: red line, placebo; blue line, 0.3 mg/kg sifalimumab; green line, 1 mg/kg sifalimumab; orange line, 3 mg/kg sifalimumab; black line, 10 mg/kg sifalimumab; pink line, 30 mg/kg sifalimumab.

Sifalimumab treatment in a responding systemic lupus erythematosus patient. Effect of sifalimumab treatment in a single systemic lupus erythematosus patient who responded to 10 mg/kg drug treatment. (a) Immunohistochemical analyses of paired biopsies (days 0 predosing and day 14 postdosing) from lesional skin obtained from this patient. BDCA2 is a specific marker for plasmacytoid dendritic cells, and CD83 is a marker for myeloid dendritic cells. IP10 is a type I interferon-inducible protein whose overexpression that was observed in lesional skin at day 0 was decreased at day 14. There is a concordant reduction of IP10 protein expression and mRNA expression in the lesional skin of this patient with sifalimumab treatment. (b) Skin lesion clearance in this patient following sifalimumab treatment (day 0 predosing; days 7, 14, and 28 postdosing).
Results from this phase 1 trial showing dose-dependent inhibition of the type I interferon signature in the WB of SLE patients confirmed the mechanism of action of sifalimumab. The combination of concordant inhibition of the type I interferon signature in WB and involved skin and a reduction of type I interferon-inducible protein expression in skin in a phase 1a trial support further testing of sifalimumab in SLE. Genentech has recently shown in a phase I trial a similar dose-dependent inhibition of the type I interferon signature in the peripheral blood of SLE patients that followed either a single dose or repeat doses of rontalizumab, a humanized IgG1 monoclonal antibody that also inhibits human IFNα [50]. These results con firmed the potential usefulness of using the type I interferon signature as a PD marker to evaluate activity of anti-IFNα therapy in SLE. Furthermore, these studies showed that expression of the type I interferon signature in WB reflects involved tissue in SLE, and raised the possibility of testing the type I interferon signature as a potential predictive biomarker to identify a subset of SLE patients who may preferentially respond to anti-IFNα treatment.
Abbreviations
- dsDNA:
-
double-stranded DNA
- ELISA:
-
enzy me-linked immunosorbent assays
- IFN:
-
interferon
- IL:
-
interleukin
- PCR:
-
polymerase chain reaction
- PD:
-
pharmacodynamic
- RT:
-
reverse transcriptase
- SLE:
-
systemic lupus erythematosus
- TNF:
-
tumor necrosis factor
- WB:
-
whole blood.
References
American College of Rheumatology Ad Hoc Committee on Systemic Lupus Erythematosus Guidelines. Guidelines for Referral and Management of systemic lupus erythematosus in adults. Arthritis Rheum. 1999, 42: 1785-1796. 10.1002/1529-0131(199909)42:9<1785::AID-ANR1>3.0.CO;2-#.
Petri M: Long-term outcomes in lupus. Am J Managed Care. 2001, 7: S480-S485.
Bengtsson AA, Sturfelt G, Truedsson L, Blomberg J, Alm G, Vallin H, Rönnblom L: Activation of type I interferon system in systemic lupus erythematosus correlates with disease activity but not with antiretroviral antibodies. Lupus. 2000, 9: 664-671. 10.1191/096120300674499064.
Rönnblom L, Alm GV: Systemic lupus erythematosus and the type I interferon system. Arthritis Res Ther. 2003, 5: 68-75. 10.1186/ar625.
Banchereau J, Pascual V, Palucka AK: Autoimmunity through cytokine-induced dendritic cell activation. Immunity. 2004, 20: 539-550. 10.1016/S1074-7613(04)00108-6.
Baechler EC, Gregersen PK, Behrens TW: The emerging role of interferon in human systemic lupus erythematosus. Curr Opin Immunol. 2004, 16: 801-807. 10.1016/j.coi.2004.09.014.
Dall'era MC, Cardarelli PM, Preston BT, Witte A, Davis JC: Type I interferon correlates with clinical and serologic manifestations of systemic lupus erythematosus. Ann Rheum Dis. 2005, 64: 1692-1697. 10.1136/ard.2004.033753.
Crow M: Interferon-α: a new target for therapy in systemic lupus erythematosus?. Arthritis Rheum. 2003, 48: 2396-2401. 10.1002/art.11226.
Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, Harley JB: Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003, 349: 1526-1533. 10.1056/NEJMoa021933.
Pascual V, Farkas L, Banchereau J: Systemic lupus erythematosus: all roads lead to type I interferons. Curr Opin Immunol. 2006, 18: 676-682. 10.1016/j.coi.2006.09.014.
Rönnblom L, Pascual V: The innate immune system in SLE: type I interferons and dendritic cells. Lupus. 2008, 17: 394-399. 10.1177/0961203308090020.
Theofilopoulos AN, Baccala R, Beutler B, Kono DH: Type I Interferons (a/b) in immunity and autoimmunity. Annu Rev Immunol. 2005, 23: 307-336. 10.1146/annurev.immunol.23.021704.115843.
Mailliard RB, Son YI, Redlinger R, Coates PT, Giermasz A, Morel PA, Storkus WJ, Kalinski P: Dendritic cells mediate NK cell help for Th1 and CTL responses: two-signal requirement for the induction of NK cell helper function. J Immunol. 2003, 171: 2366-2373.
Biron CA: Interferons α and β as immune regulators - a new look. Immunity. 2001, 14: 661-664. 10.1016/S1074-7613(01)00154-6.
Bussfeld D, Nain M, Hoffman P, Gemsa D, Sprenger H: Selective induction of the monocyte-attracting chemokines MCP-1 and IP-10 in vesicular stomatitis virus-infected human monocytes. J Interferon Cytokine Res. 2000, 20: 615-621. 10.1089/107999000414781.
Pogue SL, Preston BT, Stalder J, Bebbington CR, Cardarelli PM: The receptor for type I IFNs is highly expressed on peripheral blood B cells and monocytes and mediates a distinct profile of differentiation and activation of these cells. J Interferon Cytokine Res. 2004, 24: 131-139. 10.1089/107999004322813372.
Salazar-Mather TP, Hokeness KL: Calling in the troops: regulation of inflammatory cell trafficking through innate cytokine/chemokine networks. Viral Immunol. 2003, 16: 291-306. 10.1089/088282403322396109.
Criswell LA: The genetic contribution to systemic lupus erythematosus. Bull NYU Hosp Jt Dis. 2008, 66: 176-183.
Sigurdsson S, Göring HH, Kristjansdottir G, Milani L, Nordmark G, Sandling JK, Eloranta ML, Feng D, Sangster-Guity N, Gunnarsson I, Svenungsson E, Sturfelt G, Jönsen A, Truedsson L, Barnes BJ, Alm G, Rönnblom L, Syvänen AC: Comprehensive evaluation of the genetic variants of interferon regulatory factor 5 (IRF5) reveals a novel 5 bp length polymorphism as strong risk factor for systemic lupus erythematosus. Hum Mol Genet. 2008, 17: 872-881. 10.1093/hmg/ddm359.
Niewold TB, Swedler WI: Systemic lupus erythematosus arising during interferon-alpha therapy for cryoglobulinemic vasculitis associated with hepatitis C. Clin Rheumatol. 2005, 24: 178-181. 10.1007/s10067-004-1024-2.
Ioannou Y, Isenberg DA: Current evidence for the induction of autoimmune rheumatic manifestations by cytokine therapy. Arthritis Rheum. 2000, 43: 1431-1442. 10.1002/1529-0131(200007)43:7<1431::AID-ANR3>3.0.CO;2-E.
Mathian A, Weinberg A, Gallegos M, Banchereau J, Koutouzov F: IFNα induces early lethal lupus in preautoimmune (New Zealand Black × New Zealand White) F1 but not in BALB/c mice. J Immunol. 2005, 174: 2499-2506.
Santiago-Raber ML, Baccala R, Haraldsson KM, Choubey D, Stewart TA, Kono DH, Theofilopoulis AN: Type I interferon receptor deficiency reduces lupuslike disease in NZB mice. J Exp Med. 2003, 197: 777-788. 10.1084/jem.20021996.
Zagury D, Le Buanec H, Mathian A, Larcier P, Burnett R, Amoura Z, Emilie D, Peltre G, Bensussan A, Bizzini B, Gallo RC, Koutouzov S: IFNα kinoid vaccine-induced neutralizing antibodies prevent clinical manifestations in a lupus flare murine model. Proc Natl Acad Sci USA. 2009, 106: 5294-5299. 10.1073/pnas.0900615106.
Sotiriou C, Pusztai L: Gene-expression signatures in breast cancer. N Engl J Med. 2009, 360: 790-800. 10.1056/NEJMra0801289.
Siena S, Sartore-Bianchi A, Di Nicolantonio F, Balfour J, Bardelli A: Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J Natl Cancer Inst. 2009, 101: 1308-1324. 10.1093/jnci/djp280.
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR: MicroRNA expression profiles classify human cancers. Nature. 2005, 435: 834-838. 10.1038/nature03702.
Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, Baehner FL, Walker MG, Watson D, Park T, Hiller W, Fisher ER, Wickerham DL, Bryant J, Wolmark N: A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004, 351: 2817-2826. 10.1056/NEJMoa041588.
Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, Behrens TW: Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. 2003, 100: 2610-2615. 10.1073/pnas.0337679100.
Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V: Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003, 197: 711-723. 10.1084/jem.20021553.
Han GM, Chen SL, Shen N, Ye S, Bao CD, Gu YY: Analysis of gene expression profiles in human systemic lupus erythematosus using oligonucleotide microarray. Genes Immun. 2003, 4: 177-186. 10.1038/sj.gene.6363966.
Kirou KA, Lee C, George S, Louca K, Papagiannis IG, Peterson MG, Ly N, Woodward RN, Fry KE, Lau AY, Prentice JG, Wohlgemuth JG, Crow MK: Coordinate overexpression of interferon-alpha-induced genes in systemic lupus erythematosus. Arthritis Rheum. 2004, 50: 3958-3967. 10.1002/art.20798.
Kirou KA, Lee C, George S, Louca K, Peterson MG, Crow MK: Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. 2005, 52: 1491-1503. 10.1002/art.21031.
Feng X, Wu H, Grossman JM, Hanvivadhanakul P, FitzGerald JD, Park GS, Dong X, Chen W, Kim MH, Weng HH, Furst DE, Gorn A, McMahon M, Taylor M, Brahn E, Hahn BH, Tsao BP: Association of increased interferon-inducible gene expression with disease activity and lupus nephritis in patients with systemic lupus erythematosus. Arthritis Rheum. 2006, 54: 2951-2962. 10.1002/art.22044.
Yao Y, Higgs BW, Morehouse C, de los Reyes M, Brohawn P, Trigona W, White W, Zhang J, White B, Coyle AJ, Kiener PA, Jallal B: Development of potential pharmacodynamic and diagnostic markers for anti-IFN-α monoclonal antibody trials in systemic lupus erythematosus. Human Genomics Proteomics. 2008, 2009: article 374312-doi:10.4061/2009/374312.
Blomberg S, Eloranta ML, Cederblad B, Nordlin K, Alm GV, Rönnblom L: Presence of cutaneous interferon-alpha producing cells in patients with systemic lupus erythematosus. Lupus. 2001, 10: 484-490. 10.1191/096120301678416042.
Farkas L, Beiske K, Lund-Johansen F, Brandtzaeg P, Jahnsen FL: Plasmacytoid dendritic cells (natural interferon-alpha/beta-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol. 2001, 159: 237-243.
Yao Y, Richman L, Higgs BW, Morehouse CA, de los Reyes M, Brohawn P, Zhang J, White B, Coyle AJ, Kiener PA, Jallal B: Neutralization of IFN-α/β-inducible genes and downstream effect in a phase i trial of an anti-IFN-α monoclonal antibody in SLE. Arthritis Rheum. 2009, 60: 1785-1796. 10.1002/art.24557.
Okamoto H, Katsumata Y, Nishimura K, Kamatani N: Interferon-inducible protein 10/CXCL10 is increased in cerebrospinal fluid of patients with central nervous system lupus. Arthritis Rheum. 2004, 50: 3731-3732. 10.1002/art.20598.
Nzeusseu Toukap A, Galant C, Theate I, Maudoux AL, Lories RJ, Houssiau FA, Lauwerys BR: Identification of distinct gene expression profiles in the synovium of patients with systemic lupus erythematosus. Arthritis Rheum. 2007, 56: 1579-1588. 10.1002/art.22578.
Hua J, Kirou K, Lee C, Crow MK: Functional assay of type I interferon in systemic lupus erythematosus plasma and association with anti-RNA binding protein autoantibodies. Arthritis Rheum. 2006, 54: 1906-1916. 10.1002/art.21890.
Walsh RJ, Kong SW, Yao Y, Jallal B, Kiener PA, Pinkus JL, Beggs AH, Amato AA, Greenberg SA: Type I interferon-inducible gene expression in blood is present and reflects disease activity in dermatomyositis and polymyositis. Arthritis Rheum. 2007, 56: 3784-3792. 10.1002/art.22928.
Kraan van der Pouw TC, Wijbrandts CA, van Baarsen LG, Voskuyl AE, Rustenburg F, Baggen JM, Ibrahim SM, Fero M, Dijkmans BA, Tak PP, Verweij CL: Rheumatoid arthritis subtypes identified by genomic profiling of peripheral blood cells: assignment of a type I interferon signature in a subpopulation of patients. Ann Rheum Dis. 2007, 66: 1008-1014. 10.1136/ard.2006.063412.
Båve U, Nordmark G, Lövgren T, Rönnelid J, Cajander S, Eloranta ML, Alm GV, Rönnblom L: Activation of the type I interferon system in primary Sjogren's syndrome: a possible etiopathogenic mechanism. Arthritis Rheum. 2005, 52: 1185-1195. 10.1002/art.20998.
Zheng L, Zhang Z, Yu C, Tu L, Zhong L, Yang C: Association between IFN-alpha and primary Sjogren's syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009, 107: e12-e18. 10.1016/j.tripleo.2008.09.015.
Duan H, Fleming J, Pritchard DK, Amon LM, Xue J, Arnett HA, Chen G, Breen P, Buckner JH, Molitor JA, Elkon KB, Schwartz SM: Combined analysis of monocyte and lymphocyte messenger RNA expression with serum protein profiles in patients with scleroderma. Arthritis Rheum. 2008, 58: 1465-1474. 10.1002/art.23451.
Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanoudou D, Tawil R, Barohn RJ, Saperstein DS, Briemberg HR, Ericsson M, Park P, Amato AA: Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol. 2005, 57: 664-678. 10.1002/ana.20464.
Whitfield ML, Finlay DR, Murray JI, Troyanskaya OG, Chi JT, Pergamenschikov A, McCalmont TH, Brown PO, Botstein D, Connolly MK: Systemic and cell type-specific gene expression patterns in scleroderma skin. Proc Natl Acad Sci USA. 2003, 100: 12319-12324. 10.1073/pnas.1635114100.
Wallace DJ, Petri M, Olsen N, Kirou K, Dennis G, Yao Y, Jallal B, Coyle A, Zeng L: MEDI-545, an anti-interferon alpha monoclonal antibody, shows evidence of clinical activity in systemic lupus erythematous [abstract 1325]. Arthritis Rheum. 2007, 56: S526-S527.
McBride JM, Wallace DJ, Yao Z, Morimoto A, Jiang J, Maciuca R, McLean I, Drappa J: Dose-dependent modulation of interferon regulated genes with administration of single and repeat doses of Rontalizumab in a phase I, placebo controlled, double blind, dose escalation study in SLE [abstract 2072]. Arthritis Rheum. 2009, 60: S775-S776.
Acknowledgements
The authors would like to thank Chris Morehouse, Philip Brohawn and Wei Zhu for providing technical assistance, and Wendy White and Jiaqi Huang for critical review of the manuscript.
This article is part of Arthritis Research & Therapy Volume 12 Supplement 1: The role of IFN alpha in autoimmune disease. The full contents of the supplement are available online at http://arthritis-research.com/supplements/12/S1. Publication of the supplement has been supported with funding from MedImmune, LLC.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
All authors are employees of MedImmune.
Rights and permissions
About this article
Cite this article
Yao, Y., Higgs, B.W., Richman, L. et al. Use of type I interferon-inducible mRNAs as pharmacodynamic markers and potential diagnostic markers in trials with sifalimumab, an anti-IFNα antibody, in systemic lupus erythematosus. Arthritis Res Ther 12 (Suppl 1), S6 (2010). https://doi.org/10.1186/ar2887
Published:
DOI: https://doi.org/10.1186/ar2887