
Abstract
Patients with systemic lupus erythematosus (SLE) have ongoing interferon-α (IFN-α) production and serum IFN-α levels are correlated with both disease activity and severity. Recent studies of patients with SLE have demonstrated the presence of endogenous IFN-α inducers in such individuals, consisting of small immune complexes (ICs) containing IgG and DNA. These ICs act specifically on natural IFN-α-producing cells (NIPCs), often termed plasmacytoid dendritic cells (PDCs). Given the fact that the NIPC/PDC has a key role in both the innate and adaptive immune response, as well as the many immunoregulatory effects of IFN-α, these observations might be important for the understanding of the etiopathogenesis of SLE. In this review we briefly describe the biology of the type I IFN system, with emphasis on inducers, producing cells (especially NIPCs/PDCs), IFN-α actions and target immune cells that might be relevant in SLE. On the basis of this information and results from studies in SLE patients, we propose a hypothesis that explains how NIPCs/PDCs become activated and have a pivotal etiopathogenic role in SLE. This hypothesis also indicates new therapeutic targets in this autoimmune disease.
Similar content being viewed by others


Introduction
Systemic lupus erythematosus (SLE) is a genetically complex autoimmune disease, characterized by the occurrence of many different autoantibodies, the formation of immune complexes (ICs), and inflammation in different organs. Studies in both mice and humans have demonstrated several genetic susceptibility loci involved in immune activation and regulation, as well as clearance of apoptotic cells [1, 2]. Among the cells in the immune system, the B cells have a crucial role as producers of the autoantibodies, which are typically directed to nucleic acid and associated proteins. The B cells in SLE patients have several abnormalities that might account for the ongoing autoantibody production observed in these patients [3]. The B cell response is clearly antigen-driven and several lupus autoantigens are located in apoptotic bodies and apoptotic blebs [4, 5]. It is unknown why the immune response is directed mainly towards apoptotic cell material, but SLE patients have both increased apoptosis and a defective clearance of such material [6, 7]. Consequently, apoptotic bodies and nucleosomes are accessible to the immune system in SLE patients for longer than in normal individuals, which might contribute to the autoimmune response [8]. In addition, abnormal T cell activation, complement deficiency and the production of several cytokines might be critical for the initiation and maintenance of the autoimmune reaction [9–12].
Increased serum levels of many cytokines have been noted in SLE patients, reflecting the activation of the immune system and inflammation in this disease. In the present review we focus on the type I interferon (IFN) system in SLE, because emerging data suggest that IFN-α and the natural IFN-α-producing cells (NIPCs), often termed plasmacytoid dendritic cells (PDCs), have a pivotal role in the etiopathogenesis of SLE.
IFN-α and SLE
Raised serum levels of IFN-α in SLE patients have been noted for more than 20 years [13], and these levels are correlated with both disease activity and severity [14]. There is also a significant association between IFN-α levels and several markers of immune activation that are considered to be of fundamental importance in the disease process, such as circulating interleukin-10 (IL-10), complement activation and anti-double-stranded DNA (dsDNA) antibody titers [14]. Among SLE symptoms, there is a clear association between high serum IFN-α levels and fever as well as skin rashes [14]. It is also of interest that several signs and symptoms in SLE mimic those in influenza or during IFN-α therapy, for instance fever, fatigue, myalgia, arthralgia, and leukopenia. SLE patients without measurable serum IFN-α levels also seem to have a pathological IFN-α production, because their blood leukocytes display increased amounts of the IFN-α-inducible protein MxA [15]. Interestingly, gene array expression profiles of blood cells from SLE patients recently demonstrated a clear activation of IFN-α-regulated genes [16, 17].
A causative role for IFN-α in the initiation of the autoimmune disease process is suggested more directly by the observation that patients with non-autoimmune disorders who are treated with IFN-α can develop antinuclear antibodies, anti-dsDNA antibodies, and occasionally also SLE [18, 19]. Such observations obviously further raise the question of whether the type I IFN system could be involved in the etiopathogenesis of naturally occurring SLE.
The type I IFN system
The type I IFN system comprises the inducers of type I IFN synthesis, the type I IFN genes and proteins, the cells producing type I IFNs, and the target cells affected by the IFNs. The human type I IFN gene family contains a total of 15 functional genes, 13 encoding IFN-α subtypes and one each for IFN-β and -ω [20]. The genes and their products have several common features in structure and function; for example, the type I IFNs are typically induced by virus or dsRNA and interact with the same receptor, the IFN-α/β receptor (IFNAR) [21]. However, there are also clear differences between, for example, IFN-α and IFN-β at the post-IFNAR level [22]. The type I IFNs are produced by many cell types exposed to certain RNA viruses and dsRNA in vitro. In contrast, human leukocytes can produce IFN-α when exposed to a much wider variety of agents, including viruses, bacteria, protozoa, and certain cell lines [23].
The major IFN-α-producing cells (IPCs) were early on designated NIPCs, and several studies of these cells (reviewed in [23]) suggested that NIPCs were either a unique new hemopoietic cell population or dendritic cells (DCs). When the presence of several surface markers on NIPCs was investigated directly, it was found that they expressed CD4, CD36, CD40, CD44, CD45RA, and CD83, for example, but lacked CD80, CD86, and CD11c [24], thus presenting a phenotype similar to a previously identified DC precursor [25]. These DCs were later characterized further and are now also referred to as PDCs or precursors of type 2 DCs [26–28]. They have a high expression of the IL-3 receptor (CD123) [26] and were recently found to express two unique markers termed blood dendritic cell antigens; BDCA-2 and BDCA-4 [29]. The BDCA-2 molecule represents a novel endocytic type II C-type lectin, which might function as an antigen-capturing molecule.
The NIPCs/PDCs also express the Toll-like receptors (TLRs) 1, 6, 7, 9, and 10 [30], of which TLR9 is obviously crucial for activation of the cells by CpG-containing DNA motifs [31]. Although the NIPC/PDC population constitutes only about 0.1% of the peripheral blood mononuclear cells (PBMCs), each cell has the capacity to produce as many as 109 IFN-α molecules in 12 hours. The type I IFNs have mainly been regarded as antiviral proteins, because they are produced during viral infections and induce viral resistance in target cells. However, these IFNs also exert prominent immunoregulatory effects and might act as key cytokines, not only in the innate immune system but also in adaptive immune responses.
Immunomodulatory effects by type I IFNs
Type I IFNs have a large number of different effects on the immune system and most of these promote a strong immune response (reviewed in [32–34], for example). Several of these effects are highly relevant for the understanding of observed alterations of the immune system in SLE patients. Thus, IFN-α caused a stimulation of T helper type 1 (Th1)-type T cell and B cell responses, stimulation of CTL responses, proliferation of memory CD8+ T cells, and differentiation and increased antigen-presenting activity of type 1 DCs. It was shown, for instance, that type I IFN in mice was a potent enhancer of the primary antibody response to a soluble antigen; all subclasses were stimulated, with both long-lasting antibody responses and development of memory [35]. Part of this effect could be through effects on DCs [35], and autoantigen-loaded DCs might in fact precipitate autoimmune diseases [36]. Several other effects of type I IFN can be relevant in promotion of autoimmunity by IFN-α, such as stimulation of differentiation T cells, inhibition of apoptosis associated with activation, and induction of Fas-ligand-mediated apoptosis [34, 37, 38]. Type I IFN can also promote the survival and differentiation of B cells and enhance B cell antigen receptor (BCR)-dependent responses by lowering their threshold of activation [39, 40].
Relevant in the SLE context is also the observation that DCs activated by IFN-α can induce CD40-independent immunoglobulin class switching in B cells through the upregulation of BLys and APRIL ('a proliferation-inducing ligand') [41]. In addition, IFN-α-activated monocytes in SLE patients can act as antigen-presenting cells (APCs) [42]. Although most of these immunostimulatory activities of IFN-α remain to be demonstrated in humans in vivo, they suggest that IFN-α can have an important role in autoimmune processes. Clearly, other cytokines produced by NIPCs/PDCs, such as IL-12, as well as cytokines induced secondarily by IFN-α, such as IL-15 [43], can also be important.
The type I IFN system in SLE patients
Patients with SLE have a more than 70-fold decrease in the number of NIPCs/PDCs in blood [44], a finding recently confirmed in pediatric SLE patients [42]. However, the residual NIPCs/PDCs are functionally normal with the capacity to produce 5-10 pg of IFN-α per cell after activation. Furthermore, exposure of SLE-PBMCs to IFN-α/γ and granulocyte/macrophage colony-stimulating factor (GM-CSF) in vitro markedly increased the number of NIPCs/PDCs, further arguing against a NIPC/PDC defect in SLE. Instead, the smaller number of circulating NIPCs/PDCs in SLE might be caused by recruitment of these cells to tissues; this premise was supported by the finding of cells actively producing IFN-α in skin biopsies from SLE patients [45]. Furthermore, cells with typical NIPC/PDC phenotype have recently been identified in cutaneous lupus erythematosus lesions [46].
The NIPCs/PDCs do not normally produce measurable amounts of IFN-α unless stimulated by microorganisms or their constituents [23]. However, we made the interesting initial observation that several serum samples from SLE patients caused the production of IFN-α by PBMCs in vitro when used as culture medium supplement [44]. These results prompted a further investigation of this potential endogenous IFN-α-inducing factor in SLE (SLE-IIF), and it was shown to consist of small ICs (size 300–1000 kDa) that contained, as essential components, DNA and IgG with anti-DNA specificity [47]. In some patients with active disease, high levels of SLE-IIF were seen with the same IFN-α-inducing capacity in vitro as herpes simplex virus. SLE-IIF was mimicked by human anti-dsDNA monoclonal or polyclonal antibodies from SLE patients combined with plasmid DNA [48], and furthermore specifically activated NIPCs/PDCs to IFN-α synthesis. However, methylation of the CpG dinucleotides in the plasmid DNA totally inhibited the IFN-α production. This indicates that unmethylated CpG-containing DNA might be involved in triggering the IFN-α production in NIPCs/PDCs. However, using oligodeoxyribonucleotide (ODN) sequences originally cloned from SLE serum we could demonstrate that unmethylated CpG motifs are not obligatory for interferogenic activity and that DNA sequences with the capacity to induce IFN-α production therefore should be common in eukaryotic genomes [49].
Apoptotic cells are one obvious source of interferogenic DNA motifs, and recently we showed that all investigated cell lines formed IFN-α-inducing complexes when triggered to apoptosis and combined with IgG prepared from SLE sera [50]. In this experimental system, the specificity of the SLE autoantibodies was associated with the occurrence of antibodies against ribonucleoprotein in the SLE serum. The results therefore suggest that, in addition, RNA in ICs can act as an IFN-α inducer, and further characterization of the interferogenic material released by apoptotic cells revealed that although it is mainly sensitive to ribonuclease treatment, a significant portion is also destroyed by deoxyribonuclease [51]. Consequently, we propose that there exist two different IFN-α inducers in SLE, one being complexes between DNA and anti-DNA antibodies, and the other being complexes of RNA and anti-ribonucleoprotein/RNA antibodies, the latter being present mainly at the tissue level and not in blood. The failure to find RNA-containing IFN-α inducers in SLE blood might simply be due to rapid degradation by ribonucleases. Clearly, the precise composition of the IFN-inducing complexes remains to be determined.
Obviously, the nucleic acid might be associated with binding proteins, such as histones for DNA, as well as with SS-A/Ro, SS-B/La, and Sm for RNA. It is relevant here that autoantibodies against such proteins are common in SLE and it is well known that the removal of immune complexes is deficient in SLE [52]. In addition, the clearance of apoptotic cells by macrophages is deficient and could be linked to increased apoptosis [6, 7]. Such defects will increase IC levels, resulting in NIPC/PDC activation and IFN-α production.
Activation and regulation of NIPCs/PDCs in SLE
The NIPCs/PDCs express molecules that can detect danger signals and foreign antigens. As mentioned, they express the pattern recognition molecule TLR9, which interacts with and mediates responses to unmethylated CpG-DNA [31]. The induction of IFN-α might also require TLR9, because a new highly efficient IFN-α-inducing ODN required unmethylated CpG [53]. Furthermore, the poor IFN-inducing ability of other potent immunostimulatory ODNs was strongly increased when PDCs were co-stimulated by CD40L [54]. Consequently, several different signals via cell-membrane molecules might be required to initiate IFN-α gene expression. It is here relevant that NIPCs/PDCs express FcγRIIa ([51]; U Båve, M Magnusson, M-L Eloranta, A Perers, GV Alm and L Rönnblom, unpublished work) and that the antibodies in SLE-IIF are essential for IFN-α production [48]. The direct involvement of FcRγII in the stimulation of NIPCs/PDCs by SLE-IIF [55], or by the combination of apoptotic cells and SLE autoantibodies (U Båve, M Magnusson, M-L Eloranta, A Perers, GV Alm and L Rönnblom, unpublished work), was shown by means of blocking anti-FcRγII antibodies.
It is known that FcγRII can provide intracellular signals and internalize ICs [56, 57] and such material might be targeted to cytoplasmic compartments [58]. Thus, internalization of the IFN-α inducer could be an essential step and there might exist intracellular recognition structures for nucleic acid (DNA and RNA) motifs. The exact recognition and activation mechanisms for the different IFN-α inducers in SLE patients are unclear at present, and other TLRs than TLR9 might be involved. Thus, NIPCs/PDCs also express TLR1, 6, 7, and 10 but not the dsRNA-binding receptor TLR3 [30], and ligation of TLR7 by the drug imiquimod can elicit IFN-α production [59]. Several intracellular pathways might therefore lead to IFN-α gene expression.
IFN-α/β gene transcription in NIPCs/PDCs were, early on, shown to be dependent on de novo protein synthesis [60], and the presence of cytokines such as type I IFN, IFN-γ, IL-3, and GM-CSF increased the IFN-α production caused by viral inducers [61]. In addition, the induction of IFN-α production triggered by SLE-IIF, or the combination of apoptotic cells and autoantibodies, was markedly dependent on priming with especially IFN-α/β [48, 62]. Such priming is important for the viral induction of many IFN-α genes and might involve an initial activation of some IFN-α or IFN-β gene expression because of activation of pre-existing transcription factors [63]. This IFN then causes the synthesis of further transcription factors, such as interferon regulatory factor-5 (IRF-5) and IRF-7, that become activated and promote the expression of a wider spectrum of IFN-α genes. It is not known whether a similar mechanism is necessary for the activation of IFN-α gene expression in NIPCs/PDCs by the endogenous SLE-related IFN-α inducers.
Certain cytokines have a negative impact on NIPCs/PDCs, and IL-10 has been shown to be a potent inhibitor of IFN-α production caused by different IFN-α inducers, such as virus, SLE-IIF and the combination of apoptotic cells with antibodies [48, 62, 64]. In addition, TNF-α inhibited the action of these inducers [62]; this observation is interesting because it can explain why a blockade of TNF-α by anti-TNF-α antibodies or soluble TNF-α receptors in human patients can result in autoimmune side effects, including SLE [65, 66]. We therefore propose that such side effects are due to an increased activity of NIPCs/PDCs, which promotes the development of autoimmunity.
Antigen presentation by NIPCs/PDCs and monocyte-derived DCs in SLE
The population of NIPCs/PDCs in blood is immature. These cells can differentiate into mature PDCs in vitro, with the ability to stimulate T cells [26, 67, 68]. Such PDCs were originally shown to promote Th2 development preferentially [69], but subsequent work has demonstrated that they can for instance stimulate the development of CD8+ T cells that produce IL-10 and IFN-γ and have suppressive activity [70]. Furthermore, the NIPCs/PDCs can drive a potent Th1 development when they are induced to produce IL-12 and IFN-α by CD40 ligation combined with stimulation by virus or CpG-DNA [54, 71]. The absence of such viral or bacterial stimulants might account for the lack of detectable IFN-α production in situ by the many PDCs found infiltrating the nasal mucosa in allergic rhinitis [72]. In contrast, the presence of endogenous IFN-α inducers explains the presence of activated NIPCs/PDCs and IFN-α production in SLE. However, the full extent of activation of NIPCs/PDCs in vivo must be elucidated. That includes the different cytokines that are produced, and also the production of IFN-α subtypes. Furthermore, it remains to be determined whether NIPCs/PDCs are efficient APCs in vivo and whether the actual IPCs can develop into APCs. However, IFN-α can stimulate the development of efficient type 1 DCs that promote Th1 development [35, 73]. In this way the IFN-α produced by NIPCs/PDCs can generally promote the presentation of antigens for T cells. Indeed, an increased proportion of functionally active monocyte-derived DCs has been noted in the blood of SLE patients, and in addition the IFN-α present in SLE serum can stimulate the development of monocytes to DCs in vitro [42].
The type I IFN system in the etiopathogenesis of SLE
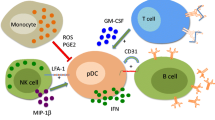
There are several intriguing observations on the type I IFN system that suggest a key role for NIPCs/PDCs, and the IFN-α that they produce, in the etiopathogenesis of SLE. They include the observed ability of IFN-α to cause autoimmunity (including SLE), evidence of ongoing IFN-α production in SLE, evidence that NIPCs/PDCs are the source of the IFN-α, the ability of SLE-derived ICs containing nucleic acid to induce IFN-α in NIPCs/PDCs, the finding that this nucleic acid can be generated from normal dying cells, and the special requirements to trigger IFN-α gene expression in NIPCs/PDCs. We have used this information to formulate a hypothesis about the role of the type I IFN system in the etiopathogenesis of SLE, which has been presented previously at various stages of refinement [47, 48, 74, 75]. This hypothesis is summarized in Fig. 1a, and more details are given in Fig. 1a,1b,1c,1d.

The central role of the type I interferon (IFN) system in the etiopathogenesis of systemic lupus erythematosus (SLE). (a) A schematic overview of IFN-α inducers and target cells. Initially, IFN-α is produced by the natural IFN-α producing cell (NIPC)/plasmacytoid dendritic cell (PDC) as a consequence of viral or bacterial infections. The IFN-α produced promotes DC1 development, T cell activation and autoantibody production by B cells. DNA or RNA and associated proteins, generated from apoptotic or necrotic cells, and autoantibodies form immune complexes (ICs) that act as endogenous IFN-α inducers and cause a prolonged IFN-α production. This IFN-α further stimulates the autoimmune response with more autoantibody production, IC formation, and co-stimulation of NIPCs/PDCs; finally, a vicious circle is created with an ongoing IFN-α production sustaining the autoimmune process. (b) Induction of IFN-α production in NIPCs/PDCs. Viruses, bacterial components, CpG-DNA and interferogenic ICs (IICs) can all trigger NIPCs/PDCs to produce IFN-α. FcγRIIa is necessary for the activation of NIPCs/PDCs by IICs. In addition, these cells express Toll-like receptor 9 (TLR9), mediating IFN-α synthesis induced by CpG-DNA, but the role of this receptor for the response to IICs is unknown. TLR7 activation by imiquimod also induces IFN-α production, but the function of TLR1, 6, and 10 in IFN-α production by NIPCs/PDCs is unknown. Ligation of CD40 enhances IFN-α synthesis and can also cause interleukin-12 (IL-12) production. In contrast, the ligation of blood dendritic cell antigen-2 (BDCA-2) by a monoclonal antibody inhibits the IFN-α production, but the natural ligand is unknown. IFNAR, IFN-α/β receptor. (c) Maturation of dendritic cells (DCs) and activation of T cells. The IFN-α produced induces the maturation of PDCs and the differentiation of monocytes to type 1 DCs; both cell types express the co-stimulatory molecules CD80 and CD86. These cells subsequently activate autoreactive T helper (Th) cells with specificity for processed antigens in IICs, for example. The cytokines IL-12 and IFN-α promote the Th1 response and prevent apoptosis in activated T cells. IL-12R, IL-12 receptor; TCR, T cell antigen receptor. (d) Production of autoantibodies by B cells. Activated Th cells provide help to B cells with reactivity to autoantigens in IICs, and these B cells are stimulated by IFN-α to prolonged survival and enhanced response to B cell antigen receptor (BCR) ligation. IFN-α also upregulates BLyS and APRIL ('a proliferation-inducing ligand') on DCs, which further promotes the B cell response and elicits CD40-independent Ig class switching and plasmacytoid differentiation. Autoantibody production is facilitated by the ability of DNA/RNA-containing autoantigens to activate B cells directly by simultaneous binding to BCR and TLR9. The autoantibodies produced bind to DNA and RNA and form more IICs, which trigger the continuous IFN-α production that is the fuel in the autoimmune process.
A critical first event in the autoimmune process is the formation of autoantibodies reactive with autoantigens that contain nucleic acid (RNA and DNA), because they form ICs that serve as endogenous IFN-α inducers. Such autoantibodies might be produced as a consequence of viral or bacterial infections inducing the synthesis of IFN-α and other adjuvant cytokines. The NIPCs/PDCs (Fig. 1b) are here a main producer of such cytokines, but other cells might also be involved, depending on the type of infection. Once interferogenic ICs (the endogenous IFN-α inducers) have formed, they replace the original exogenous bacterial/viral IFN-α inducers and continuously trigger IFN-α production in NIPCs/PDCs. The stimulatory effects of IFN-α on key cells in the immune system can counteract the maintenance of self-tolerance in several ways, as outlined above. The IFN-α produced triggers the maturation of DCs with the capacity to activate naive autoimmune T cells (Fig. 1c), although necrotic cells alone could have an adjuvant action on type 1 DCs [76]. These events are facilitated by the fact that antigen presentation and the production of cytokines such as type I IFNs occur in similar, if not identical, DCs. Activated T cells subsequently trigger the production of autoantibodies by B cells, an event promoted by IFN-α-induced upregulation of BLyS and APRIL on DCs [41] (Fig. 1c). In this context it is important to note that B cells can become stimulated by chromatin–IgG complexes by the dual engagement of IgM and TLR9 receptors [77]. This would be expected to favor the production of antibodies that can form immunostimulatory IFN-α-inducing immune complexes (Fig. 1c).
The endogenous IFN-α inducers are present for a prolonged time in SLE patients owing to impaired clearance [52], and the resulting IFN-α production sustains the autoimmune process, with the generation of more autoantibodies and IFN-α inducers. Increased apoptosis and deficient clearance of apoptotic material in SLE [7] can contribute by providing more autoantigens. In this way, a process resembling a vicious circle is established (Fig. 1a) that maintains the autoimmune process by continuously exposing the immune system to endogenous IFN-α inducers. Epitope spreading is expected to occur with time, involving the production of antibodies against autoantigens that are not associated with material containing nucleic acids.
The activity of this vicious circle in tissues can be augmented by several cytokines and chemokines that recruit new NIPCs/PDCs. The mechanisms for the migration of NIPCs/PDCs in vivo in SLE remain to be determined, for instance whether SDF-1 and PDC-expressed CXCR4 (chemokine [CXC motif], receptor 4) are important. Furthermore, the priming of NIPCs/PDCs by IFN-α and by IL-3 and GM-CSF is probably important for the activation of their IFN-α production. The formation of the endogenous IFN-α inducers is increased by production of more autoantibodies, and by exposure to ultraviolet light or infections that generate more apoptotic or necrotic material with IFN-α-inducing activity. Conversely, the activity of the disease process might be decreased by nucleases that degrade the IFN-α inducer [47], or by the scavenging of IC and apoptotic material by macrophages [7, 52]. The NIPC/PDC population might also be exhausted because these cells are infrequent and their production of IFN-α is transient [78]. Finally, some cytokines, especially IL-10 and TNF-α (see above), can inhibit the IFN-α production by NIPCs/PDCs and might therefore constitute a beneficial negative feedback mechanism in SLE. In SLE patients with a low production of IFN-α and low activity in the immune system, the vicious circle might be reactivated by, for example, infections that cause new IFN-α production. The activation of the autoimmune process by this IFN-α can explain relapses of SLE seen during infections.
Possible new therapeutic targets in SLE
Chloroquine is used both for therapy and to maintain remissions in SLE patients. This drug is known to inhibit IFN-α production by NIPCs/PDCs in vitro by the inhibition of endosomal acidification/maturation [23]. The proposed role of the type I IFN system in SLE suggests further therapeutic targets for the inhibition of the IFN-α production. For instance, the endogenous IFN-α inducers could be degraded by nucleases, or their activation of NIPCs/PDCs through the FcγRII could be blocked. The actions of the IFN-α produced could furthermore be inhibited by neutralizing anti-IFN-α antibodies [79], antibodies blocking the anti-IFNAR [80], or soluble IFNAR. It is also possible to target the NIPCs/PDCs and inhibit their production of IFN-α. Thus, antibodies binding the BDCA-2 molecules specifically expressed by PDCs abolished the IFN-α production triggered by SLE-related inducers [29]. Some of these approaches are being considered by the pharmaceutical industry and the results of future clinical trials will be of great interest because they can provide direct evidence for the relevance of the type I IFN system in SLE and other autoimmune diseases, and also provide more efficient therapy.
Conclusion
We have argued for a pivotal etiopathogenic role for the type I IFN system in SLE, in which endogenous inducers cause an ongoing production of IFN-α by NIPCs/PDCs. This IFN-α can promote the development of autoimmunity by multiple actions on cells of the immune system, causing autoimmune disease in genetically predisposed individuals. The endogenous IFN-α inducers contain nucleic acids (RNA or DNA) and probably also proteins, and originate from apoptotic or necrotic cells. They are present as complexes with autoantibodies. The activation of the type I IFN system can be maintained by a process resembling a vicious circle, in which the continuous generation of these endogenous IFN-α inducers is especially important. However, the activity of this vicious circle can be regulated in several ways. One important goal in the search for a better treatment of SLE is therefore to learn how this IFN-α production can be therapeutically controlled.
Abbreviations
- APC:
-
antigen-presenting cell
- APRIL:
-
a proliferation-inducing ligand
- BDCA:
-
blood dendritic cell antigen
- GM-CSF:
-
granulocyte/macrophage colony-stimulating factor
- IC:
-
immune complex
- IFN:
-
interferon
- IFNAR:
-
IFN-α/β receptor
- IL:
-
interleukin
- IRF:
-
interferon regulatory factor
- NIPC:
-
natural IFN-α-producing cell
- ODN:
-
oligodeoxyribonucleotide
- PBMC:
-
peripheral blood mononuclear cell
- PDC:
-
plasmacytoid dendritic cell
- SLE:
-
systemic lupus erythematosus
- SLE-IIF:
-
IFN-α-inducing factor in SLE
- Th1:
-
T helper type 1
- TLR:
-
Toll-like receptor.
References
Kelly JA, Moser KL, Harley JB: The genetics of systemic lupus erythematosus: putting the pieces together. Genes Immun. 2002, 3 (Suppl): S71-S85. 10.1038/sj.gene.6363885.
Tsao BP: An update on genetic studies of systemic lupus erythematosus. Curr Rheumatol Rep. 2002, 4: 359-367.
Grammer AC, Lipsky PE: CD154-CD40 interactions mediate differentiation to plasma cells in healthy individuals and persons with systemic lupus erythematosus. Arthritis Rheum. 2002, 46: 1417-1429. 10.1002/art.10287.
Casciola-Rosen LA, Anhalt G, Rosen A: Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994, 179: 1317-1330. 10.1084/jem.179.4.1317.
Cocca BA, Cline AM, Radic MZ: Blebs and apoptotic bodies are B cell autoantigens. J Immunol. 2002, 169: 159-166.
Grondal G, Traustadottir KH, Kristjansdottir H, Lundberg I, Klareskog L, Erlendsson K, Steinsson K: Increased T-lymphocyte apoptosis/necrosis and IL-10 producing cells in patients and their spouses in Icelandic systemic lupus erythematosus multicase families. Lupus. 2002, 11: 435-442. 10.1191/0961203302lu223oa.
Herrmann M, Voll RE, Kalden JR: Etiopathogenesis of systemic lupus erythematosus. Immunol Today. 2000, 21: 424-426. 10.1016/S0167-5699(00)01675-3.
Chernysheva AD, Kirou KA, Crow MK: T cell proliferation induced by autologous non-T cells is a response to apoptotic cells processed by dendritic cells. J Immunol. 2002, 169: 1241-1250.
Kammer GM, Perl A, Richardson BC, Tsokos GC: Abnormal T cell signal transduction in systemic lupus erythematosus. Arthritis Rheum. 2002, 46: 1139-1154. 10.1002/art.10192.
Walport MJ: Complement and systemic lupus erythematosus. Arthritis Res. 2002, 4 (Suppl): S279-S293. 10.1186/ar586.
Woo C, Kirou KA, Koshy M, Berger D, Crow MK: New pieces to the SLE cytokine puzzle. Arthritis Rheum. 1999, 42: 871-881. 10.1002/1529-0131(199905)42:5<871::AID-ANR5>3.0.CO;2-J.
Dean GS, Tyrrell-Price J, Crawley E, Isenberg DA: Cytokines and systemic lupus erythematosus. Ann Rheum Dis. 2000, 59: 243-251. 10.1136/ard.59.4.243.
Ytterberg SR, Schnitzer TJ: Serum interferon levels in patients with systemic lupus erythematosus. Arthritis Rheum. 1982, 25: 401-406.
Bengtsson A, Sturfelt G, Truedsson L, Blomberg J, Alm G, Vallin H, Rönnblom L: Activation of type I interferon system in systemic lupus erythematosus correlates with disease activity but not antiretroviral antibodies. Lupus. 2000, 9: 664-671. 10.1191/096120300674499064.
von Wussow P, Jakschies D, Hochkeppel H, Horisberger M, Hartung K, Deicher H: MX homologous protein in mononuclear cells from patients with systemic lupus erythematosus. Arthritis Rheum. 1989, 32: 914-918.
Crow MK, George S, Paget SA, Ly N, Woodward R, Fry K, Chan A, Prentice J, Wohlgemuth J: Expression of an interferon-alpha gene program in SLE. Arthritis Rheum. 2002, 46 (Suppl): S281.
Baechler EC, Batliwall FM, Karypis G, Gaffney P, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, Behrens TW: Interferon-inducible gene expression signature in peripheral blood cells of patients with severe SLE. Arthritis Rheum. 2002, 46 (Suppl): S281.
Rönnblom LE, Alm GV, Öberg KE: Possible induction of systemic lupus erythematosus by interferon-α treatment in a patient with a malignant carcinoid tumour. J Intern Med. 1990, 227: 207-210.
Ioannou Y, Isenberg DA: Current evidence for the induction of autoimmune rheumatic manifestations by cytokine therapy. Arthritis Rheum. 2000, 43: 1431-1442. 10.1002/1529-0131(200007)43:7<1431::AID-ANR3>3.0.CO;2-E.
Díaz MO: The human type I interferon gene cluster. Semin Virol. 1995, 6: 143-149. 10.1006/smvy.1995.0019.
Mogensen KE, Lewerenz M, Reboul J, Lutfalla G, Uze G: The type I interferon receptor: structure, function, and evolution of a family business. J Interferon Cytokine Res. 1999, 19: 1069-1098. 10.1089/107999099313019.
Doly J, Civas A, Navarro S, Uze G: Type I interferons: expression and signalization. Cell Mol Life Sci. 1998, 54: 1109-1121. 10.1007/s000180050240.
Fitzgerald-Bocarsly P: Human natural interferon-α producing cells. Pharmac Ther. 1993, 60: 39-62. 10.1016/0163-7258(93)90021-5.
Svensson H, Johannisson A, Nikkilä T, Alm GV, Cederblad B: The cell surface phenotype of human natural interferon-α producing cells as determined by flow cytometry. Scand J Immunol. 1996, 44: 164-172. 10.1046/j.1365-3083.1996.d01-289.x.
O'Doherty U, Peng M, Gezelter S, Swiggard WJ, Betjes M, Bhardwaj N, Steinman RM: Human blood contains two subsets of dendritic cells, one immunologically mature and the other immature. Immunology. 1994, 82: 487-493.
Olweus J, BitMansour A, Warnke R, Thompson PA, Carballido J, Picker LJ, Lund-Johansen F: Dendritic cell ontogeny: a human dendritic cell lineage of myeloid origin. Proc Natl Acad Sci USA. 1997, 94: 12551-12556. 10.1073/pnas.94.23.12551.
Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, Antonenko S, Liu YJ: The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999, 284: 1835-1837. 10.1126/science.284.5421.1835.
Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, Colonna M: Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med. 1999, 5: 919-923. 10.1038/11360.
Dzionek A, Sohma Y, Nagafune J, Cella M, Colonna M, Facchetti F, Gunther G, Johnston I, Lanzavecchia A, Nagasaka T, Okada T, Vermi W, Winkels G, Yamamoto T, Zysk M, Yamaguchi Y, Scmitz J: BDCA-2, a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon-α/β induction. J Exp Med. 2001, 194: 1823-1834. 10.1084/jem.194.12.1823.
Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, Endres S, Hartmann G: Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002, 168: 4531-4537.
Wagner H: Interactions between bacterial CpG-DNA and TLR9 bridge innate and adaptive immunity. Curr Opin Microbiol. 2002, 5: 62-69. 10.1016/S1369-5274(02)00287-4.
Biron CA: Interferons α and β as immune regulators – a new look. Immunity. 2001, 14: 661-664. 10.1016/S1074-7613(01)00154-6.
Belardelli F, Ferrantini M: Cytokines as a link between innate and adaptive antitumor immunity. Trends Immunol. 2002, 23: 201-208. 10.1016/S1471-4906(02)02195-6.
Akbar AN, Lord JM, Salmon M: IFN-α and IFN-β: a link between immune memory and chronic inflammation. Immunol Today. 2000, 21: 337-342. 10.1016/S0167-5699(00)01652-2.
Le Bon A, Schiavoni G, D'Agostino G, Gresser I, Belardelli F, Tough DF: Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity. 2001, 14: 461-470. 10.1016/S1074-7613(01)00126-1.
Ludewig B, Junt T, Hengartner H, Zinkernagel RM: Dendritic cells in autoimmune diseases. Curr Opin Immunol. 2001, 13: 657-662. 10.1016/S0952-7915(01)00275-8.
Marrack PK, J Mitchell T: Type I interferons keep activated T cells alive. J Exp Med. 1999, 189: 521-529. 10.1084/jem.189.3.521.
Kirou KA, Vakkalanka RK, Butler MJ, Crow MK: Induction of Fas ligand-mediated apoptosis by interferon-α. Clin Immunol. 2000, 95: 218-226. 10.1006/clim.2000.4866.
Ruuth K, Carlsson L, Hallberg B, Lundgren E: Interferon-α promotes survival of human primary B-lymphocytes via phosphatidylinositol 3-kinase. Biochem Biophys Res Commun. 2001, 284: 583-586. 10.1006/bbrc.2001.5025.
Braun D, Caramalho I, Demengeot J: IFN-α/β enhances BCR-dependent B cell responses. Int Immunol. 2002, 14: 411-419. 10.1093/intimm/14.4.411.
Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, Cerutti A: DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol. 2002, 3: 822-829. 10.1038/ni829.
Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J: Induction of dendritic cell differentiation by IFN-α in systemic lupus erythematosus. Science. 2001, 294: 1540-1543. 10.1126/science.1064890.
Mattei F, Schiavoni G, Belardelli F, Tough DF: IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J Immunol. 2001, 167: 1179-1187.
Cederblad B, Blomberg S, Vallin H, Perers A, Alm GV, Ronnblom L: Patients with systemic lupus erythematosus have reduced numbers of circulating natural interferon-α-producing cells. J Autoimmun. 1998, 11: 465-470. 10.1006/jaut.1998.0215.
Blomberg S, Eloranta ML, Cederblad B, Nordlind K, Alm GV, Rönnblom L: Presence of cutaneous interferon-α producing cells in patients with systemic lupus erythematosus. Lupus. 2001, 10: 484-490. 10.1191/096120301678416042.
Farkas L, Beiske K, Lund-Johansen F, Brandtzaeg P, Jahnsen FL: Plasmacytoid dendritic cells (natural interferon-α/β-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol. 2001, 159: 237-243.
Vallin H, Blomberg S, Alm GV, Cederblad B, Rönnblom L: Patients with systemic lupus erythematosus (SLE) have a circulating inducer of interferon-alpha (IFN-α) production acting on leucocytes resembling immature dendritic cells. Clin Exp Immunol. 1999, 115: 196-202. 10.1046/j.1365-2249.1999.00772.x.
Vallin H, Perers A, Alm GV, Ronnblom L: Anti-double-stranded DNA antibodies and immunostimulatory plasmid DNA in combination mimic the endogenous IFN-α inducer in systemic lupus erythematosus. J Immunol. 1999, 163: 6306-6313.
Magnusson M, Magnusson S, Vallin H, Rönnblom L, Alm GV: Importance of CpG dinucleotides in activation of natural interferon-α producing cells by a lupus-related oligodeoxynucleotide. Scand J Immunol. 2001, 54: 543-550. 10.1046/j.1365-3083.2001.01018.x.
Båve U, Alm GV, Rönnblom L: The combination of apoptotic U937 cells and lupus IgG is a potent IFN-α inducer. J Immunol. 2000, 165: 3519-3526.
Rönnblom LE, Båve U, Lövgren T, Eloranta ML, Alm GV: Complexes of SLE-IgG and nucleic acid from apoptotic or necrotic cells trigger IFN-alpha production by plasmacytoid dendritic cells (PDC) in a CD32 dependent fashion. Arthritis Rheum. 2002, 46 (Suppl): S131.
Davies KA, Robson MG, Peters AM, Norsworthy P, Nash JT, Walport MJ: Defective Fc-dependent processing of immune complexes in patients with systemic lupus erythematosus. Arthritis Rheum. 2002, 46: 1028-1038. 10.1002/art.10189.
Krug A, Rothenfusser S, Hornung V, Jahrsdorfer B, Blackwell S, Ballas ZK, Endres S, Krieg AM, Hartmann G: Identification of CpG oligonucleotide sequences with high induction of IFN-α/β in plasmacytoid dendritic cells. Eur J Immunol. 2001, 31: 2154-2163. 10.1002/1521-4141(200107)31:7<2154::AID-IMMU2154>3.0.CO;2-U.
Krug A, Towarowski A, Britsch S, Rothenfusser S, Hornung V, Bals R, Giese T, Engelmann H, Endres S, Krieg AM, Hartmann G: Toll-like receptor expression reveals CpG DNA as a unique microbial stimulus for plasmacytoid dendritic cells which synergizes with CD40 ligand to induce high amounts of IL-12. Eur J Immunol. 2001, 31: 3026-3037. 10.1002/1521-4141(2001010)31:10<3026::AID-IMMU3026>3.0.CO;2-H.
Batteux F, Palmer P, Daeron M, Weill B, Lebon P: FCγRII (CD32)-dependent induction of interferon-alpha by serum from patients with lupus erythematosus. Eur Cytokine Netw. 1999, 10: 509-514.
Ravetch JV, Bolland S: IgG Fc receptors. Annu Rev Immunol. 2001, 19: 275-290. 10.1146/annurev.immunol.19.1.275.
Dijstelbloem HM, van de Winkel JG, Kallenberg CG: Inflammation in autoimmunity: receptors for IgG revisited. Trends Immunol. 2001, 22: 510-516. 10.1016/S1471-4906(01)02014-2.
Dhodapkar KM, Krasovsky J, Williamson B, Dhodapkar MV: Anti-tumor monoclonal antibodies enhance cross-presentation of cellular antigens and the generation of myeloma-specific killer T cells by dendritic cells. J Exp Med. 2002, 195: 125-133. 10.1084/jem.20011097.
Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S: Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002, 3: 196-200. 10.1038/ni758.
Cederblad B, Gobl AE, Alm GV: The induction of interferon-α and interferon-β mRNA in human natural interferon-producing blood leukocytes requires de novo protein synthesis. J Interferon Res. 1991, 11: 371-377.
Cederblad B, Alm GV: Interferons and the colony-stimulating factors IL-3 and GM-CSF enhance the IFN-α response in human blood leucocytes induced by Herpes simplex virus. Scand J Immunol. 1991, 34: 549-555.
Båve U, Vallin H, Alm GV, Rönnblom L: Activation of natural interferon-α producing cells by apoptotic U 937 cells combined with lupus IgG and its regulation by cytokines. J Autoimmun. 2001, 17: 71-80. 10.1006/jaut.2001.0519.
Barnes B, Lubyova B, Pitha PM: On the role of IRF in host defense. J Interferon Cytokine Res. 2002, 22: 59-71. 10.1089/107999002753452665.
Payvandi F, Amrute S, Fitzgerald-Bocarsly P: Exogenous and endogenous IL-10 regulate IFN-α production by peripheral blood mononuclear cells in response to viral stimulation. J Immunol. 1998, 160: 5861-5868.
Shakoor N, Michalska M, Harris CA, Block JA: Drug-induced systemic lupus erythematosus associated with etanercept therapy. Lancet. 2002, 359: 579-580. 10.1016/S0140-6736(02)07714-0.
Charles PJ, Smeenk RJ, De Jong J, Feldmann M, Maini RN: Assessment of antibodies to double-stranded DNA induced in rheumatoid arthritis patients following treatment with infliximab, a monoclonal antibody to tumor necrosis factor alpha: findings in open-label and randomized placebo-controlled trials. Arthritis Rheum. 2000, 43: 2383-2390. 10.1002/1529-0131(200011)43:11<2383::AID-ANR2>3.0.CO;2-D.
Grouard G, Rissoan MC, Filgueira L, Durand I, Banchereau J, Liu YJ: The enigmatic plasmacytoid T cells develop into dendritic cells with interleukin (IL)-3 and CD40-ligand. J Exp Med. 1997, 185: 1101-1111. 10.1084/jem.185.6.1101.
Shortman K, Liu YJ: Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002, 2: 151-161. 10.1038/nri746.
Rissoan MC, Soumelis V, Kadowaki N, Grouard G, Briere F, de Waal Malefyt R, Liu YJ: Reciprocal control of T helper cell and dendritic cell differentiation. Science. 1999, 283: 1183-1186. 10.1126/science.283.5405.1183.
Gilliet M, Liu YJ: Generation of human CD8 T regulatory cells by CD40 ligand-activated plasmacytoid dendritic cells. J Exp Med. 2002, 195: 695-704. 10.1084/jem.20011603.
Cella M, Facchetti F, Lanzavecchia A, Colonna M: Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent Th1 polarization. Nat Immunol. 2000, 1: 305-310. 10.1038/79747.
Jahnsen FL, Lund-Johansen F, Dunne JF, Farkas L, Haye R, Brandtzaeg P: Experimentally induced recruitment of plasmacytoid (CD123high) dendritic cells in human nasal allergy. J Immunol. 2000, 165: 4062-4068.
Santini SM, Lapenta C, Logozzi M, Parlato S, Spada M, Di Pucchio T, Belardelli F: Type I interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in Hu-PBL-SCID mice. J Exp Med. 2000, 191: 1777-1788. 10.1084/jem.191.10.1777.
Rönnblom L, Alm GV: An etiopathogenetic role for the type I interferon system in systemic lupus erythematosus. Trends Immunol. 2001, 22: 427-431. 10.1016/S1471-4906(01)01955-X.
Rönnblom L, Alm GV: A pivotal role for the natural interferon α-producing cells (plasmacytoid dendritic cells) in the pathogenesis of lupus. J Exp Med. 2001, 194: F59-F63. 10.1084/jem.194.12.f59.
Gallucci S, Matzinger P: Danger signals: SOS to the immune system. Curr Opin Immunol. 2001, 13: 114-119. 10.1016/S0952-7915(00)00191-6.
Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A: Chromatin–IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002, 416: 603-607. 10.1038/416603a.
Gobl AE, Funa K, Alm GV: Different induction patterns of mRNA for IFN-α and -β in human mononuclear leukocytes after in vitro stimulation with herpes simplex virus-infected fibroblasts and Sendai virus. J Immunol. 1988, 140: 3605-3609.
Chuntharapai A, Lai J, Huang X, Gibbs V, Kim KJ, Presta LG, Stewart TA: Characterization and humanization of a monoclonal antibody that neutralizes human leukocyte interferon: a candidate therapeutic for IDDM and SLE. Cytokine. 2001, 15: 250-260. 10.1006/cyto.2001.0934.
Benizri E, Gugenheim J, Lasfar A, Eid P, Blanchard B, Lallemand C, Tovey MG: Prolonged allograft survival in cynomolgus monkeys treated with a monoclonal antibody to the human type I interferon receptor and low doses of cyclosporine. J Interferon Cytokine Res. 1998, 18: 273-284.
Acknowledgements
We thank all colleagues who contributed to the results that form the basis of this review, especially Brita Cederblad, Maija-Leena Eloranta, Helena Vallin, Stina Blomberg, Ullvi Båve, Tanja Lövgren, Mattias Magnusson, Anders Perers, Anne Riesenfeld, and Lotta Sjöberg in our laboratory, as well as Anders Bengtsson and Gunnar Sturfelt at the University of Lund. Financial support was provided by the Swedish Medical Research Council, The Swedish Rheumatism Foundation, the 80-years foundation of King Gustaf V, the Åke Wiberg foundation, the Nanna Svartz foundation and Magnus Bergvall foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None declared.
Rights and permissions
About this article
Cite this article
Rönnblom, L., Alm, G.V. Systemic lupus erythematosus and the type I interferon system. Arthritis Res Ther 5, 68 (2003). https://doi.org/10.1186/ar625
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar625