
Abstract
With little in the way of effective therapeutic strategies to target the innate immune response, a better understanding of the critical pathways regulating neutrophil and macrophage responses in inflammation is key to the development of novel therapies. Hypoxia inducible factor (HIF) was originally identified as a central transcriptional regulator of cellular responses to oxygen deprivation. However, the HIF signalling pathway now appears, in myeloid cells at least, to be a master regulator of both immune cell function and survival. As such, understanding the biology of HIF and its regulators may provide new approaches to myeloid-specific therapies that are urgently needed.
Similar content being viewed by others

Introduction
Despite the evolution of respiratory and cardiovascular systems in multicellular higher organisms, the presence of physiological oxygen gradients within and across tissues is well described. At sites of tissue injury and inflammation, oxygen gradients become exaggerated – and it is within relatively oxygen-deplete tissue environments that myeloid cells are required to migrate and function. These sites are typified by empyemas, healing wounds and inflamed joints, where oxygen tensions in the range of 0 to 3 kPa are well documented [1]. It therefore makes sense that myeloid cells have adapted to function at these sites of relative tissue hypoxia, although subversion of this response may also be important in the persistent inflammation associated with inflammatory arthritides, notably rheumatoid arthritis where tissue hypoxia is also linked to disease severity and progression.
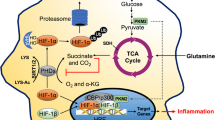
Hypoxia inducible factor (HIF), a transcriptional regulator of cellular responses to oxygen deprivation, plays a crucial role in the regulating myeloid cell function in hypoxia and in inflammation more broadly. The roles of HIF in regulating key myeloid cell functions and signalling pathways are discussed in the present review and are summarized in Figure 1.

Central role of hypoxia inducible factor in the regulation of myeloid cell-mediated inflammation. Under conditions of reduced oxygenation, hydroxylase inhibition and the presence of bacteria/bacterial lipopolysaccharide (LPS), hypoxia inducible factor (HIF) is stabilized and modulates the expression of hypoxia response element (HRE)-responsive genes – resulting in the upregulation of myeloid cell glycolysis, microbicidal proteases, phagocytosis and vascular permeability, and consequently enhanced macrophage and neutrophil recruitment, bacterial killing and persistent myeloid cell-mediated inflammation. PHD, prolyl hydroxylase domain-containing enzyme; FIH, factor inhibiting HIF; IKKB, IκB kinase beta; SLC11a1, phagocyte-specific solute particle carrier 11A1 protein.
Adaptation of myeloid cells to hypoxia
The major pathway for sustainable production of ATP utilizes oxygen in the mitochondrial electron transport system, the process known as oxidative phosphorylation. Within the majority of cells there is a critical intracellular oxygen partial pressure required for respiration (the Pasteur point), below which cells produce ATP through the nonoxygen-requiring process of glycolysis, resulting in the accumulation of lactic acid. The relative importance of these aerobic and anaerobic pathways is highly dependent on the cell systems examined. Myeloid cells are unique in that they have adapted to operate by anaerobic metabolism, even when transiting oxygen-replete areas, with neutrophils incorporating 85% of their glucose uptake into lactate even under resting aerobic conditions [2]. As such, mitochondrial inhibitors have been shown to have no effect on inflammatory responses, in contrast to glycolytic inhibitors that significantly reduce the intracellular ATP concentrations and functional capacity of these cells [2]. With this in mind, the enhanced phagocytic capacity of neutrophils cultured in hypoxia [3] and the profound effects of hypoxia on tissue macrophage phagocytosis [4], chemokine receptor expression [5] and β2-integrin-mediated adhesion in vitro [6] are less surprising.
Neutrophils are programmed to undergo apoptosis constitutively following their release from the bone marrow into the circulation. Neutrophil apoptosis is critical for the resolution of inflammation, with direct effects of apoptosis on neutrophil function and indirect effects on macrophage release of proinflammatory and anti-inflammatory cytokines [7]. Indirect evidence that neutrophil apoptosis occurs in vivo has now been supported by work highlighting the potential of driving neutrophil apoptosis as a therapeutic strategy in a range of murine models, including arthritis [8]. Neutrophil apoptosis is modulated by chemokines (IL-8, granulocyte–macrophage colony-stimulating factor, TNFα) and mediators induced by pathogens (bacterial lipopolysaccharide) [9, 10], providing a mechanism by which the tissue environment can modulate the longevity of neutrophils. In addition to modulation of neutrophil apoptosis by secreted factors, we and other workers have described the profound regulation of neutrophil apoptosis by physiological hypoxia [11, 12]. This is in direct contrast to the effects of hypoxia on most other primary cell types where an induction of cell death is described [13], and is reversible, with neutrophils recovered to a normoxic environment being able to regain their full apoptotic potential.
Moreover, direct hypoxic neutrophil survival requires active protein synthesis and is independent of the phosphatidylinositol 3-kinase pathway so fundamental to the functional competence of these cells. This pathway can, however, be enhanced in a phosphatidylinositol 3-kinase-dependent manner; an effect at least partially dependent on the hypoxic release of the novel neutrophil survival factor macrophage inflammatory protein 1β [11, 12]. In macrophages, whilst acute hypoxia has been shown to induce apoptosis, repeated exposure of RAW264.7 macrophages to hypoxia can result in the selection of an apoptosis-resistant population [14]. Myeloid cells would therefore appear to have adapted to facilitate their persistence at sites of inflammation, where other cell types have a reduced lifespan. This prolongation of functional longevity may, however, prove to be of detriment to the host organism in the context of autoimmune disease. Hypoxia has itself been shown to be critical in regulating the pro-apoptotic and anti-apoptotic effects of rheumatoid synovial fluid upon neutrophils [15], and is therefore a potentially important regulator of neutrophil function and lifespan within the inflamed joint in vivo.
The mechanisms by which hypoxia and, more specifically, the HIF pathway have been shown to modulate myeloid cell function remain to be fully elucidated and represent an area of active research interest. The better characterized pathways are detailed below (and summarized in Figure 1) but remain largely incomplete and only partially understood. Whilst much work has focused recently on the importance of the HIF pathway, it is likely that this will not reflect the sole mechanism for the functional regulation of these cells. For example, there is evidence of hypoxic regulation of HIF-independent transcription factors ATF4 and Egr-1 [16] and of the bHLH transcription factor inhibitor Id2 described in vitro in hypoxia-exposed monocyte-derived macrophages [17].
Oxygen sensing and the hypoxia inducible factor hydroxylase pathway
Over the past 20 years it has become apparent that all cells have an intrinsic ability to sense and adapt to tissue oxygen levels through the oxygen-sensitive transcription factor, HIF. HIF is a heterodimeric beta helix–loop–helix protein composed of an unstable oxygen-sensitive alpha subunit and a constitutively expressed stable beta subunit (aryl hydrocarbon nuclear translocator) [18, 19]. Three HIFα subunits have been identified to date in higher organisms (HIF-1α, HIF-2α and HIF-3α) [20–22], with most detailed information available for HIF-1α and HIF-2α. Differential cellular expression of HIFα genes, with genetic conservation in eukaryotes and multiple splice variants of HIF-3α, support an essential and nonoverlapping role for the HIFs, although the precise nature of their functional diversity remains to be fully characterized.
All HIFα subunits are subject to regulation in two ways: first, proteasomal degradation following hydroxylation of two highly conserved prolyl residues (Pro-402 and Pro-564) by members of the prolyl hydroxylase domain-containing family (PHD) [23, 24]; and secondly, transcriptional inactivation following asparaginyl hydroxylation by factor inhibiting HIF [25–27]. Prolyl hydroxylation enables proteosomal degradation of the HIFα subunit through high-affinity binding to the von Hippel–Lindau (VHL) E3 ubiquitin ligase [18, 24, 28]. Four PHD proteins that can hydroxylate HIF have currently been identified (PHD1, PHD2 and PHD3, and a recently described fourth enzyme P4H-TM), with all four displaying wide tissue expression but in a differential cellular localization – nuclear (PHD1), cytoplasmic (PHD2), nuclear and cytoplasmic (PHD3), and endoplasmic reticulum (P4H-TM) [29, 30].
PHDs and factor inhibiting HIF all display an absolute requirement for dioxygen, Fe(II) and 2-oxoglutarate, with PHD1 and PHD3 also regulated by ubiquitination as targets for the E3 ubiquitin ligases Siah1a and Siah2 [31]. At sites of reduced oxygen tension, therefore, reduced PHD and factor inhibiting HIF hydroxylase activity permits stabilization and transcriptional activation of HIF, resulting in the modulation of multiple HIF effector genes, which contain hypoxia response elements [22], to facilitate the cellular adaptive responses to hypoxia. These responses include the function of enhancing local oxygen delivery by promoting erythropoiesis and angiogenesis and by the metabolic adaptation to oxygen deprivation through the upregulation of glycolytic enzymes and glucose transporters. In excess of 60 HIF target genes have been identified to date, with gene expression profiling confirming significant overlap between HIF-1 and HIF-2 regulated genes. With a degree of nonredundancy of function, and differential basal and cell-specific expression of the HIFα isoforms described, however, it is likely that the regulation of gene expression by the HIF pathway is complex, and that characterizing the regulation of the relative changes in expression between the isoforms may be important in understanding the subversion of physiological hypoxic responses in disease states.
With an oxygen concentration that enables a half-maximal catalytic rate for oxygen well above tissue oxygen concentrations, the hydroxylase enzymes are well placed to function over all physiologically relevant oxygen tensions [32, 33], enabling the HIF system to operate as a highly efficient oxygen sensor in vivo. The HIF-dependent hypoxic induction of PHD2 and PHD3 mRNA and protein [34] allows further adaptation to oxygen thresholds within individual cells, since this is dependent on previous oxygen exposure, and may therefore explain the ability of the HIF pathway to respond to the wide variety of tissue oxygen tensions in vivo in a cell-specific way. In addition to the oxygen-dependent regulation of hydroxylase activity, the metabolic intermediates fumarate and succinate have also been shown to modulate hydroxylase activity and HIF signalling, as have the intracellular availability of iron and ascorbate and the local concentration of reactive oxygen species. A role for sumoylation and histone acetylase inhibition [35] has also been postulated but remains controversial, with reports of both increased and decreased HIF stability following HIF sumoylation [36, 37]. Whilst oxygen sensing remains the fundamental regulator of HIF signalling, it is clear from the above that modulation of HIF activity by intermediates may play a physiological role at sites of inflammation that are characterized by low levels of glucose and high levels of reductive metabolites.
Hypoxia inducible factor and myeloid cell function
In addition to the key role of HIF in regulating the cellular responses to hypoxia, work from Randall Johnson's group has shown that HIF also plays a fundamental role in regulating inflammation. Using myeloid-targeted HIF-1α knockout mice, they described a critical role for HIF-1α in regulating neutrophil and mononuclear cell glycolysis [38]. In HIF-1α-deficient myeloid cells, this resulted in a reduction in ATP pools, accompanied by profound impairment of cell aggregation, motility, invasiveness, and bacterial killing. In vivo this correlated with the ablation of sodium dodecyl sulphate-induced cutaneous inflammation and a reduction in synovial infiltration, pannus formation and cartilage destruction in an immune complex-mediated inflammatory arthritis model.
Further studies by this group subsequently demonstrated the importance of HIF in the regulation of phagocytic bactericidal capacity in vivo [39], with decreased bactericidal activity and exaggerated systemic spread of infection in conditional HIF-1α knockouts compared with littermate controls. Importantly, using human pathogens (Group A Streptococcus, methicillin-resistant Staphylococcus aureus, Pseudomonas aeruginosa, and Salmonella species), they also showed induction of HIF-1α expression and transcriptional activity in macrophages that was independent of oxygen tension.
Interestingly there was a divergence in myeloid cell functional regulation, with HIF-1α regulating production of nitric oxide, the granule proteases cathepsin G and neutrophil elastase, and the antimicrobial peptide cathelicidin, but not endothelial transmigration or respiratory burst activity. Bacterial lipopolysaccharide was also shown to directly increase HIF-1α transcription and to decrease PHD2-mediated and PHD3-mediated HIF-1α degradation in macrophages in a Toll-like receptor 4-dependent fashion. HIF-1α deletion in these macrophages was subsequently shown to be protective against bacterial lipopolysaccharide-induced mortality and to be associated with the downregulation of cytokines in these cells – including TNFα, IL-1, IL-4, IL-6 and IL-12, which are implicated in the pathogenesis of sepsis syndrome [40].
The importance of the differential regulation of these functional responses by HIF-1α remains to be explored. Furthermore, the relative contributions of HIF-2 and the different isoforms of HIF-3 to these functional pathways have yet to be clarified. Work looking at differential expression of the HIF isoforms in vitro and in vivo in monocytes, monocyte-derived macrophages and tumour-associated macrophages is also complex, with Burke and colleagues describing the preferential induction of HIF-1 in human macrophages in vitro following hypoxic stimulation, and in vivo in different tumour sites [17]. In contrast, work by Talks and colleagues describes HIF-2 as the predominant isoform in differentiated promonocytic cells [41]. More recently, Elbarghati and colleagues have shown the regulation of both HIF-1 and HIF-2 by hypoxia in human monocyte-derived macrophages [16], and postulated the enhanced stability of HIF-2 over HIF-1 in the context of more prolonged hypoxic exposure. Interestingly, in macrophages isolated from rheumatoid joints, HIF-1 has been described previously as being the predominant isoform [42].
To enable neutrophils to migrate from the oxygen-replete circulation to the site of tissue damage, the neutrophils undergo a process of selectin-mediated rolling and β2-integrin-mediated adhesion [43]. This process of diapedesis is itself modified by HIF-1α expression, with HIF-1 being a transcriptional regulator of CD18, the β2-integrin beta subunit [44]. Once neutrophils have migrated down an oxygen gradient to the site of tissue damage, it is the regulation of their function longevity that is thought to be critical for the resolution of inflammatory responses. Given the profound survival effect of hypoxia on neutrophils aged in vitro [11, 12], and the critical role for HIF in regulating cellular responses to hypoxia, we studied the importance of HIF itself in regulating neutrophil apoptosis. We showed a marked reduction in survival of bone-marrow-derived HIF-1α-deficient neutrophils compared with controls, following their culture under hypoxic conditions [45]. Together these data highlight the importance of HIF-1α in coordinating appropriate and effective innate immune responses, but also identify a potential role for dysregulation of HIF in conditions of inappropriate or persistent inflammation.
Direct evidence that the HIF pathway regulates innate immune responses in vivo in humans is provided by a series of experiments in which we isolated peripheral blood neutrophils from individuals with germline mutations in the vhl gene. Since these individuals retain one intact copy of the vhl allele, this enabled us to study the effects of heterozygous VHL expression in human neutrophils [3]. We described a partial hypoxic phenotype, which manifests as a reduction of constitutive rates of neutrophil apoptosis, as enhanced neutrophil susceptibility to hydroxylase inhibition and as enhanced neutrophil phagocytosis of heat-killed bacteria under normoxic conditions [3]. Further studies of individuals, for example, with homozygous mutations in the vhl alleles with Chuvash polycythaemia may further clarify the importance of the HIF pathway in the pathogenesis of human disease in vivo.
NF-κB, HIF-1α and innate immunity
The first evidence of a direct interaction between NF-κB and HIF signalling pathways was provided by a search for non-HIF substrates of the HIF hydroxylase enzymes by Cockman and colleagues [46]. They describe the efficient hydroxylation of asparginyl residues within the ankyrin repeat domain of the IκB proteins p105 (NFKB1) and IκBα, but no functional consequence of this interaction with respect to NF-κB-dependent transcription. A possible role in the stoichiometric competition between HIF and other ankyrin repeat domain-containing proteins was subsequently raised. Concurrently, Cummins and colleagues proposed a model of hypoxic de-repression of NF-κB activity through a reduction in PHD1-dependent hydroxylation of the IκB kinase beta (IKKβ) classical pathway regulator, although again the cellular consequences of this association were not defined and only modest NF-κB activation was described [47]. HIF-1α has also been shown, however, to promote the expression of NF-κB-regulated inflammatory cytokines [40], and loss of HIF-1α results in the downregulation of hypoxia-induced NF-κB message in murine bone-marrow-derived neutrophils [45]. Whilst these data clearly highlight crosstalk between the HIF and NF-κB pathways, the consequences of these associations both in vitro and in vivo and the variation between the cell types studied makes functional interpretation of these existing data difficult.
Given the complex nature of the relationship between HIF-1α and NF-κB and the central role of both the HIF and NF-κB signalling pathways in the regulation of innate immune responses, Rius and colleagues investigated the consequence of IKKβ deficiency for the induction of HIF-1α target genes and HIF-1α accumulation in macrophages using mice deficient in IKKβ [48]. They show that loss of IKKβ results in the defective induction of HIF target genes, with IKKβ essential for HIF-1α accumulation following macrophage exposure to bacteria. NF-κB activation without hypoxic inhibition of the prolyl hydroxylase enzymes, however, was insufficient for HIF-1α protein accumulation. Of note, IKKβ was not required for the hypoxic induction of HIF-2α protein in bone-marrow-derived macrophages. Taken together these results propose IKKβ to be an important link between hypoxic signalling, innate immunity and inflammation with NF-κB, a critical transcriptional activator of HIF-1α. With no described effect on HIF-2α signalling, the biological importance of this association may, however, in part depend on the dominant HIF subunit specific to the cell type and on the physiological or disease conditions in which that cell is functioning.
Novel hypoxia inducible factor targets in the innate immune response
Anaemia of chronic disease following the sequestration of iron in the reticuloendothelial system has been recognized as a clinically important entity for many decades [49]. Whilst the increased iron retention by inflammatory macrophages has also been well characterized [50], direct links between critical signalling pathways involved in innate immune responses and iron homeostasis have only recently been revealed.
Work by Peyssonnaux and colleagues initially described the key iron regulator hepcidin to be regulated by the HIF/VHL pathway [51]. Hepcidin is a small, acute-phase peptide synthesized by the liver that limits iron export from macrophages through the inhibition of the major iron exporter ferroportin [52]. Hepcidin is itself downregulated in conditions of chronic anaemia and hypoxia. Using mice with HIF-1α inactivated in hepatocytes alone, the authors showed an HIF-1α dependence for hepcidin downregulation following a diet-induced iron deficiency [51]. They were subsequently able to show (indirectly) that HIF-1α binds to and negatively trans-activates the hepcidin promoter. Importantly, however, HIF-1α deletion alone was not sufficient to fully compensate for iron-deficient loss of hepcidin, inferring that other factors may also be involved – a potential role for HIF-2 in this response again remains to be addressed.
Subsequent to that work, Tacchini and colleagues looked at the effect of inflammatory and anti-inflammatory signals on HIF-1-mediated transferrin receptor (TfR1) expression in macrophages [53]. TfR1 represents one of three major pathways required for the macrophage acquisition of iron [54]. Its role in inflammatory iron sequestration is somewhat controversial given the reported post-transcriptional downregulation of TfR1 following prolonged in vitro exposure of macrophages (10 to 24 hours) to bacterial lipopolysaccharide IFNγ [55]. Work by this group and others, however, describes an initial early induction of TfR1 (30 min) that involves the successive activation of NF-κB and HIF-1 signalling pathways [53]. Furthermore this induction of TfR1 was functionally important since it was associated with a greater uptake of transferrin-bound iron by the RAW264.7 macrophages. This would thus represent an early transient macrophage response to inflammatory stimuli that would precede the role of hepcidin in maintaining the iron sequestration by macrophages, again demonstrating a dual regulation by NF-κB and HIF inflammatory pathways.
Whilst hypoxia and the HIF/hydroxylase pathway in the regulation of myeloid cell function and survival is of direct functional importance in regulation of innate immune responses, a role for HIF in the regulation of heritable macrophage resistance to intracellular pathogens would clearly be important for an individual's overall disease risk. A genetic basis for the protection of organisms from infection by intra-cellular pathogens remains a relatively new concept, and was first supported by work in mice through the cloning of a locus that encodes a phagocyte-specific solute particle carrier 11A1 protein (SLC11a1) and protects in-bred mice from infection by intracellular pathogens [56]. Despite this observation, no identifiable mutations in human SLC11a1 have been reported despite obvious functional differences in individual resistance to infection and inflammatory disease. Bayele and colleagues proposed that quantitative differences in SLC11A1 transcription may underlie human disease susceptibility [57]. They subsequently described the regulation of allele expression by a Z-DNA-forming microsatellite within the SLC11A1 promoter following its binding to HIF-1α. The functional importance of this association was shown by an attenuation of macrophage responsiveness to Salmonella typhimurium following the targeted deletion of HIF-1α in murine macrophages. As such the authors propose that HIF-1 may influence the heritable variation in innate resistance to infection and inflammation through the regulation of gene expression phenotypes. This would indeed make HIF the true master regulator of innate immune myeloid cell-mediated responses.
Conclusion
With a predominantly glycolytic metabolism, myeloid cells are well placed to function within the oxygen-deplete environments in which they find themselves at sites of tissue injury and infection. The importance of HIF in mediating their transcriptional responses to relative physiological oxygen tensions is now well described, with the oxygen-sensitive hydroxylase enzymes critical in this response. Increasing evidence for the oxygen-independent regulation of myeloid cell function by the HIF pathway, and in particular HIF-1α, now lends credence to a fundamental role of the HIF pathway in regulating innate immune responses. This evidence would appear to range from an inherited predisposition to inflammation and microbial resistance to the regulation of bacterial killing, migration and apoptosis. This has important therapeutic implications both with respect to the enhanced ability of organisms to effectively remove injurious stimuli and with respect to the persistence of exaggerated myeloid cell-mediated inflammatory responses associated acutely with respiratory distress syndromes and more chronically with diseases such as rheumatoid arthritis and chronic obstructive pulmonary disease.
Clearly whilst the enhanced activity of HIF may thus be important in the acute infectious disease setting, facilitating microbial recognition and clearance by myeloid cells, persistent activity of HIF may equally be detrimental in the development of inflammatory diseases, particularly – as in arthritis, where hypoxia and inflammation co-exist. Any novel therapeutic strategies will thus have to be very selective for the disease pathways they are to target and, as a result, are more likely to involve critical regulators of the HIF pathway unique to that disease condition and considerably downstream of the global regulator itself. This makes the further understanding of the HIF hydroxylase pathway and its interactions with other key regulators of the innate immune response – for instance, the NF-κB pathway – critical for the development of novel therapeutic strategies.
Note
This review is part of a series on Hypoxia edited by Ewa Paleolog.
Other articles in this series can be found at http://arthritis-research.com/series/ar_Hypoxia
Abbreviations
- HIF:
-
hypoxia inducible factor
- IFN:
-
interferon
- IKKβ:
-
IκB kinase beta
- IL:
-
interleukin
- NF:
-
nuclear factor
- PHD:
-
prolyl hydroxylase domain-containing enzyme
- SLC11a1:
-
phagocyte-specific solute particle carrier 11A1 protein
- TfR1:
-
transferrin receptor
- TNF:
-
tumour necrosis factor
- VHL:
-
von Hippel–Lindau.
References
Hunt TK, Twomey P, Zederfeldt B, Englebert Dunphy J: Respiratory gas tensions and pH in healing wounds. Am J Surg. 1967, 114: 302-307. 10.1016/0002-9610(67)90388-1.
Borregaard N, Herlin T: Energy metabolism of human neutrophils during phagocytosis. J Clin Invest. 1982, 70: 550-557. 10.1172/JCI110647.
Walmsley SR, Cowburn AS, Clatworthy MR, Morrell NW, Roper EC, Singleton V, Maxwell P, Whyte M, Chilvers ER: Neutrophils from patients with heterozygous germline mutations in the von Hippel Lindau protein (VHL) display delayed apoptosis and enhanced bacterial phagocytosis. Blood. 2006, 108: 3176-3178. 10.1182/blood-2006-04-018796.
Anand RJ, Gribar SC, Li J, Kohler JW, Branca MF, Dubowski T, Sodhi CP, Hackam DJ: Hypoxia causes an increase in phagocytosis by macrophages in a HIF-1α-dependent manner. J Leukoc Biol. 2007, 82: 1257-1265. 10.1189/jlb.0307195.
Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, Bernasconi S, Saccani S, Nebuloni M, Vago L, Mantovani A, Melillo G, Sica A: Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med. 2003, 198: 1391-1402. 10.1084/jem.20030267.
Arnould T, Michiels C, Remacle J: Increased PMN adherence on endothelial cells after hypoxia: involvement of PAF, CD18/CD11b, and ICAM-1. Am J Physiol. 1993, 264: C1102-C1110.
Bianchi SM, Dockrell DH, Renshaw SA, Sabroe I, Whyte MK: Granulocyte apoptosis in the pathogenesis and resolution of lung disease. Clin Sci (Lond). 2006, 110: 293-304.
Rossi AG, Sawatzky DA, Walker A, Ward C, Sheldrake TA, Riley NA, Caldicott A, Martinez-Losa M, Walker TR, Duffin R, Gray M, Crescenzi E, Martin MC, Brady HJ, Savill JS, Dransfield I, Haslett C: Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med. 2006, 12: 1056-1064. 10.1038/nm1468.
Cox G, Gauldie J, Jordana M: Bronchial epithelial cell-derived cytokines (G-CSF and GM-CSF) promote the survival of peripheral blood neutrophils in vitro. Am J Respir Cell Mol Biol. 1992, 7: 507-513.
Haslett C: Resolution of acute inflammation and the role of apoptosis in the tissue fate of granulocytes [editorial]. Clin Sci. 1992, 83: 639-648.
Hannah S, Mecklenburgh K, Rahman I, Bellingan GJ, Greening A, Haslett C, Chilvers ER: Hypoxia prolongs neutrophil survival in vitro. FEBS Lett. 1995, 372: 233-237. 10.1016/0014-5793(95)00986-J.
Mecklenburgh KI, Walmsley SR, Cowburn AS, Wiesener M, Reed BJ, Upton PD, Deighton J, Greening AP, Chilvers ER: Involvement of a ferroprotein sensor in hypoxia-mediated inhibition of neutrophil apoptosis. Blood. 2002, 100: 3008-3016. 10.1182/blood-2002-02-0454.
Rosenbaum DM, Michaelson M, Batter DK, Doshi P, Kessler JA: Evidence for hypoxia-induced, programmed cell death of cultured neurons. Ann Neurol. 1994, 36: 864-870. 10.1002/ana.410360610.
Yun JK, McCormick TS, Villabona C, Judware RR, Espinosa MB, Lapetina EG: Inflammatory mediators are perpetuated in macrophages resistant to apoptosis induced by hypoxia. Proc Natl Acad Sci USA. 1997, 94: 13903-13908. 10.1073/pnas.94.25.13903.
Cross A, Barnes T, Bucknall RC, Edwards SW, Moots RJ: Neutrophil apoptosis in rheumatoid arthritis is regulated by local oxygen tensions within joints. J Leuk Biol. 2006, 80: 521-528. 10.1189/jlb.0306178.
Elbarghati L, Murdoch C, Lewis CE: Effects of hypoxia on transcription factor expression in human monocytes and macrophages. Immunobiol. 2008, 213: 899-908. 10.1016/j.imbio.2008.07.016.
Burke B, Giannoudis A, Corke KP, Gill D, Wells M, Ziegler-Heitbrock L, Lewis CE: Hypoxia-induced gene expression in human macrophages. Am J Pathol. 2003, 163: 1233-1243.
Jaakkola P, Mole DR, Tian Y-M, Wilson M, Gielbert J, Gaskell SJ, Kriegsheim Av, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ: Targeting of HIFα to the von Hippel–Lindau ubiquitylation complex by O2 regulated prolyl hydroxylation. Science. 2001, 292: 468-472. 10.1126/science.1059796.
Wang GL, Jiang B-H, Rue EA, Semenza GL: Hypoxia-inducible factor 1 is a basic–helix–loop–helix–PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995, 92: 5510-5514. 10.1073/pnas.92.12.5510.
Hogenesch JB, Chan WK, Jackiw VH, Brown RC, Gu Y-Z, Pray-Grant M, Perdew GH, Bradfield CA: Characterisation of a subset of the basic helix–loop–helix–PAS superfamily that interacts with components of the dioxin signalling pathway. J Biol Chem. 1997, 272: 8581-8593. 10.1074/jbc.272.13.8581.
Gu YZ, Moran SM, Hogenesch JB, Wartman L, Bradfield CA: Molecular characterisation and chromosomal localisation of a third a-class hypoxia inducible factor subunit, HIF3α. Gene Expr. 1998, 7: 205-213.
Wenger RH: Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2 regulated gene expression. FASEB J. 2002, 16: 1151-1162. 10.1096/fj.01-0944rev.
Masson N, William C, Maxwell P, Pugh CW, Ratcliffe PJ: Independent function of two destruction domains in hypoxia-inducible factor-α chains activated by prolyl hydroxylation. EMBO J. 2001, 20: 5197-5206. 10.1093/emboj/20.18.5197.
Yu F, White SB, Zhao Q, Lee FS: Dynamic, site-specific interaction of hypoxia-inducible factor-1α with the von Hippel–Lindau tumour suppressor protein. Cancer Res. 2001, 61: 4136-4142.
Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML: Asparagine hydroxylation of the HIF transactivation domain: a hypoxic switch. Science. 2002, 295: 858-861. 10.1126/science.1068592.
Mahon PC, Hirota k, Semenza GL: FIH-1: a novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15: 2675-2680. 10.1101/gad.924501.
Sang N, Fang J, Srinivas V, Leshchinsky I, Caro J: Carboxyl-terminal transactivation activity of hypoxia-inducible factor 1α is governed by a von Hippel–Lindau protein-independent, hydroxylation-regulated association with p300/CBP. Mol Cell Biol. 2002, 22: 2984-2992. 10.1128/MCB.22.9.2984-2992.2002.
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG: HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2sensing. Science. 2001, 292: 464-468. 10.1126/science.1059817.
Metzen E, Berchner-Pfannschmidt U, Stengel P, Marxsen JH, Stolze I, Klinger M, Huang WQ, Wotzlaw C, Hellwig-Bürgel T, Jelkmann W, Acker H, Fandrey J: Intracellular localisation of human HIF-1 alpha hydroxylases: implications for oxygen sensing. J Cell Sci. 2003, 116: 1319-1326. 10.1242/jcs.00318.
Koivunen P, Tiainen P, Hyvärinen J, Williams KE, Sormunen R, Klaus SJ, Kivirikko KI, Myllyharju J: An endoplasmic reticulum transmembrane prolyl 4-hydroxylase is induced by hypoxia and acts on hypoxia-inducible factor alpha. J Biol Chem. 2007, 282: 30544-30552. 10.1074/jbc.M704988200.
Nakayama K, Few IJ, Hagensen M, Skalas M, Habelhah H, Bhoumik A, Kadoya T, Erdjument-Bromage H, Tempst P, Frappell PB, Bowtell DD, Ronai Z: Siah2 regulates stability of prolyl-hydroxylases, controls HIF1α abundance, and modulates physiological responses to hypoxia. Cell. 2004, 117: 941-952. 10.1016/j.cell.2004.06.001.
Tuckerman JR, Zhao Y, Hewitson KS, Tian Y-M, Pugh CW, Ratcliffe PJ, Mole DR: Determination and comparison of specific activity of the HIF-prolyl hydroxylases. FEBS Lett. 2004, 576: 145-150. 10.1016/j.febslet.2004.09.005.
Stiehl DP, Wirthner R, Koditz J, Spielmann P, Camenisch G, Wenger RH: Increased prolyl 4-hydroxylase domain proteins compensate for decreased oxygen levels. J Biol Chem. 2006, 281: 23482-23491. 10.1074/jbc.M601719200.
Appelhoff RJ, Tian Y-M, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, Gleadle JM: Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004, 279: 38458-38465. 10.1074/jbc.M406026200.
Kong X, Lin Z, Liang D, Fath D, Sang N, Caro J: Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteosomal degradation of hypoxia-inducible factor 1 alpha. Mol Cell Biol. 2006, 26: 2019-2028. 10.1128/MCB.26.6.2019-2028.2006.
Carbia-Nagashima A, Gerez J, Perez-Castro C, Paez-Pereda M, Silberstein S, Stalla GK, Holsboer F, Arzt E: RSUME, a small RWD-containing protein, enhances SUMO conjugation and stabilizes HIF-1 alpha during hypoxia. Cell. 2007, 131: 309-323. 10.1016/j.cell.2007.07.044.
Cheng J, Kang X, Zhang S, Yeh ET: SUMO-specific protease 1 is essential for stabilization of HIF1 alpha during hypoxia. Cell. 2007, 131: 584-595. 10.1016/j.cell.2007.08.045.
Cramer T, Yaminishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber H-P, Ferrara N, Johnson RS: HIF-1α is essential for myeloid cell-mediated inflammation. Cell. 2003, 112: 645-657. 10.1016/S0092-8674(03)00154-5.
Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, Johnson RS: HIF-1α expression regulates the bactericidal capacity of phagocytes. J Clin Invest. 2005, 115: 1806-1815. 10.1172/JCI23865.
Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V: Essential role of hypoxia inducible factor-1 α in development of lipopolysaccharide-induced sepsis. J Immunol. 2007, 178: 7516-7519.
Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL: The expression and distribution of the hypoxia-inducible factors HIF-1α and HIF-2α in normal human tissues, cancers and tumor-associated macrophages. Am J Pathol. 2000, 157: 411-421.
Hollander AP, Corke KP, Freemont AJ, Lewis CE: Expression of hypoxia-inducible factor 1α by macrophages in the rheumatoid synovium: implications for targeting of therapeutic genes to the inflamed joint. Arthritis Rheum. 2001, 44: 1540-1544. 10.1002/1529-0131(200107)44:7<1540::AID-ART277>3.0.CO;2-7.
Carlos TM, Harlan JM: Leukocyte–endothelial adhesion molecules. Blood. 1994, 84: 2068-2101.
Kong T, Eltzschig HK, Karhausen J, Colgan SP, Shelley CS: Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of β2 integrin gene expression. Proc Natl Acad Sci USA. 2004, 101: 10440-10445. 10.1073/pnas.0401339101.
Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, Sobolewski A, Condliffe A, Cowburn AS, Johnson N, Chilvers ER: The role of HIF-1α and NF-κB in hypoxia-induced survival in human and murine neutrophils. J Exp Med. 2005, 201: 105-115. 10.1084/jem.20040624.
Cockman ME, Lancaster DE, Stolze IP, Hewitson KS, McDonough MA, Coleman ML, Coles CH, Yu X, Hay RT, Ley SC, Pugh CW, Oldham NJ, Masson N, Schofield CJ, Ratcliffe P: Posttranslational hydroxylation of ankyrin repeats in IκB proteins by the hypoxiinducible factor (HIF) asparginyl hydroxylase, factor inhibiting HIF (FIH). Proc Natl Acad Sci USA. 2006, 103: 14767-14772. 10.1073/pnas.0606877103.
Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE, Moynagh P, Pouyssegur J, Taylor CT: Prolyl hydroxylase-1 negatively regulates IκB-kinase-β, giving insight into hypoxia-induced NFkB activity. Proc Natl Acad Sci USA. 2006, 103: 18154-18159. 10.1073/pnas.0602235103.
Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M: NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature. 2008, 453: 807-811. 10.1038/nature06905.
Ganz T: Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood. 2003, 102: 783-788. 10.1182/blood-2003-03-0672.
Gallí A, Bergamaschi G, Recalde H, Biasiotto G, Santambrogio P, Boggi S, Levi S, Arosio P, Cazzola M: Ferroportin gene silencing induces iron retention and enhances ferritin synthesis in human macrophages. Br J Haematol. 2004, 127: 598-603. 10.1111/j.1365-2141.2004.05238.x.
Peyssonnaux C, Zinkernagel AS, Schuepbach RA, Rankin E, Vaulont S, Haase VH, Nizet V, Johnson RS: Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J Clin Invest. 2007, 117: 1926-1932. 10.1172/JCI31370.
Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J: Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004, 306: 2090-2093. 10.1126/science.1104742.
Tacchini L, Gammella E, De Ponti C, Recalcati S, Cairo G: Role of HIF-1 and NF-κB transcription factors in the modulation of transferrin receptor by inflammatory and anti-inflammatory signals. J Biol Chem. 2008, 283: 20674-20686. 10.1074/jbc.M800365200.
Knutson M, Wessling-Resnick M: Iron metabolism in the reticuloendothelial system. Crit Rev Biochem Mol Biol. 2003, 38: 61-88. 10.1080/713609210.
Ludwiczek S, Aigner E, Theurl I, Weiss G: Cytokine-mediated regulation of iron transport in human monocytic cells. Blood. 2003, 101: 4148-4154. 10.1182/blood-2002-08-2459.
Vidal SM, Malo D, Vogan K, Skamene E, Gros P: Natural resistance to infection with intracellular parasites: isolation of a candidate for Bcg. Cell. 1993, 73: 469-485. 10.1016/0092-8674(93)90135-D.
Bayele HK, Peyssonnaux C, Giatromanolaki A, Arrais-Silva WW, Mohamed HS, Collins H, Giorgio S, Koukourakis M, Johnson RS, Blackwell JM, Nizet V, Srai SKS: HIF-1 regulates heritable variation and allele expression phenotypes of the macrophage immune response gene SLC11A1 from a Z-DNA-forming microsatellite. Blood. 2007, 15: 3039-3048. 10.1182/blood-2006-12-063289.
Acknowledgements
SRW is funded by a Wellcome Trust Intermediate Fellowship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Walmsley, S.R., Chilvers, E.R. & Whyte, M.K. Hypoxia. Hypoxia, hypoxia inducible factor and myeloid cell function. Arthritis Res Ther 11, 219 (2009). https://doi.org/10.1186/ar2632
Published:
DOI: https://doi.org/10.1186/ar2632