
Abstract
Rheumatoid arthritis is a multisystemic auto-inflammatory disease affecting up to 1% of the population and leading to the destruction of the joints. Evidence exists for the involvement of the innate as well as the adaptive immune systems in the pathology of the disease. The success of anti-tumour necrosis factor-α indicates the importance of pro-inflammatory mediators produced by innate immune cells in rheumatoid arthritis progression. Therefore, considerable efforts have been made in elucidating the signalling pathways leading to the expression of those mediators. This review will concentrate on the role of signalling pathways in innate immune cells in the context of rheumatoid arthritis.
Similar content being viewed by others

Introduction
The immune system evolved as a mechanism to protect organisms from infection by pathogenic organisms and other harmful substances. In general, the immune system is capable of recognising invading pathogens and their products as well as endogenous danger signals [1]. This recognition results in the initiation of an immune response, which will under normal circumstances eliminate the insult without further damage to the host. However, it is now well recognised that defects in regulating inflammation can lead to an excessive response to infectious agents, such as sepsis, or auto-inflammatory diseases, such as rheumatoid arthritis (RA).
In the context of RA numerous cellular mechanisms and signalling pathways drive the chronic inflammation observed in this disease, and current evidence suggests an involvement of the innate as well as the adaptive immune systems in RA pathology. The importance of the adaptive immune response is supported by rodent models of disease, such as collagen-induced arthritis (CIA), that are mainly Th1- and/or Th17-driven [2]. Mice lacking IL-23 do not develop CIA [3] and CCR6-expressing Th17 cells are preferentially recruited to inflamed joints [4]. In humans, the efficacy of anti-CD20 (Rituximab) and anti-CTLA4 (Abatacept) antibodies in RA treatment suggest a function for activated B and T cells in RA [5, 6]. Moreover, a role for CD4+ T cells in RA pathogenesis is inferred by the strong HLA-DR association [7].
During the progression of RA the production of cytokines, chemokines and matrix metalloproteinases by mainly innate immune cells leads to the destruction of cartilage and bone. Currently, the most successful RA therapeutics are the biologicals Infliximab, Etanercept and Adalimumab [8], which block tumour necrosis factor (TNF)α, a cytokine produced mainly by macrophages [9]. The importance of TNFα in disease pathogenesis has also been shown in murine models of the disease [10, 11]. Given the success of anti-TNFα therapy, there has been a great deal of interest in elucidating the pathways driving the production of this cytokine as well as other inflammatory mediators in RA. Other innate immune cells that may have a role in RA include neutrophils [12], mast cells [13] and natural killer cells [14]. They have been shown to be present in high numbers and widely distributed in synovial fluid and tissues. These cells are able to produce several cytokines that may be involved in the pathogenesis of disease, but their contribution to pathogenesis is poorly understood.
This review will describe inflammatory signalling mechanisms in innate immune cells, and concentrate on the emerging evidence implicating certain signalling pathways in driving the continuous production of pro-inflammatory mediators in the RA joint.
The danger signal hypothesis
The main role of toll-like receptors (TLRs) is considered to be the recognition and response to microbial pathogens, but they have also been reported to recognise endogenous ligands (reviewed in [15–20]). Endogenous ligands are thought to be released during necrotic cell death induced by tissue damage, stress factors or infection, resulting in the release of cell components that initiate an inflammatory response [21]. The contents released from necrotic cells may activate TLRs, generating further inflammation and thus more necrosis. This cycle of inflammation may explain the chronic inflammatory state found in autoimmune diseases such as RA. Indeed, endogenous TLR ligands, such as hyaluronan oligosaccharides, fibronectin fragments, heat shock proteins, antibody-DNA complexes and high mobility group box (HMGB)-1, have all been identified in the RA joint [22–25] and several studies emphasise a role for TLRs in the promotion of systemic lupus erythematosus, asthma, Crohn's disease, multiple sclerosis, type 1 diabetes, and RA [18].
TLR signalling driving inflammation in RA
Given the existing evidence of an involvement of TLRs in the pathogenesis of RA and other inflammatory diseases, a great deal of interest exists in understanding the molecular basis of the signalling pathways induced by these receptors, with the hope of identifying therapeutic targets.
Due to their structural similarities TLRs share certain signalling pathways with the IL-1R family [26]. TLR and IL-1R signalling is initiated by ligand-induced hetero- or homodimerisation of the receptors or association with accessory proteins [27]. The signal is transduced by the intracellular Toll/IL-1 receptor (TIR) domain, present in TLRs as well as IL-1Rs, through the recruitment of TIR domain-containing adaptor molecules [28]. The TLRs use distinct combinations of these adaptors to turn on the common TLR/IL-1R pathway as well as pathways unique to TLRs, leading to the activation of transcription factors (Figure 1).

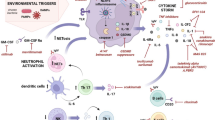
TLR signalling pathways. For simplicity reasons the signalling pathways induced by toll-like receptor (TLR)4, which utilises all four known adaptor proteins, is shown. Following stimulation and dimerisation, the IL-1R and TLR signalling pathways, with the exception of TLR3, recruit the adaptor molecule MyD88 and induce nuclear factor (NF)-κB and mitogen-activated protein kinases (MAPKs) through IL-1R associated kinase (IRAK)-4, IRAK-1 and TNF receptor associated factor (TRAF)-6. In addition, a MyD88-independent signalling pathway is utilised by TLR3 and TLR4, which depends on the adaptor molecule TRIF (TIR-domain-containing adapter-inducing interferon-β) and leads to the induction of interferon regulatory factors (IRFs) and a late activation of NF-κB. Signalling through TLR4 results in phosphoprylation and activation of protein tyrosine kinases (TKs). The Tec family member Btk interacts with the Toll/IL-1 receptor (TIR) domains of TLRs, MyD88 and Mal (MyD88 adaptor like protein). Once activated, Btk phoshporylates Mal and activates NF-κB and/or p38 MAPK. Src family kinases (SFKs; for example, Hck) are known to function upstream of both Pyk2 and Syk kinases, respectively, in TLR signalling. TLRs mediate phosphatidylinositol-3-kinase (PI3K) activation that suppresses p38 MAPK and NF-κB. Inhibition of these signalling cascades by PI3K is possibly mediated by protein kinase B (PKB), and limits the production of inflammatory cytokines. IKK = IkappaB kinase; RANTES, Regulated on activation, normal T expressed and secreted; TBK, TANK-binding kinase; TNF, tumour necrosis factor; TRAM, TRIF-related adaptor molecule.
MyD88 dependent TLR/IL-1R signalling
IL-1Rs and all TLRs, with the exception of TLR3, share a common signalling pathway that depends on the adaptor molecule MyD88 (Myeloid differentiation primary response gene 88) [28–31]. It has originally been identified as a protein induced during myeloid differentiation [32] but has since been shown to be recruited to IL-1Rs and most TLRs through its carboxyl terminal TIR domain [28, 30]. In addition, MyD88 contains an amino-terminal death domain that is responsible for the recruitment of downstream signalling mediators, including IL-1R associated kinase (IRAK)-1, IRAK4 and TNF receptor associated factor (TRAF)6 to the receptor complex [28, 30]. Ultimately, this leads to the activation of mitogen-activated protein kinases (MAPKs) as well as nuclear factor (NF)-κB and the transcription of inflammatory mediators such as TNFα [26] and the stabilisation of inflammatory response protein mRNAs through the AU-rich elements in the 3' untranslated region [33].
The essential role of MyD88 in IL-1R/TLR signal transduction has been demonstrated in MyD88-deficient mice. In response to IL-1R and IL-18R stimulation, MyD88-deficient macrophages show a loss in NF-κB and MAPK activation, as well as TNFα and IL-6 production [29]. This has also been observed for most TLRs, with the exception of TLR3 and TLR4 [34–37]; TLR3 does not utilise MyD88 for signal transduction while TLR4 recruits additional adaptor molecules that are responsible for MyD88 independent signalling. Subsequently, TIR domain homology searches led to the discovery of Mal (MyD88 adaptor like protein; also termed TIRAP [38, 39]). Mal-deficient mice show a reduction in TLR4- and TLR2-induced NF-κB activation [40, 41]. To date, Mal is thought to function as a sorting adaptor for TLR2 and TLR4, recruiting MyD88 to the receptor complex in the plasma membrane, through its ability to interact with phosphatidylinositol-4,5-bisphosphate [42] (Figure 1).
Evidence obtained in murine and human models indicate the involvement of the MyD88-dependent signalling pathway in the pathology of RA. TLR2 knockout and MyD88 knockout mice are protected from streptococcal cell wall-induced joint inflammation since these animals do not develop joint swelling [43, 44]. Furthermore, intra-articular administration of peptidoglycan or lipopolysaccharide, the ligands for TLR2 and TLR4, respectively, results in destructive arthritis in mice, which is also dependent on MyD88 [45, 46]. The IL-1R antagonist (IL-1RA) knockout mice model displays uncontrolled IL-1 signalling and leads to the development of chronic arthritis [47]. The arthritis observed in those mice is markedly reduced when backcrossed to TLR4-deficient but not TLR2-deficient mice, suggesting a TLR4-specific function in this model [48]. Furthermore, blocking TLR4 signalling with a naturally occurring antagonist in mice with CIA leads to reduced disease severity, even when administered after disease onset [49].
In humans, stimulation of TLR2- and TLR9-expressing RA synovial fibroblasts with peptidoglycan leads to the expression of matrix metalloproteinases and secretion of IL-6 and IL-8, while no activation has been observed in response to the TLR9 ligand CpG oligodeoxynucleotides [50]. Strengthening the role of TLR4 signalling in RA pathogenesis is the observation that serum and synovial fluid from RA patients stimulated TLR4 expressing CHO cells to up-regulate CD25 [51]. In accordance with this study are results obtained in RA synovial membrane cultures, where the over-expression of a dominant negative construct of MyD88 or Mal inhibits the spontaneous release of cytokines and matrix metalloproteinases [52, 53]. Based on these results, increased efforts have been made to identify potential endogenous TLR ligands in the joints of RA patients. Indeed, it has been shown that conditioned medium from RA synovial membrane cultures activates human macrophages in a MyD88- and Mal-dependent manner, further strengthening the involvement of an endogenous TLR ligand driving RA pathology [52, 53]. In addition to endogenous TLR ligands, exogenous ligands derived from infections might potentially also play a role in RA, although no ligand has so far been defined.
TRIF dependent TLR signalling
In addition to the MyD88-dependent TLR signalling pathway, which is shared with the IL-1Rs, TLRs also induce MyD88 independent signalling cascades. Stimulation of cells with double-stranded RNA or lipopolysaccharide (TLR3- and TLR4-ligands, respectively) results in the activation of interferon regulatory factors (IRFs). This is due to the presence of additional TLR adaptor molecules, which have been identified through TIR domain homology searches and include: TRIF (TIR-domain-containing adapter-inducing IFN-β; also termed TICAM-1), TRAM (TRIF-related adaptor molecule; also termed TICAM2) and SARM (TIR domain-sterile alpha and HEAT/Armadillo motif) [54].
Stimulation of TLR3 or TLR4 results in the recruitment of TRIF, and in the case of TLR4 also TRAM [55–57]. The dissociation of TRIF activates a complex consisting of the kinases IkappaB kinase (IKK)i and TANK-binding kinase (TBK)-1 as well as the scaffolding protein TRAF3 [58], which ultimately leads to the activation of IRF-3 and IRF-7 and the expression of IFN-inducible genes such as those encoding IFN-β, IP10 (inducible protein 10) and RANTES (Regulated on activation, normal T expressed and secreted) [26, 59, 60]. Moreover, TRIF recruitment has also been shown to be responsible for MyD88-independent activation of NF-κB. However, the exact mechanism of NF-κB activation by TRIF is still unclear. Some find that binding of receptor interacting protein (RIP)-1 to the RIHM (RIP interacting homology motif) domain of TRIF leads to the induction of NF-κB, while others suggest that an autocrine effect of TNFα, initially induced through IRF-3, is responsible for NF-κB activation [61, 62].
TRAM is structurally related to Mal and has therefore been suggested to function as a sorting adaptor, recruiting TRIF to TLR4 [42, 56]. In this context, TRAM has been shown to be recruited to the plasma membrane by myristoylation [63]. However, a recent study provides evidence that TRAM recruitment is subsequent to the endocytosis of the TLR4 complex [64]. Therefore, TRAM provides a mechanism that allows sequential activation of MyD88-dependent signalling while TLR4 is located in the plasma membrane, followed by TRIF-dependent signalling after TLR4 internalisation [64] (Figure 1).
SARM is the least investigated TLR adaptor molecule. So far, no activation function could be assigned to it. However, recent data describe SARM as an inhibitor of TRIF-dependent signalling [65]. SARM has been shown to interact with TRIF and expression of SARM in HEK293 cells led to the inhibition of TRIF-dependent, but not MyD88-dependent, NF-κB activation [65].
Some evidence indicates the involvement of the TRIF-dependent signalling pathway in the pathology of RA due to TLR3 stimulation. RNA released from necrotic synovial fluid cells has been shown to activate RA synovial fibroblasts via TLR3 [66]. Interestingly, RA synovial fibroblasts have been shown to respond to the TLR3 stimulation by producing TNFα while primary human skin fibroblasts do not [67]. This shows that TLR3 is functional in the inflamed synovium and that TLR3 stimulation could potentially result in the production of TNFα in the RA joint.
Other signalling pathways induced by TLRs
Up until now the focus of TLR signalling research has been on delineating the membrane-proximal adaptor molecules utilised. But determining the downstream pathways engaged is important in understanding TLR specificity, as well as providing therapeutic targets.
The involvement of protein tyrosine kinases (TKs) in TLR signalling was appreciated even before the TLRs themselves were discovered [68], but with dozens of TKs found in mammalian cells the identities of the molecules involved in TLR signalling have only been revealed recently [69]. There is good evidence to suggest that Hck [70, 71], Btk [72–75], Bmx [76, 77], Syk [78, 79] and Pyk2 [80–82] are involved in TLR signalling, even though the evidence can be hard to come by due to extensive redundancies found in TKs (Figure 1). The mechanisms by which these kinases operate in TLR signalling pathways still need to be resolved. Concomitantly, a number of TKs have been implicated in the negative regulation of TLR signalling. For example, members of the TAM receptor family inhibit both MyD88 and TRIF pathways by induction of suppressor of cytokine signalling (SOCS)-1 and -3 [83–85]. In light of these links between TKs and TLR signalling, the recently discovered TK inhibitors such as Dasatinib may potentially be useful in blocking harmful effects of TLR signalling in chronic inflammation [86].
Phosphatidylinositol-3-kinases (PI3Ks) belong to a large family of lipid signalling kinases that phosphorylate phosphoinositides and control numerous cellular functions, such as proliferation, survival and migration [87]. They consist of a catalytic 110 kDa subunit and a tightly associated 85 kDa regulatory subunit. PI3Ks become activated in response to numerous TLR stimuli, including lipopolysaccharide, peptido-glycan and CpG-DNA, and subsequently induce the phos-phorylation of Akt/protein kinas B [88, 89]. Current data suggest that the activation of PI3Ks, following TLR stimulation, leads to the inhibition of MAPKs and NF-κB as observed using chemical inhibitors or over-expression systems [88] (Figure 1). In the context of RA it is interesting to note that p110γ-knockout mice are resistant to models of RA and that the administration of PI3Kγ inhibitors restrain the progression of inflammation and joint damage [87]. However, the reduced incidence and severity of RA observed in PI3K-knockout mice is most likely due to its role in the T- and B-cell compartment rather than in innate immune cells [87].
It is likely that several TLRs are stimulated in the RA joint due to the release of numerous 'danger signals' following cell necrosis. The induction of TLR signalling pathways would subsequently lead to the expression of pro-inflammatory mediators, including cytokines and chemokines. These mediators, discussed in the next section, are able to feedback upon the macrophage to form an autocrine inflammatory loop, potentiating disease.
Activation of macrophages by cytokines
Several cytokines have a direct effect on monocytes/macrophages in the context of RA (Table 1) and exert pathological effects during disease progression. One such example is IL-15, which exhibits pro-inflammatory activity both in vitro and in CIA, and when blocked will reduce the incidence of disease [90]. However, this review focuses on six of those cytokines for which involvement in RA is well characterised: TNFα, IL-1, IL-10, macrophage migration inhibitory factor (MIF), IL-17, and receptor activator for NF-κB (RANK).
Interleukin-1
The IL-1 family of cytokines plays a significant role in RA and includes IL-1α, IL-1β, IL-1RA, IL-18, IL-33, and IL-1F5, 6, 7, 8, 9 and 10. IL-1β is a potent pro-inflammatory cytokine with roles in bone erosion and cartilage degradation, rather than in synovitis. In a streptococcal cell wall-induced arthritis model, IL-1-/- mice showed reduced late cellular infiltration and cartilage damage while joint swelling is unaffected [91]. Also, by crossing IL-1-/- mice with the TNFα-transgenic model of arthritis, Zwerina and colleagues [92] showed that IL-1 is essential for TNFα-mediated cartilage damage and has a partial role in TNFα-mediated bone damage. IL-1β is able to activate macrophages to induce the production of cytokines, reactive oxygen intermediates and prostaglandin (Table 1). Signalling is mediated through the dimerisation of two receptors, IL-1RI and IL-1R-AcP. A third receptor, IL-1RII, can also bind IL-1β but cannot mediate signalling due to a small cytoplasmic tail and acts as a decoy [93]. IL-1RA can also bind these receptors and acts as a competitive inhibitor. In the case of RA, IL-1β is more plentiful than IL-1RA, inducing a pro-inflammatory state [94]. The intracellular signalling cascade of IL-1 is similar to that of the MyD88-dependant TLR cascade discussed previously and involves the induction of IRAK1, IRAK4, MyD88 and transforming growth factor-activated kinase (TAK)1 [26]. NF-κB mediates multiple gene transcription events, and in the context of IL-1β is able to activate another transcription factor, ESE-1, which modulates several pro-inflammatory genes [95].
Tumour necrosis factor alpha
TNFα is considered to be the principal inflammatory cytokine in RA and is the major factor involved in inducing and maintaining synovitis. It is commonly found at high levels in RA patients and, as such, has been targeted successfully to alleviate disease symptoms. TNFα is a cytokine that both activates and can be produced by macrophages and therefore forms an autocrine inflammatory effect. As well as its well-documented effects (Figure 1), TNFα has been shown to affect both major histocompatibility complex and Fcγ receptor (FcγR) expression. TNFα is able to reduce the expression of HLA-DR on myeloid RA cells where this is brought back to normal upon the addition of anti-TNFα. TNF treatment of healthy monocytes also reduced HLA-DR and a mixed lymphocyte reaction [96]. TNFα is able to reduce the expression of all activating FcγRs in vitro where anti-TNFα can increase FcγRIIa and IIIa. However, in RA patients, anti-TNFα therapy is accompanied by an initial reduction in FcγRI but increases back to normal after therapy is finished [97].
Intra-cellular signalling is mediated through TNF-R1 and TNF-R2, which upon binding of TNFα will recruit several signalling molecules [98]. TRAF2 is recruited to the receptor and in conjunction with TAK1 is able to activate a signalling cascade resulting in JNK and c-Jun activation. RIP is recruited to this receptor complex, which in turn can activate the IKK signalosome to activate NF-κB. IKK2 and the p50 subunit of NF-κB have been shown to be essential for this process, while IKK1 is not [99, 100]. TRADD (TNFR-associated via death domain) and FADD (Fas-associated protein with death domain) are also recruited to the receptor signalling complex to induce apoptosis. NF-κB inducing kinase is another TNF receptor-associated factor described in TNFα induction, but has proven to be non-essential [101]. Zwerina and colleagues [102] recently showed that p38 MAPK was essential for TNFα-mediated bone degradation through affecting osteo-clast differentiation, but did not specify if this involves the activation of macrophages.
Macrophage migration inhibitory factor
MIF is able to activate and recruit macrophages during RA. It is the essential factor in RA fibroblast-conditioned medium for TNFα induction in monocytes [103]. This is mediated through CD74, the subsequent engagement of p38 MAPK, ERK, Src kinase, phospholipase A2 and PI3K pathways [104–108], and the binding of NF-κB and AP-1 to DNA to effect gene transcription [109]. MIF has been shown to be an endogenous antagonist of glucocorticoids (reviewed in [110, 111]); by inhibiting the latter, MIF enhances the inflammatory state through p38 MAPK and MAPK phosphatase 1 (MKP1) [112], which in turn deactivates p38, JNK and ERK. MKP1-deficiency has been associated with exacerbation of CIA [113], potentially by affecting MIF signalling. MIF is also able to negatively regulate p53 [114] through cyclooxygenase 2 [115] and the PI3K/Akt pathway [116] to arrest cell-apoptosis. Finally, MIF recruits monocytes/macrophages to the site of inflammation through CCL2 induction [117], or acting as a chemokine ligand on endothelial cell surfaces by directly binding to CXCR2 [118].
IL-10: anti- or pro-inflammatory in rheumatoid arthritis?
IL-10 is widely considered to be a powerful anti-inflammatory cytokine that is able to suppress the production of TNFα, IL-6 and IL-1 from macrophages. Its role in RA disease-associated macrophages, however, is controversial. Human IL-10 has little effect when used to alleviate disease in RA patients. On the contrary, circulating monocytes have been shown to upregulate the expression of FcγRI and FcγRIIa in response of IL-10 [119, 120], which may potentially enhance disease. IL-10 has also been shown to upregulate various genes associated with pro-inflammatory function, as well as the IFN-γ-inducible genes [121]. In response to IL-10, RA macrophages upregulate TNF receptor (TNFR)1 and TNFR2 mRNA and produce elevated levels of IL-1β and IL-6 in response to TNFα and macrophage-colony stimulating factor [122]. However, others suggest that IL-10 upregulates the soluble form of the TNFR rather than the membrane bound form, which in turn would inhibit inflammation [123]. To confound the matter further, it has been shown repeatedly that in whole RA synovial cultures the addition of IL-10 suppresses the level of TNFα and IL-1β two- to three-fold [124] (reviewed in [125]), in sharp contrast to the RA macrophage phenotype. IL-10 treatment of CIA mice has also been shown to inhibit disease progression [126]. Overall, this suggests that arthritic macrophages may have altered signalling patterns when compared to other cell types, and IL-10 may have both anti- and pro-inflammatory functions in RA.
In macrophages the principal intracellular mediator of suppressive effects of IL-10 is STAT3 [127]. IL-10 binds to the IL-10R1/IL-10R2 receptor complex and recruits both Jak1 and Tyk2 to activate STAT3 [125]. The tyrosine residues contained within the YXXQ-STAT3-docking site on IL-10R1 were found to be essential for this interaction [128]. The mechanism of IL-10 suppressive activity is unclear [125]. IL-10 has been reported to reduce the activity of the IKK signalosome and induce the translocation of NF-κB p50:p50 homodimers, resulting in a suppression of NF-κB-mediated gene transcription [129]. However, our own studies have found no effect on the activation of NF-κB [125, 130]. It should also be noted that IL-10 is able to strongly induce the expression of SOCS-3, a classic suppressor of cytokine signalling [121]. However, the role for SOCS-3 in mediating the effect of IL-10 is undetermined as the cytokine is still functional in SOCS-3-/- mice [131].
Interleukin-17
CD4+ helper T cells that secrete IL-17 are at the centre of an orchestra of cellular interactions that mediate acute inflammation in many autoimmune diseases. This has renewed the interest in understanding signalling by the IL-17 receptor, which is found in macrophages [132] and synovial fibroblasts [133]. As with other cytokines, the IL-17 receptor is a complex of at least two separate proteins, IL-17RA and IL-17RC [134]. These, together with IL-17RB, RD and RE, form a distinct receptor superfamily with little similarity to other cytokine receptors. Likewise, there are no fewer than six members in the IL-17 cytokine family (reviewed in [135]). Two of them, IL-17A and IL-17F, are secreted by Th17 cells. A third one, IL-17E or IL-25, is associated with Th2 responses. The functions of the other family members are unknown at the moment.
It has been suggested that the IL-17 receptor chains are pre-assembled before ligand binding [136], but the details are murky. So are the intracellular signalling pathways utilized by IL-17R. IL-17RA has a long cytoplasmic tail, but factors that engage this tail are unknown, with the exception of TRAF6, which is needed for IL-17 signalling [137]. But as IL-17RA has little similarity to the TNF receptor superfamily, the structural basis of this interaction is unclear at this stage. Activation of NF-κB and MAPK pathways lead to transcription and mRNA stabilisation of pro-inflammatory molecules; transcription factors such as AP-1 and c/EPB are also important in mediating the full activities of IL-17 (reviewed in [138]).
RANK/RANKL/osteoprotegerin
Bone destruction in arthritic conditions can be directly attributed to osteoclasts, a specialised lineage of macrophages involved in normal bone development and remodelling. In RA they are found to be overactive, and this can largely be attributed to the pro-inflammatory milieu found in RA joints, which includes excessive RANK ligand (RANKL)-RANK signalling.
RANKL is a member of the TNF superfamily and, correspondingly, RANK belongs to the TNFR superfamily. The discovery that RANKL-RANK signalling is the key molecular event in osteoclast differentiation by several independent groups in the late 1990s [139–142] gave birth to the field of osteoimmunology. RANKL is normally expressed in osteoblasts and stromal cells, but in pro-inflammatory environments, as in RA, RANKL expression is elevated and spreads, particularly to activated T cells [139, 143, 144]. This increases the maturation and activity of osteoclasts, thus tipping the bone metabolic balance in favour of destruction. A further level of regulation is provided by osteoprotegerin, which acts as a soluble decoy receptor of RANKL, and thus is an effective inhibitor of RANK signalling and osteoclast differentiation [145, 146]. Given the importance of bone destruction as a cause of morbidity in RA, the RANK-RANKL-osteoprotegerin triad is now a target for therapeutic intervention.
Characterisation of the signalling pathways utilised by RANKL-RANK is aided by their similarities to other TNF superfamily members. In essence, RANK and RANKL are trimeric molecules; upon ligand binding multiple TRAFs are recruited, with TRAF6 being the critical adaptor as its absence incapacitates osteoclasts [147, 148]. TRAF6, together with Gab2 [149], triggers NF-kB, Akt and Jnk pathways. Ultimately, expression of osteoclastogenic genes is switched on by a cascade of transcription factors that include NF-κB, NFATc1, c-Fos [150, 151] and osterix [152].
T cell-mediated activation of the innate immune cells
The role of T cells in RA pathogenesis has been questioned by some, but as cited earlier it is now accepted that autoreactive T cell activation is essential in the development of the full blown disease both in human and animal models. However, it is unclear what the relative contributions from T cell-derived versus TLR- or cytokine-derived signals are. Perhaps T cells prime the initial inflammation; once tissue damage is occurring other signals take over in the maintenance and amplification of inflammation at disease sites.
The signals innate immune cells receive from arthritogenic T cells are still being worked out. Some of the soluble factors, such as IL-17 and RANK, have already been covered, but they clearly are not the whole story. For example, RA T cells have been shown to induce TNFα production in monocytes in a direct cell-contact, PI3K-dependent manner [153–155]. Separately, but perhaps in a related finding, it was found that T cells generate microparticles that can promote cytokine production in macrophages [156]. The molecular basis of these phenomena is still being worked out; molecules such as CD40L and membrane bound-TNFα have been implicated [157, 158]. Modelling this interaction in vitro is difficult as the outcome is highly dependent on the status of both T cells and macrophages [155, 158]. It is likely that in vivo studies will be needed to resolve this question.
Conclusion
RA is an auto-inflammatory disease where multiple mechanisms of the immune system play a role in its pathology. Current evidence in human suggests a strong influence of innate immune cells, such as macrophages and synovial fibroblasts, in the progression of disease, as they produce large amounts of pro-inflammatory mediators leading to the destruction of the cartilage and bone.
Given the success of anti-TNFα therapy, there has been a great deal of interest in elucidating the pathways driving the production of this cytokine as well as other inflammatory mediators in RA. However, the continuous and systemic blockade of a cytokine results in unwanted side effects, such as increased infections. Current research on a new generation of anti-inflammatory drugs has focused on blocking intracellular signalling pathways, for example, NF-κB/IKK2 and p38 MAPK. However, no compounds have succeeded in the clinic so far. A major problem could be that both these kinases are ubiquitously expressed, which may lead to side effects. Therefore, there is a need for more specific targets that would either affect specific parts of the immune response, act only in specific tissues/cells or would actually lead to the complete resolution of the chronic inflammation. The use of specific blocking antibodies as well as emerging technologies such as small interfering RNA will expand our knowledge on particular signalling transducers in the context of the disease. Therefore, further elucidation of the signalling pathways driving chronic inflammation in disease-relevant models could potentially lead to the identification of therapeutic targets.
Note
The Scientific Basis of Rheumatology: A Decade of Progress
This article is part of a special collection of reviews, The Scientific Basis of Rheumatology: A Decade of Progress, published to mark Arthritis Research & Therapy's 10th anniversary.
Other articles in this series can be found at: http://arthritis-research.com/sbr
Abbreviations
- CIA:
-
collagen-induced arthritis
- FcγR:
-
Fcγ receptor
- IFN:
-
interferon
- IKK:
-
IkappaB kinase
- IL:
-
interleukin
- IL-1RA:
-
IL-1R antagonist
- IRAK:
-
IL-1R associated kinase
- IRF:
-
interferon regulatory factor
- MAPK:
-
mitogen-activated protein kinase
- MIF:
-
macrophage migration inhibitory factor
- NF:
-
nuclear factor
- PI3K:
-
phosphatidylinositol-3-kinase
- RA:
-
rheumatoid arthritis
- RANK:
-
receptor activator for NF-κB
- RANKL:
-
RANK ligand
- RIP:
-
receptor interacting protein
- SARM:
-
TIR domain-sterile alpha and HEAT/Armadillo motif
- SOCS:
-
suppressor of cytokine signalling
- TAK:
-
transforming growth factor-activated kinase
- TIR:
-
Toll/IL-1 receptor
- TK:
-
tyrosine kinase
- TLR:
-
toll-like receptor
- TNF:
-
tumour necrosis factor
- TNFR:
-
TNF receptor
- TRAF:
-
TNF receptor associated factor
- TRAM:
-
TRIF-related adaptor molecule
- TRIF:
-
TIR-domain-containing adapter-inducing interferon-β.
References
Janeway CA: How the immune system protects the host from infection. Microbes Infect. 2001, 3: 1167-1171. 10.1016/S1286-4579(01)01477-0.
Cho YG, Cho ML, Min SY, Kim HY: Type II collagen autoimmunity in a mouse model of human rheumatoid arthritis. Autoimmun Rev. 2007, 7: 65-70. 10.1016/j.autrev.2007.08.001.
Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ: Divergent pro- and anti-inflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003, 198: 1951-1957. 10.1084/jem.20030896.
Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, Yamaguchi T, Nomura T, Ito H, Nakamura T, Sakaguchi N, Sakaguchi S: Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med. 2007, 204: 2803-2812. 10.1084/jem.20071397.
Kremer JM, Westhovens R, Leon M, Di Giorgio E, Alten R, Steinfeld S, Russell A, Dougados M, Emery P, Nuamah IF, Williams GR, Becker JC, Hagerty DT, Moreland LW: Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig. N Engl J Med. 2003, 349: 1907-1915. 10.1056/NEJMoa035075.
Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T: Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004, 350: 2572-2581. 10.1056/NEJMoa032534.
Seldin MF, Amos CI, Ward R, Gregersen PK: The genetics revolution and the assault on rheumatoid arthritis. Arthritis Rheum. 1999, 42: 1071-1079. 10.1002/1529-0131(199906)42:6<1071::AID-ANR1>3.0.CO;2-8.
Feldmann M, Brennan FM, Foxwell BM, Taylor PC, Williams RO, Maini RN: Anti-TNF therapy: where have we got to in 2005?. J Autoimmun. 2005, 25 (Suppl): 26-28. 10.1016/j.jaut.2005.09.006.
Feldmann M, Brennan FM, Maini RN: Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996, 14: 397-440. 10.1146/annurev.immunol.14.1.397.
Inglis JJ, Criado G, Medghalchi M, Andrews M, Sandison A, Feldmann M, Williams RO: Collagen-induced arthritis in C57BL/6 mice is associated with a robust and sustained T-cell response to type II collagen. Arthritis Res Ther. 2007, 9: R113-10.1186/ar2319.
Williams RO, Feldmann M, Maini RN: Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc Natl Acad Sci USA. 1992, 89: 9784-9788. 10.1073/pnas.89.20.9784.
Edwards SW, Hallett MB: Seeing the wood for the trees: the forgotten role of neutrophils in rheumatoid arthritis. Immunol Today. 1997, 18: 320-324. 10.1016/S0167-5699(97)01087-6.
Woolley DE: The mast cell in inflammatory arthritis. N Engl J Med. 2003, 348: 1709-1711. 10.1056/NEJMcibr023206.
Dalbeth N, Callan MF: A subset of natural killer cells is greatly expanded within inflamed joints. Arthritis Rheum. 2002, 46: 1763-1772. 10.1002/art.10410.
Tsan MF, Gao B: Endogenous ligands of Toll-like receptors. J Leukoc Biol. 2004, 76: 514-519. 10.1189/jlb.0304127.
Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A: Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol Rev. 2005, 204: 27-42. 10.1111/j.0105-2896.2005.00239.x.
Brentano F, Kyburz D, Schorr O, Gay R, Gay S: The role of Toll-like receptor signalling in the pathogenesis of arthritis. Cell Immunol. 2005, 233: 90-96. 10.1016/j.cellimm.2005.04.018.
Drexler SK, Sacre SM, Foxwell BM: Toll-like receptors: a new target in rheumatoid arthritis?. Expert Rev Clin Immunol. 2006, 2: 585-599. 10.1586/1744666X.2.4.585.
Marshak-Rothstein A: Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006, 6: 823-835. 10.1038/nri1957.
Marshak-Rothstein A, Rifkin IR: Immunologically active autoantigens: the role of toll-like receptors in the development of chronic inflammatory disease. Annu Rev Immunol. 2007, 25: 419-441. 10.1146/annurev.immunol.22.012703.104514.
Searle J, Kerr JF, Bishop CJ: Necrosis and apoptosis: distinct modes of cell death with fundamentally different significance. Pathol Annu. 1982, 17: 229-259.
Schett G, Redlich K, Xu Q, Bizan P, Groger M, Tohidast-Akrad M, Kiener H, Smolen J, Steiner G: Enhanced expression of heat shock protein 70 (hsp70) and heat shock factor 1 (HSF1) activation in rheumatoid arthritis synovial tissue. Differential regulation of hsp70 expression and hsf1 activation in synovial fibroblasts by proinflammatory cytokines, shear stress, and antiinflammatory drugs. J Clin Invest. 1998, 102: 302-311. 10.1172/JCI2465.
Scott DL, Delamere JP, Walton KW: The distribution of fibronectin in the pannus in rheumatoid arthritis. Br J Exp Pathol. 1981, 62: 362-368.
Yu D, Rumore PM, Liu Q, Steinman CR: Soluble oligonucleosomal complexes in synovial fluid from inflamed joints. Arthritis Rheum. 1997, 40: 648-654. 10.1002/art.1780400409.
Sakaguchi S, Negishi H, Asagiri M, Nakajima C, Mizutani T, Takaoka A, Honda K, Taniguchi T: Essential role of IRF-3 in lipopolysaccharide-induced interferon-beta gene expression and endotoxin shock. Biochem Biophys Res Commun. 2003, 306: 860-866. 10.1016/S0006-291X(03)01049-0.
Akira S, Takeda K: Toll-like receptor signalling. Nat Rev Immunol. 2004, 4: 499-511. 10.1038/nri1391.
Wesche H, Korherr C, Kracht M, Falk W, Resch K, Martin MU: The interleukin-1 receptor accessory protein (IL-1RAcP) is essential for IL-1-induced activation of interleukin-1 receptor-associated kinase (IRAK) and stress-activated protein kinases (SAP kinases). J Biol Chem. 1997, 272: 7727-7731. 10.1074/jbc.272.12.7727.
Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z: MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity. 1997, 7: 837-847. 10.1016/S1074-7613(00)80402-1.
Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S: Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998, 9: 143-150. 10.1016/S1074-7613(00)80596-8.
Burns K, Martinon F, Esslinger C, Pahl H, Schneider P, Bodmer JL, Di Marco F, French L, Tschopp J: MyD88, an adapter protein involved in interleukin-1 signaling. J Biol Chem. 1998, 273: 12203-12209. 10.1074/jbc.273.20.12203.
Janssens S, Beyaert R: A universal role for MyD88 in TLR/IL-1R-mediated signaling. Trends Biochem Sci. 2002, 27: 474-482. 10.1016/S0968-0004(02)02145-X.
Lord KA, Hoffman-Liebermann B, Liebermann DA: Nucleotide sequence and expression of a cDNA encoding MyD88, a novel myeloid differentiation primary response gene induced by IL6. Oncogene. 1990, 5: 1095-1097.
Clark AR, Dean JL, Saklatvala J: Post-transcriptional regulation of gene expression by mitogen-activated protein kinase p38. FEBS Lett. 2003, 546: 37-44. 10.1016/S0014-5793(03)00439-3.
Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A: The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001, 410: 1099-1103. 10.1038/35074106.
Heil F, Ahmad-Nejad P, Hemmi H, Hochrein H, Ampenberger F, Gellert T, Dietrich H, Lipford G, Takeda K, Akira S, Wagner H, Bauer S: The Toll-like receptor 7 (TLR7)-specific stimulus loxoribine uncovers a strong relationship within the TLR7, 8 and 9 subfamily. Eur J Immunol. 2003, 33: 2987-2997. 10.1002/eji.200324238.
Schnare M, Holt AC, Takeda K, Akira S, Medzhitov R: Recognition of CpG DNA is mediated by signaling pathways dependent on the adaptor protein MyD88. Curr Biol. 2000, 10: 1139-1142. 10.1016/S0960-9822(00)00700-4.
Takeuchi O, Kaufmann A, Grote K, Kawai T, Hoshino K, Morr M, Muhlradt PF, Akira S: Cutting edge: preferentially the R-stereoisomer of the mycoplasmal lipopeptide macrophage-activating lipopeptide-2 activates immune cells through a toll-like receptor 2- and MyD88-dependent signaling pathway. J Immunol. 2000, 164: 554-557.
Fitzgerald KA, Palsson-McDermott EM, Bowie AG, Jefferies CA, Mansell AS, Brady G, Brint E, Dunne A, Gray P, Harte MT, McMurray D, Smith DE, Sims JE, Bird TA, O'Neill LA: Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 2001, 413: 78-83. 10.1038/35092578.
Horng T, Barton GM, Medzhitov R: TIRAP: an adapter molecule in the Toll signaling pathway. Nat Immunol. 2001, 2: 835-841. 10.1038/ni0901-835.
Horng T, Barton GM, Flavell RA, Medzhitov R: The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature. 2002, 420: 329-333. 10.1038/nature01180.
Yamamoto M, Sato S, Hemmi H, Sanjo H, Uematsu S, Kaisho T, Hoshino K, Takeuchi O, Kobayashi M, Fujita T, Takeda K, Akira S: Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature. 2002, 420: 324-329. 10.1038/nature01182.
Kagan JC, Medzhitov R: Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell. 2006, 125: 943-955. 10.1016/j.cell.2006.03.047.
Joosten LA, Koenders MI, Smeets RL, Heuvelmans-Jacobs M, Helsen MM, Takeda K, Akira S, Lubberts E, Loo van de FA, Berg van den WB: Toll-like receptor 2 pathway drives streptococcal cell wall-induced joint inflammation: critical role of myeloid differentiation factor 88. J Immunol. 2003, 171: 6145-6153.
Pierer M, Rethage J, Seibl R, Lauener R, Brentano F, Wagner U, Hantzschel H, Michel BA, Gay RE, Gay S, Kyburz D: Chemokine secretion of rheumatoid arthritis synovial fibroblasts stimulated by toll-like receptor 2 ligands. J Immunol. 2004, 172: 1256-1265.
Liu ZQ, Deng GM, Foster S, Tarkowski A: Staphylococcal peptidoglycans induce arthritis. Arthritis Res. 2001, 3: 375-380. 10.1186/ar330.
Choe JY, Crain B, Wu SR, Corr M: Interleukin 1 receptor dependence of serum transferred arthritis can be circumvented by toll-like receptor 4 signaling. J Exp Med. 2003, 197: 537-542. 10.1084/jem.20021850.
Horai R, Saijo S, Tanioka H, Nakae S, Sudo K, Okahara A, Ikuse T, Asano M, Iwakura Y: Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med. 2000, 191: 313-320. 10.1084/jem.191.2.313.
Abdollahi-Roodsaz S, Joosten LA, Koenders MI, Devesa I, Roelofs MF, Radstake TR, Heuvelmans-Jacobs M, Akira S, Nicklin MJ, Ribeiro-Dias F, Berg van den WB: Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest. 2008, 118: 205-216. 10.1172/JCI32639.
Abdollahi-Roodsaz S, Joosten LA, Roelofs MF, Radstake TR, Matera G, Popa C, Meer van der JW, Netea MG, Berg van den WB: Inhibition of Toll-like receptor 4 breaks the inflammatory loop in autoimmune destructive arthritis. Arthritis Rheum. 2007, 56: 2957-2967. 10.1002/art.22848.
Kyburz D, Rethage J, Seibl R, Lauener R, Gay RE, Carson DA, Gay S: Bacterial peptidoglycans but not CpG oligodeoxynucleotides activate synovial fibroblasts by toll-like receptor signaling. Arthritis Rheum. 2003, 48: 642-650. 10.1002/art.10848.
Roelofs MF, Joosten LA, Abdollahi-Roodsaz S, van Lieshout AW, Sprong T, Hoogen van den FH, Berg van den WB, Radstake TR: The expression of toll-like receptors 3 and 7 in rheumatoid arthritis synovium is increased and costimulation of toll-like receptors 3, 4, and 7/8 results in synergistic cytokine production by dendritic cells. Arthritis Rheum. 2005, 52: 2313-2322. 10.1002/art.21278.
Sacre SM, Andreakos E, Kiriakidis S, Amjadi P, Lundberg A, Giddins G, Feldmann M, Brennan F, Foxwell BM: The Toll-like receptor adaptor proteins MyD88 and Mal/TIRAP contribute to the inflammatory and destructive processes in a human model of rheumatoid arthritis. Am J Pathol. 2007, 170: 518-525. 10.2353/ajpath.2007.060657.
Sacre SM, Drexler SK, Andreakos E, Feldmann M, Brennan FM, Foxwell BM: Could toll-like receptors provide a missing link in chronic inflammation in rheumatoid arthritis? Lessons from a study on human rheumatoid tissue. Ann Rheum Dis. 2007, 66 (Suppl 3): iii81-86. 10.1136/ard.2007.079012.
O'Neill LA, Bowie AG: The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007, 7: 353-364. 10.1038/nri2079.
Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T: TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat Immunol. 2003, 4: 161-167. 10.1038/ni886.
Oshiumi H, Sasai M, Shida K, Fujita T, Matsumoto M, Seya T: TIR-containing adapter molecule (TICAM)-2, a bridging adapter recruiting to toll-like receptor 4 TICAM-1 that induces interferon-beta. J Biol Chem. 2003, 278: 49751-49762. 10.1074/jbc.M305820200.
Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S: Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003, 301: 640-643. 10.1126/science.1087262.
Häcker H, Redecke V, Blagoev B, Kratchmarova I, Hsu LC, Wang GG, Kamps MP, Raz E, Wagner H, Häcker G, Mann M, Karin M: Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature. 2006, 439: 204-207. 10.1038/nature04369.
Kawai T, Takeuchi O, Fujita T, Inoue J, Muhlradt PF, Sato S, Hoshino K, Akira S: Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol. 2001, 167: 5887-5894.
Honda K, Taniguchi T: IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006, 6: 644-658. 10.1038/nri1900.
Cusson-Hermance N, Khurana S, Lee TH, Fitzgerald KA, Kelliher MA: Rip1 mediates the Trif-dependent toll-like receptor 3- and 4-induced NF-{kappa}B activation but does not contribute to interferon regulatory factor 3 activation. J Biol Chem. 2005, 280: 36560-36566. 10.1074/jbc.M506831200.
Covert MW, Leung TH, Gaston JE, Baltimore D: Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science. 2005, 309: 1854-1857. 10.1126/science.1112304.
Rowe DC, McGettrick AF, Latz E, Monks BG, Gay NJ, Yamamoto M, Akira S, O'Neill LA, Fitzgerald KA, Golenbock DT: The myristoylation of TRIF-related adaptor molecule is essential for Toll-like receptor 4 signal transduction. Proc Natl Acad Sci USA. 2006, 103: 6299-6304. 10.1073/pnas.0510041103.
Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R: TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. 2008, 9: 361-368. 10.1038/ni1569.
Carty M, Goodbody R, Schroder M, Stack J, Moynagh PN, Bowie AG: The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat Immunol. 2006, 7: 1074-1081. 10.1038/ni1382.
Brentano F, Schorr O, Gay RE, Gay S, Kyburz D: RNA released from necrotic synovial fluid cells activates rheumatoid arthritis synovial fibroblasts via Toll-like receptor 3. Arthritis Rheum. 2005, 52: 2656-2665. 10.1002/art.21273.
Lundberg AM, Drexler SK, Monaco C, Williams LM, Sacre SM, Feldmann M, Foxwell BM: Key differences in TLR3/poly I:C signaling and cytokine induction by human primary cells: a phenomenon absent from murine cell systems. Blood. 2007, 110: 3245-3252. 10.1182/blood-2007-02-072934.
Novogrodsky A, Vanichkin A, Patya M, Gazit A, Osherov N, Levitzki A: Prevention of lipopolysaccharide-induced lethal toxicity by tyrosine kinase inhibitors. Science. 1994, 264: 1319-1322. 10.1126/science.8191285.
Page TH, Smolinska MJ, Gillespie J, Urbaniak AM, Foxwell BM: Tyrosine kinases and inflammatory signalling. Curr Mol Med. 2008, in press.
Smolinska MJ, Horwood NJ, Page TH, Smallie T, Foxwell BM: Chemical inhibition of Src family kinases affects major LPS-activated pathways in primary human macrophages. Mol Immunol. 2008, 45: 990-1000. 10.1016/j.molimm.2007.07.026.
English BK, Ihle JN, Myracle A, Yi T: Hck tyrosine kinase activity modulates tumor necrosis factor production by murine macrophages. J Exp Med. 1993, 178: 1017-1022. 10.1084/jem.178.3.1017.
Schmidt NW, Thieu VT, Mann BA, Ahyi AN, Kaplan MH: Bruton's tyrosine kinase is required for TLR-induced IL-10 production. J Immunol. 2006, 177: 7203-7210.
Jefferies CA, Doyle S, Brunner C, Dunne A, Brint E, Wietek C, Walch E, Wirth T, O'Neill LA: Bruton's tyrosine kinase is a Toll/interleukin-1 receptor domain-binding protein that participates in nuclear factor kappaB activation by Toll-like receptor 4. J Biol Chem. 2003, 278: 26258-26264. 10.1074/jbc.M301484200.
Horwood NJ, Mahon T, McDaid JP, Campbell J, Mano H, Brennan FM, Webster D, Foxwell BM: Bruton's tyrosine kinase is required for lipopolysaccharide-induced tumor necrosis factor alpha production. J Exp Med. 2003, 197: 1603-1611. 10.1084/jem.20021845.
Horwood NJ, Page TH, McDaid JP, Palmer CD, Campbell J, Mahon T, Brennan FM, Webster D, Foxwell BM: Bruton's tyrosine kinase is required for TLR2 and TLR4-induced TNF, but not IL-6, production. J Immunol. 2006, 176: 3635-3641.
Palmer CD, Mutch BE, Workman S, McDaid JP, Horwood NJ, Foxwell BM: Bmx tyrosine kinase regulates TLR4-induced IL-6 production in human macrophages independently of p38 MAPK and NFkapp}B activity. Blood. 2008, 111: 1781-1788. 10.1182/blood-2007-07-102343.
Palmer CD, Mutch BE, Page TH, Horwood NJ, Foxwell BM: Bmx regulates LPS-induced IL-6 and VEGF production via mRNA stability in rheumatoid synovial fibroblasts. Biochem Biophys Res Commun. 2008, 370: 599-602. 10.1016/j.bbrc.2008.03.142.
Yamada T, Fujieda S, Yanagi S, Yamamura H, Inatome R, Sunaga H, Saito H: Protein-tyrosine kinase Syk expressed in human nasal fibroblasts and its effect on RANTES production. J Immunol. 2001, 166: 538-543.
Arndt PG, Suzuki N, Avdi NJ, Malcolm KC, Worthen GS: Lipopolysaccharide-induced c-Jun NH2-terminal kinase activation in human neutrophils: role of phosphatidylinositol 3-Kinase and Syk-mediated pathways. J Biol Chem. 2004, 279: 10883-10891. 10.1074/jbc.M309901200.
Williams LM, Ridley AJ: Lipopolysaccharide induces actin reorganization and tyrosine phosphorylation of Pyk2 and paxillin in monocytes and macrophages. J Immunol. 2000, 164: 2028-2036.
Hazeki K, Masuda N, Funami K, Sukenobu N, Matsumoto M, Akira S, Takeda K, Seya T, Hazeki O: Toll-like receptor-mediated tyrosine phosphorylation of paxillin via MyD88-dependent and -independent pathways. Eur J Immunol. 2003, 33: 740-747. 10.1002/eji.200323375.
Achuthan A, Elsegood C, Masendycz P, Hamilton JA, Scholz GM: CpG DNA enhances macrophage cell spreading by promoting the Src-family kinase-mediated phosphorylation of paxillin. Cell Signal. 2006, 18: 2252-2261. 10.1016/j.cellsig.2006.05.007.
Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G: TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007, 131: 1124-1136. 10.1016/j.cell.2007.10.034.
Lu Q, Lemke G: Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science. 2001, 293: 306-311. 10.1126/science.1061663.
Camenisch TD, Koller BH, Earp HS, Matsushima GK: A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J Immunol. 1999, 162: 3498-3503.
Hantschel O, Rix U, Schmidt U, Bürckstümmer T, Kneidinger M, Schütze G, Colinge J, Bennett KL, Ellmeier W, Valent P, Superti-Furga G: The Btk tyrosine kinase is a major target of the Bcr-Abl inhibitor dasatinib. Proc Natl Acad Sci USA. 2007, 104: 13283-13288. 10.1073/pnas.0702654104.
Rommel C, Camps M, Ji H: PI3K delta and PI3K gamma: partners in crime in inflammation in rheumatoid arthritis and beyond?. Nat Rev Immunol. 2007, 7: 191-201. 10.1038/nri2036.
Fukao T, Koyasu S: PI3K and negative regulation of TLR signaling. Trends Immunol. 2003, 24: 358-363. 10.1016/S1471-4906(03)00139-X.
Kim AH, Khursigara G, Sun X, Franke TF, Chao MV: Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol. 2001, 21: 893-901. 10.1128/MCB.21.3.893-901.2001.
Ferrari-Lacraz S, Zanelli E, Neuberg M, Donskoy E, Kim YS, Zheng XX, Hancock WW, Maslinski W, Li XC, Strom TB, Moll T: Targeting IL-15 receptor-bearing cells with an antagonist mutant IL-15/Fc protein prevents disease development and progression in murine collagen-induced arthritis. J Immunol. 2004, 173: 5818-5826.
Berg van den WB, Joosten LA, Kollias G, Loo van De FA: Role of tumour necrosis factor alpha in experimental arthritis: separate activity of interleukin 1beta in chronicity and cartilage destruction. Ann Rheum Dis. 1999, 58 (Suppl 1): I40-48.
Zwerina J, Redlich K, Polzer K, Joosten L, Krönke G, Distler J, Hess A, Pundt N, Pap T, Hoffmann O, Gasser J, Scheinecker C, Smolen JS, Berg van den W, Schett G: TNF-induced structural joint damage is mediated by IL-1. Proc Natl Acad Sci USA. 2007, 104: 11742-11747. 10.1073/pnas.0610812104.
Colotta F, Re F, Muzio M, Bertini R, Polentarutti N, Sironi M, Giri JG, Dower SK, Sims JE, Mantovani A: Interleukin-1 type II receptor: a decoy target for IL-1 that is regulated by IL-4. Science. 1993, 261: 472-475. 10.1126/science.8332913.
Chikanza IC, Roux-Lombard P, Dayer JM, Panayi GS: Dysregulation of the in vivo production of interleukin-1 receptor antagonist in patients with rheumatoid arthritis. Pathogenetic implications. Arthritis Rheum. 1995, 38: 642-648. 10.1002/art.1780380511.
Grall F, Gu X, Tan L, Cho JY, Inan MS, Pettit AR, Thamrongsak U, Choy BK, Manning C, Akbarali Y, Zerbini L, Rudders S, Goldring SR, Gravallese EM, Oettgen P, Goldring MB, Libermann TA: Responses to the proinflammatory cytokines interleukin-1 and tumor necrosis factor alpha in cells derived from rheumatoid synovium and other joint tissues involve nuclear factor kappaB-mediated induction of the Ets transcription factor ESE-1. Arthritis Rheum. 2003, 48: 1249-1260. 10.1002/art.10942.
Mueller RB, Skapenko A, Grunke M, Wendler J, Stuhlmuller B, Kalden JR, Schulze-Koops H: Regulation of myeloid cell function and major histocompatibility complex class II expression by tumor necrosis factor. Arthritis Rheum. 2005, 52: 451-460. 10.1002/art.20863.
Wijngaarden S, Winkel van de JG, Bijlsma JW, Lafeber FP, van Roon JA: Treatment of rheumatoid arthritis patients with anti-TNF-alpha monoclonal antibody is accompanied by down-regulation of the activating Fcgamma receptor I on monocytes. Clin Exp Rheumatol. 2008, 26: 89-95.
Tumor Necrosis Factor Pathway. [http://stke.sciencemag.org/cgi/cm/CMP_7107]
Andreakos E, Smith C, Kiriakidis S, Monaco C, de Martin R, Brennan FM, Paleolog E, Feldmann M, Foxwell BM: Heterogeneous requirement of IkappaB kinase 2 for inflammatory cytokine and matrix metalloproteinase production in rheumatoid arthritis: implications for therapy. Arthritis Rheum. 2003, 48: 1901-1912. 10.1002/art.11044.
Campbell IK, Gerondakis S, O'Donnell K, Wicks IP: Distinct roles for the NF-kappaB1 (p50) and c-Rel transcription factors in inflammatory arthritis. J Clin Invest. 2000, 105: 1799-1806. 10.1172/JCI8298.
Smith C, Andreakos E, Crawley JB, Brennan FM, Feldmann M, Foxwell BM: NF-kappaB-inducing kinase is dispensable for activation of NF-kappaB in inflammatory settings but essential for lymphotoxin beta receptor activation of NF-kappaB in primary human fibroblasts. J Immunol. 2001, 167: 5895-5903.
Zwerina J, Hayer S, Redlich K, Bobacz K, Kollias G, Smolen JS, Schett G: Activation of p38 MAPK is a key step in tumor necrosis factor-mediated inflammatory bone destruction. Arthritis Rheum. 2006, 54: 463-472. 10.1002/art.21626.
Leech M, Metz C, Hall P, Hutchinson P, Gianis K, Smith M, Weedon H, Holdsworth SR, Bucala R, Morand EF: Macrophage migration inhibitory factor in rheumatoid arthritis: evidence of proinflammatory function and regulation by glucocorticoids. Arthritis Rheum. 1999, 42: 1601-1608. 10.1002/1529-0131(199908)42:8<1601::AID-ANR6>3.0.CO;2-B.
Amin MA, Haas CS, Zhu K, Mansfield PJ, Kim MJ, Lackowski NP, Koch AE: Migration inhibitory factor up-regulates vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 via Src, PI3 kinase, and NFkappaB. Blood. 2006, 107: 2252-2261. 10.1182/blood-2005-05-2011.
Santos LL, Lacey D, Yang Y, Leech M, Morand EF: Activation of synovial cell p38 MAP kinase by macrophage migration inhibitory factor. J Rheumatol. 2004, 31: 1038-1043.
Sampey AV, Hall PH, Mitchell RA, Metz CN, Morand EF: Regulation of synoviocyte phospholipase A2 and cyclooxygenase 2 by macrophage migration inhibitory factor. Arthritis Rheum. 2001, 44: 1273-1280. 10.1002/1529-0131(200106)44:6<1273::AID-ART219>3.0.CO;2-8.
Lue H, Kapurniotu A, Fingerle-Rowson G, Roger T, Leng L, Thiele M, Calandra T, Bucala R, Bernhagen J: Rapid and transient activation of the ERK MAPK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on JAB1/CSN5 and Src kinase activity. Cell Signal. 2006, 18: 688-703. 10.1016/j.cellsig.2005.06.013.
Mitchell RA, Metz CN, Peng T, Bucala R: Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J Biol Chem. 1999, 274: 18100-18106. 10.1074/jbc.274.25.18100.
Onodera S, Nishihira J, Koyama Y, Majima T, Aoki Y, Ichiyama H, Ishibashi T, Minami A: Macrophage migration inhibitory factor up-regulates the expression of interleukin-8 messenger RNA in synovial fibroblasts of rheumatoid arthritis patients: common transcriptional regulatory mechanism between interleukin-8 and interleukin-1beta. Arthritis Rheum. 2004, 50: 1437-1447. 10.1002/art.20190.
Morand EF, Leech M, Bernhagen J: MIF: a new cytokine link between rheumatoid arthritis and atherosclerosis. Nat Rev Drug Discov. 2006, 5: 399-410. 10.1038/nrd2029.
Ayoub S, Hickey MJ, Morand EF: Mechanisms of disease: macrophage migration inhibitory factor in SLE, RA and atherosclerosis. Nat Clin Pract Rheumatol. 2008, 4: 98-105. 10.1038/ncprheum0701.
Aeberli D, Yang Y, Mansell A, Santos L, Leech M, Morand EF: Endogenous macrophage migration inhibitory factor modulates glucocorticoid sensitivity in macrophages via effects on MAP kinase phosphatase-1 and p38 MAP kinase. FEBS Lett. 2006, 580: 974-981. 10.1016/j.febslet.2006.01.027.
Salojin KV, Owusu IB, Millerchip KA, Potter M, Platt KA, Oravecz T: Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J Immunol. 2006, 176: 1899-1907.
Leech M, Lacey D, Xue JR, Santos L, Hutchinson P, Wolvetang E, David JR, Bucala R, Morand EF: Regulation of p53 by macrophage migration inhibitory factor in inflammatory arthritis. Arthritis Rheum. 2003, 48: 1881-1889. 10.1002/art.11165.
Mitchell RA, Liao H, Chesney J, Fingerle-Rowson G, Baugh J, David J, Bucala R: Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc Natl Acad Sci USA. 2002, 99: 345-350. 10.1073/pnas.012511599.
Lue H, Thiele M, Franz J, Dahl E, Speckgens S, Leng L, Fingerle-Rowson G, Bucala R, Luscher B, Bernhagen J: Macrophage migration inhibitory factor (MIF) promotes cell survival by activation of the Akt pathway and role for CSN5/JAB1 in the control of autocrine MIF activity. Oncogene. 2007, 26: 5046-5059. 10.1038/sj.onc.1210318.
Gregory JL, Morand EF, McKeown SJ, Ralph JA, Hall P, Yang YH, McColl SR, Hickey MJ: Macrophage migration inhibitory factor induces macrophage recruitment via CC chemokine ligand 2. J Immunol. 2006, 177: 8072-8079.
Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle-Rowson G, Ghezzi P, Kleemann R, McColl SR, Bucala R, Hickey MJ, Weber C: MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007, 13: 587-596. 10.1038/nm1567.
van Roon J, Wijngaarden S, Lafeber FP, Damen C, Winkel van de J, Bijlsma JW: Interleukin 10 treatment of patients with rheumatoid arthritis enhances Fc gamma receptor expression on monocytes and responsiveness to immune complex stimulation. J Rheumatol. 2003, 30: 648-651.
Wijngaarden S, Winkel van de JG, Jacobs KM, Bijlsma JW, Lafeber FP, van Roon JA: A shift in the balance of inhibitory and activating Fcgamma receptors on monocytes toward the inhibitory Fcgamma receptor IIb is associated with prevention of monocyte activation in rheumatoid arthritis. Arthritis Rheum. 2004, 50: 3878-3887. 10.1002/art.20672.
Antoniv TT, Ivashkiv LB: Dysregulation of interleukin-10-dependent gene expression in rheumatoid arthritis synovial macrophages. Arthritis Rheum. 2006, 54: 2711-2721. 10.1002/art.22055.
Takasugi K, Yamamura M, Iwahashi M, Otsuka F, Yamana J, Sunahori K, Kawashima M, Yamada M, Makino H: Induction of tumour necrosis factor receptor-expressing macrophages by interleukin-10 and macrophage colony-stimulating factor in rheumatoid arthritis. Arthritis Res Ther. 2006, 8: R126-10.1186/ar2015.
Joyce DA, Gibbons DP, Green P, Steer JH, Feldmann M, Brennan FM: Two inhibitors of pro-inflammatory cytokine release, interleukin-10 and interleukin-4, have contrasting effects on release of soluble p75 tumor necrosis factor receptor by cultured monocytes. Eur J Immunol. 1994, 24: 2699-2705. 10.1002/eji.1830241119.
Katsikis PD, Chu CQ, Brennan FM, Maini RN, Feldmann M: Immunoregulatory role of interleukin 10 in rheumatoid arthritis. J Exp Med. 1994, 179: 1517-1527. 10.1084/jem.179.5.1517.
Williams LM, Ricchetti G, Sarma U, Smallie T, Foxwell BM: Interleukin-10 suppression of myeloid cell activation – a continuing puzzle. Immunology. 2004, 113: 281-292. 10.1111/j.1365-2567.2004.01988.x.
Saidenberg-Kermanac'h N, Bessis N, Deleuze V, Bloquel C, Bureau M, Scherman D, Boissier MC: Efficacy of interleukin-10 gene electrotransfer into skeletal muscle in mice with collagen-induced arthritis. J Gene Med. 2003, 5: 164-171. 10.1002/jgm.321.
Staples KJ, Smallie T, Williams LM, Foey A, Burke B, Foxwell BM, Ziegler-Heitbrock L: IL-10 induces IL-10 in primary human monocyte-derived macrophages via the transcription factor Stat3. J Immunol. 2007, 178: 4779-4785.
Williams LM, Sarma U, Willets K, Smallie T, Brennan F, Foxwell BM: Expression of constitutively active STAT3 can replicate the cytokine-suppressive activity of interleukin-10 in human primary macrophages. J Biol Chem. 2007, 282: 6965-6975. 10.1074/jbc.M609101200.
Driessler F, Venstrom K, Sabat R, Asadullah K, Schottelius AJ: Molecular mechanisms of interleukin-10-mediated inhibition of NF-kappaB activity: a role for p50. Clin Exp Immunol. 2004, 135: 64-73. 10.1111/j.1365-2249.2004.02342.x.
Denys A, Udalova IA, Smith C, Williams LM, Ciesielski CJ, Campbell J, Andrews C, Kwaitkowski D, Foxwell BM: Evidence for a dual mechanism for IL-10 suppression of TNF-alpha production that does not involve inhibition of p38 mitogen-activated protein kinase or NF-kappa B in primary human macrophages. J Immunol. 2002, 168: 4837-4845.
Lang R, Pauleau AL, Parganas E, Takahashi Y, Mages J, Ihle JN, Rutschman R, Murray PJ: SOCS3 regulates the plasticity of gp130 signaling. Nat Immunol. 2003, 4: 546-550. 10.1038/ni932.
Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, Mineau F, Pelletier JP: IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol. 1998, 160: 3513-3521.
Kehlen A, Thiele K, Riemann D, Langner J: Expression, modulation and signalling of IL-17 receptor in fibroblast-like synoviocytes of patients with rheumatoid arthritis. Clin Exp Immunol. 2002, 127: 539-546. 10.1046/j.1365-2249.2002.01782.x.
Toy D, Kugler D, Wolfson M, Bos Vanden T, Gurgel J, Derry J, Tocker J, Peschon J: Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol. 2006, 177: 36-39.
Weaver CT, Hatton RD, Mangan PR, Harrington LE: IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007, 25: 821-852. 10.1146/annurev.immunol.25.022106.141557.
Kramer JM, Yi L, Shen F, Maitra A, Jiao X, Jin T, Gaffen SL: Evidence for ligand-independent multimerization of the IL-17 receptor. J Immunol. 2006, 176: 711-715.
Schwandner R, Yamaguchi K, Cao Z: Requirement of tumor necrosis factor receptor-associated factor (TRAF)6 in interleukin 17 signal transduction. J Exp Med. 2000, 191: 1233-1240. 10.1084/jem.191.7.1233.
Shen F, Gaffen SL: Structure-function relationships in the IL-17 receptor: implications for signal transduction and therapy. Cytokine. 2008, 41: 92-104. 10.1016/j.cyto.2007.11.013.
Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T: Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA. 1998, 95: 3597-3602. 10.1073/pnas.95.7.3597.
Yasuda H, Shima N, Nakagawa N, Mochizuki SI, Yano K, Fujise N, Sato Y, Goto M, Yamaguchi K, Kuriyama M, Kanno T, Murakami A, Tsuda E, Morinaga T, Higashio K: Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology. 1998, 139: 1329-1337. 10.1210/en.139.3.1329.
Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Lüthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ: Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997, 89: 309-319. 10.1016/S0092-8674(00)80209-3.
Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ: Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998, 93: 165-176. 10.1016/S0092-8674(00)81569-X.
Shigeyama Y, Pap T, Kunzler P, Simmen BR, Gay RE, Gay S: Expression of osteoclast differentiation factor in rheumatoid arthritis. Arthritis Rheum. 2000, 43: 2523-2530. 10.1002/1529-0131(200011)43:11<2523::AID-ANR20>3.0.CO;2-Z.
Gravallese EM, Manning C, Tsay A, Naito A, Pan C, Amento E, Goldring SR: Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum. 2000, 43: 250-258. 10.1002/1529-0131(200002)43:2<250::AID-ANR3>3.0.CO;2-P.
Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, Boyle WJ, Simonet WS: Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12: 1260-1268. 10.1101/gad.12.9.1260.
Whyte MP, Obrecht SE, Finnegan PM, Jones JL, Podgornik MN, McAlister WH, Mumm S: Osteoprotegerin deficiency and juvenile Paget's disease. N Engl J Med. 2002, 347: 175-184. 10.1056/NEJMoa013096.
Naito A, Azuma S, Tanaka S, Miyazaki T, Takaki S, Takatsu K, Nakao K, Nakamura K, Katsuki M, Yamamoto T, Inoue J: Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells. 1999, 4: 353-362. 10.1046/j.1365-2443.1999.00265.x.
Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, Morony S, Capparelli C, Van G, Kaufman S, Heiden van der A, Itie A, Wakeham A, Khoo W, Sasaki T, Cao Z, Penninger JM, Paige CJ, Lacey DL, Dunstan CR, Boyle WJ, Goeddel DV, Mak TW: TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999, 13: 1015-1024. 10.1101/gad.13.8.1015.
Wada T, Nakashima T, Oliveirados-Santos AJ, Gasser J, Hara H, Schett G, Penninger JM: The molecular scaffold Gab2 is a crucial component of RANK signaling and osteoclastogenesis. Nat Med. 2005, 11: 394-399. 10.1038/nm1203.
Yamashita T, Yao Z, Li F, Zhang Q, Badell IR, Schwarz EM, Takeshita S, Wagner EF, Noda M, Matsuo K, Xing L, Boyce BF: NF-kappaB p50 and p52 regulate receptor activator of NF-kappaB ligand (RANKL) and tumor necrosis factor-induced osteoclast precursor differentiation by activating c-Fos and NFATc1. J Biol Chem. 2007, 282: 18245-18253. 10.1074/jbc.M610701200.
Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, Wagner EF, Mak TW, Kodama T, Taniguchi T: Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002, 3: 889-901. 10.1016/S1534-5807(02)00369-6.
Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B: The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002, 108: 17-29. 10.1016/S0092-8674(01)00622-5.
Brennan FM, Hayes AL, Ciesielski CJ, Green P, Foxwell BM, Feldmann M: Evidence that rheumatoid arthritis synovial T cells are similar to cytokine-activated T cells: involvement of phosphatidylinositol 3-kinase and nuclear factor kappaB pathways in tumor necrosis factor alpha production in rheumatoid arthritis. Arthritis Rheum. 2002, 46: 31-41. 10.1002/1529-0131(200201)46:1<31::AID-ART10029>3.0.CO;2-5.
Dayer JM: How T-lymphocytes are activated and become activators by cell-cell interaction. Eur Respir J Suppl. 2003, 44: 10s-15s. 10.1183/09031936.03.00000403b.
Brennan FM, Smith NM, Owen S, Li C, Amjadi P, Green P, Andersson A, Palfreeman AC, Hillyer P, Foey A, Beech JT, Feldmann M: Resting CD4+ effector memory T cells are precursors of bystander-activated effectors: a surrogate model of rheumatoid arthritis synovial T-cell function. Arthritis Res Ther. 2008, 10: R36-10.1186/ar2390.
Scanu A, Molnarfi N, Brandt KJ, Gruaz L, Dayer JM, Burger D: Stimulated T cells generate microparticles, which mimic cellular contact activation of human monocytes: differential regulation of pro- and anti-inflammatory cytokine production by high-density lipoproteins. J Leukoc Biol. 2008, 83: 921-927. 10.1189/jlb.0807551.
Rossol M, Meusch U, Pierer M, Kaltenhauser S, Hantzschel H, Hauschildt S, Wagner U: Interaction between transmembrane TNF and TNFR1/2 mediates the activation of monocytes by contact with T cells. J Immunol. 2007, 179: 4239-4248.
Burger D, Molnarfi N, Gruaz L, Dayer JM: Differential induction of IL-1beta and TNF by CD40 ligand or cellular contact with stimulated T cells depends on the maturation stage of human monocytes. J Immunol. 2004, 173: 1292-1297.
McInnes IB, Schett G: Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007, 7: 429-442. 10.1038/nri2094.
Acknowledgements
The authors thank Dr Lynn Williams for critical reading of the manuscript. The authors' work is generously supported by the Arthritis Research Campaign, Trustees of the Kennedy Institute of Rheumatology and the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Drexler, S.K., Kong, P.L., Wales, J. et al. Cell signalling in macrophages, the principal innate immune effector cells of rheumatoid arthritis. Arthritis Res Ther 10, 216 (2008). https://doi.org/10.1186/ar2481
Published:
DOI: https://doi.org/10.1186/ar2481