
Abstract
T-cell activation and differentiation depend on the signal strength received by the T-cell receptor and on signals provided by co-stimulatory molecules. The most prominent co-stimulatory molecule is CD28, which controls the activation of naïve and memory T cells by antigen presented on professional antigen-presenting cells. Blocking of the CD28-CD80/86 pathway has been an appealing strategy for inducing tolerance in autoimmune diseases where the disease-inducing autoantigens are not known. Although CD28 has maintained its unique position, the past decade has witnessed the recognition that a large number of regulatory molecules on T cells must be stimulated to generate a fully protective immune response. These regulatory receptors differ in their preferential expression on T-cell subsets, in the ligands that they recognize, and in the signaling pathways that they trigger. They have in common the fact that they provide information on the cellular environment in which the T-cell response occurs. By intercepting these signals, we may be able to influence disease-relevant T-cell responses in autoimmune diseases while potentially minimizing broad immunosuppression.
Similar content being viewed by others

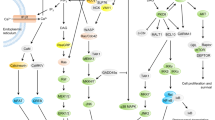
Introduction
Since its first introduction, the concept of co-stimulation has sparked the interest of autoimmunity researchers and immunologists interested in translational applications, because it holds the promise to influence immune responses without needing to define the antigen involved. The basic idea is that co-stimulatory signals decide whether an activation signal translates into an immune response or into tolerance. The concept goes back to the report by Cohn and Bretscher [1], who postulated in the 1970s that regulatory control of a system requiring high sensitivity and high specificity, such as the adaptive immune system, must rely on two independent signals.
Lafferty [2] and Jenkins and Schwartz [3] introduced this concept into T-cell immunology in the 1980s. Signal 1 is delivered by the T-cell receptor (TCR) recognizing the antigenic peptide within the context of the major histocompatibility complex (MHC) molecule. However, stimulation of the TCR alone does not induce a productive T-cell response, but renders the T cells anergic. A second signal, generally provided by molecules expressed on the antigen-presenting cell (APC), is required to optimize a T-cell response. In this concept, the APC holds a central position because of its ability to provide a co-stimulatory signal. Dendritic cells (DCs) are of particular importance in T-cell priming, but B cells and macrophages can also function as APCs. Under normal conditions, other cell types, such as epithelial or stromal cells, cannot induce an immune response because they lack the co-stimulatory ligand. Subsequent studies have shown that APCs need activation signals to provide co-stimulatory ligands, for example through the activation of Toll-like receptors [4, 5].
The model has placed co-stimulation at the central decision point in the induction of an immune response; in fact, stimulation of a naïve T-cell response is unlikely in the absence of co-stimulatory signals. It is therefore not surprising that this decision point is thought to be an optimal target for immune interventions.
The CD28-CD80/CD86 pathway is the classical co-stimulatory pathway and can be considered to be unique [6, 7]. CD28 is constitutively expressed on naïve CD4+ and CD8+ T cells. In contrast, CD80 and CD86 are inducible on DCs and other APCs upon stimulation. The best way to inhibit CD28-CD80/CD86 interaction is by means of cytotoxic T-lymphocyte antigen (CTLA)4-immunoglobulin (Ig). CTLA4 is a negative regulatory receptor that is expressed after stimulation and functions to limit T-cell responses. Because it binds to CD80 and CD86 with greater affinity than to CD28, it can outcompete CD28. A CTLA4-Ig construct with inserted mutations to inactivate the Fc element has been introduced under the generic name abatacept and was recently approved for treatment of rheumatoid arthritis (RA), clearly documenting the validity of this treatment concept [8]. It has been less effective in transplantation models, but recent data from a nonhuman primate model [9] indicate that a mutated abatacept - namely belatacept, which binds CD86 with greater affinity - has promising effects.
In essence, co-stimulation implies that naïve T-cell responses are and should be context dependent. This concept has been expanded in recent years in several dimensions. First, it is now clear that T cells sense co-stimulatory as well as co-inhibitory cues from their environment [10]. Co-inhibitory signals are equally important to our understanding of immune regulation; in fact, the information transmitted upon cell activation constitutes an integration of positive and negative signals. Negative regulatory receptors are not reviewed here, and we refer the interested reader to recent excellent reviews [10–12]; targeting these receptors will first find clinical applications in vaccine responses or in tumor immunology, and not in autoimmunity. The second development came from the insight that co-stimulatory signals also function if they do not coincide spatially or temporally with the TCR-mediated recognition of MHC-peptide complexes. Many co-stimulatory molecules are only induced subsequent to T-cell activation and function in amplification and differentiation. Finally, it is now being recognized that co-stimulatory control is not only a concept for the early stages of an immune response, when naïve T cells encounter antigen for the first time and must make the decision between being tolerant or being driven into proliferation. Context dependency of T-cell responses is evident at all stages of the immune response. For naïve T cells or central memory T cells, this context represents the recognition of ligands on APCs (Figure 1); for effector cells, regulatory signals from stromal cells gain in importance and are of relevance for autoimmune diseases where T-cell effector functions are exerted at immunoprivileged sites and induce tissue damage.

Co-stimulatory receptor-ligand pairs in the initiation of a T-cell response. CD27 and CD28 are co-stimulatory molecules that are constitutively expressed. Their ligands CD70, CD80, and CD86 are inducible on dendritic cells upon their activation. The CD154-CD40 interaction represents an important amplification loop that upregulates the expression of CD80 and CD86. MHC, major histocompatibility complex; TCR, T-cell receptor.
CD28-mediated co-stimulation
The importance of CD28-mediated signals was first emphasized by the immunodeficiency observed in CD28-deficient mice [13–17]. These mice have compromised responses to an array of immune functions, including deficient germinal center formation and immunoglobulin isotype class switching, reduced delayed hypersensitivity responses, and reduced helper cell differentiation. Also, CD8-dependent cytotoxic responses are impaired, although not completely abrogated.
CD28 is expressed on all naïve T cells, but may be lost with T-cell differentiation. This loss is frequently observed in humans and in nonhuman primates, and in particular involves the CD8+ effector cell population [18]. CD4+ T cells can also be affected, in particular in patients with autoimmune conditions. Loss of CD28 occurs normally with age, but this process may be accelerated in susceptible individuals. One contributing factor may be tumor necrosis factor (TNF), which has been shown to downregulate the number of CD28 molecules that are expressed on each individual cell and which may also in part be responsible for the complete loss of CD28 expression in some cells [19]. It should be noted that cell surface expression of CD28 in rodent models is different, and that CD28 is expressed by all murine functional T-cell subsets, irrespective of age or disease. Murine models may therefore represent an imperfect model of CD28 function in humans, with its more restrictive expression patterns on naïve and central memory CD4+ and CD8+ cells.
CD28 is stimulated by its recognition of CD80 and CD86 on APCs (Figure 1) [20, 21]. Expression of both of these ligands is not constitutive but activation induced. It is possible that CD80 and CD86 have distinct functions, and that CD86 is induced later in the immune response than is CD80. Expression of both of these molecules is dependent on various activation stimuli, such as Toll-like receptor ligands. One important amplification loop is the stimulation of CD40 on DCs by CD40 ligand expressed on activated CD4 T cells, which upregulates CD80/CD86 expression [22].
CD28 is a separate signaling unit, independent of the TCR activation complex, which is very much in line with the two-signal hypothesis. Initially, it was thought that each of these two signaling pathways would be unique and only partially overlapping, if at all, and that information and integration would occur at the level of transcription. More recent studies, however, have shown that this is not the case and that signal integration occurs early [23]. CD28 does not stimulate all signaling effectors that are activated by the TCR, but primarily provides a potent synergistic signal for transcription factors such as nuclear factor-κB (NF-κB), nuclear factor of activated T cells (NFAT), and activator protein-1 (AP1) [24–26]. CD28 therefore functions mainly as an amplifying mechanism to overcome signaling thresholds that, in principle, are obtainable by ligation of the TCR alone. CD28 stimulation is mandatorily required at low TCR occupancy, because it occurs in most antigen-specific responses. However, it may be partially or totally dispensable in responses to high-affinity antigens with high TCR occupancy or in situations where the TCR threshold is lower, as seen in effector cell populations. In this model, the contribution of CD28 co-stimulation is more qualitative than quantitative [27, 28]. This concept has been supported by gene expression studies comparing TCR-induced gene expression in the presence or absence of co-stimulation [26, 29]. CD28 co-stimulation was unable to induce transcription of any gene that could not be induced by TCR stimulation alone. However, CD28 co-stimulation was able to amplify or suppress many of the TCR-induced genes to a varying degree. CD28-mediated signaling is not just repetitive of TCR signaling, but sets a different emphasis that shifts the balance of signal events and influences feedback circuits. In this context it is important to note that anti-CD28 antibodies that bind to the laterally exposed C"D loop of the immunoglobulin-like domain of CD28 - distant from the natural ligand-binding domain - induces NF-κB signaling by itself without any need for TCR stimulation [30]. This stimulatory activity of some anti-CD28 antibodies led to the dramatic complications in the Tegenero trial [31].
CD28 signals are mediated through phosphorylated tyrosines that are not part of an ITAM (immunoreceptor tyrosine-based activation motif) motif and by proline-rich motifs in its cytoplasmic tail. CD28 signaling also does not involve the LAT-SLP76 complex, which is the basic signaling module of the TCR intracellular signaling complex. Instead, the CD28 signaling pathway primarily involves a small subset of signaling molecules that are also implicated in TCR signal transmission, in particular the phosphoinositide-3 kinase (PI3-K) pathway with downstream activation of AKT [32, 33], and activation of TEC protein kinases and of interleukin-2-inducible T-cell kinases [27, 34]. AKT activation cooperates with the TCR-induced activation of protein kinase C (PKC)θ to activate the NF-κB pathway, which is otherwise only slightly activated by TCR alone. Activation of TEC and interleukin-2-inducible T-cell kinases are crucial for the regulation of phospholipase C-γ1, contributing to intracellular calcium signal as well as activation of PKC.
Although the main function of CD28 appears to lie in lowering the TCR threshold to antigen-specific stimuli, it has a selective imprint on the transcriptional program induced by TCR-mediated activation rather than a random effect. In addition to its activation of selected transcription factors such as NF-κB, AP1, and NFAT, CD28 co-stimulation has a major impact on DNA demethylation and chromatin remodeling. Most of the biological processes that are influenced by CD28 co-stimulation pertain to T-cell clonal expansion and differentiation. Typical examples are that CD28 co-stimulation induces an anti-apoptotic program, accelerates cell cycle progression, and upregulates metabolic pathways [32, 35, 36]. This pro-proliferative activity makes sense if one considers that the major functions of CD28 co-stimulation are to allow naïve T cells to be activated and clonally expanded.
How does this appreciation of CD28-mediated co-stimulation help us to understand the action of CTLA4-Ig treatment in RA? Although CD28-mediated co-stimulation functions in central memory T-cell activation and can directly or indirectly modulate CD8+ memory T-cell responses in murine systems, CTLA4-Ig blockade should affect naïve T-cell responses more than effector T cells [37, 38]. This has implications for the safety and the side effects predicted to occur with CTLA4-Ig treatment. Immune responses to novel antigens could be importantly abrogated, as is the case in a primary exposure to infectious viruses such as new influenza strains. Indeed, a single dose of CTLA4-Ig in young healthy adults reduced vaccine responses to a peptide antigen, although it could not completely abrogate them [39]. The risk from disabling T cells with CTLA4-Ig in nonimmune patients is not known at present, but it is possible that host defenses are deficient. T-cell responses for which a memory T-cell response has already been established may be less affected, and patients may only be at moderate risk for reactivation of chronic viral infections such as herpes zoster or viral hepatitis. In the clinical studies conducted thus far, the incidence of herpes zoster reactivation has been estimated at 0.7%, which corresponds to the incidence rate in a normal population of 65-year-old individuals [40].
How then is CTLA4-Ig efficacious in RA, if it preferentially targets naïve and memory T cells? Effector T-cell populations expanded in RA are unlikely to be amenable to CTLA-4Ig, in particular because they have frequently lost CD28 expression, which is in sharp contrast to rodent systems. It is possible that CD28 blockade works upstream of these effector cells and that the disease activity in RA is dependent on the continuous recruitment of naïve and central memory cells into the effector cell pool. In this model, continuous disease activity may be dependent on progressive epitope spreading and broadening of the autoimmune response [41]. Alternatively, RA may not be the clinical consequence of the misguided T-cell response to a selective set of autoantigens, but it may represent a global defect in T-cell responsiveness that is amenable to CTLA4-Ig treatment. There is indeed evidence for this model; abnormalities that already occur in the naïve T-cell compartment have been described [42].
The CD27-CD70 pathway
CD27 is a member of the TNF receptor family that shares several features with CD28 [43]. It is also constitutively expressed on naïve and central memory T cells, and it is progressively lost with differentiation into effector cells. CD27 activity is induced by engagement of CD70, a molecule that is found on activated T cells, B cells, and DCs. Expression of CD70 is strictly controlled and occurs transiently after activation. CD27 signaling can lower the threshold of the TCR response to low-affinity antigens, it enhances proliferation and survival, and it promotes differentiation [44, 45]. Like many other TNF-like receptors, CD27 signaling is TNF receptor-associated factor (TRAF) dependent and primarily targets the NF-κB and c-Jun amino-terminal kinase/p38 pathways.
The importance of the CD27-CD70 pathway to autoimmunity is reflected in several murine models [46–48]. In the physiological setting, CD27 stimulation is strictly controlled by CD70 expression. Constitutive expression of CD70 under the control of the CD19 promoter in transgenic mice leads to a persistent stimulation of CD27. These mice exhibit a rapid differentiation of CD4+and CD8+ cells into effector cells, presumably due to the recognition of autoantigens. T-cell responses to exogenous antigen are initially increased. However, mice eventually develop a progressive depletion of the naïve T-cell pool and immune deficiency. In this context, it is of particular interest that patients with systemic lupus erythematosus (SLE) have a selective demethylation of the CD70 promoter that leads to aberrant CD70 expression [49]. Also, in T cells from patients with RA, the regulation of CD70 expression is altered. Upon activation, CD4+ effector T cells from patients with RA express CD70 for a prolonged period of time, providing signals to CD27+ naïve T cells and facilitating their activation and proliferation [50]. In analogy to the mouse model, the over-expression of CD70 has the potential to recruit and activate low-affinity T cells and to amplify T-cell responses. Whether blocking of the CD27-CD70 pathway is efficacious in RA or SLE is not known, but this intervention has been shown to be beneficial in several animal models of autoimmunity, including the autoimmune encephalomyelitis model [47].
The CD40-CD154 pathway
CD40 and its ligand, CD40L or CD154, were first identified as instrumental in T-cell-dependent B-cell activation [51]. Antibody-mediated triggering of CD40 on B cells, in conjunction with B-cell receptor stimulation, induces B-cell proliferation and is essential for germinal center formation and isotype switching. CD40 is constitutively expressed on B cells; its ligand CD154 is induced on CD4+ T cells upon activation. Expression of CD154 on CD8+ T cells is infrequent, but it appears to be of importance in germinal center formation in the synovial tissue [52]. The bearing of the CD40-CD154 receptor-ligand interaction for T-cell activation was subsequently recognized; inhibition of this pathway greatly reduced the activation of naïve T cells to antigen presented by DCs, leading to the definition of CD154 as a co-stimulatory molecule. More appropriately, the pathway is now recognized as a mechanism to activate APCs and to enhance their potential to activate T cells. CD154-mediated CD40 stimulation provides an important feedback mechanism for the initial co-stimulatory pathway of CD28-CD80/CD86. Initial suboptimal stimulation of CD4+ T cells induces CD154 to engage CD40 on DCs, which then upregulates the expression of CD80 and CD86, facilitating further co-stimulation through CD28. CD40-activated DCs can stimulate CD8+ T cells without further T-cell help [53, 54].
Therapeutic targeting of this pathway had beneficial effects in rodent autoimmune and transplant models [9]. Anti-CD154 antibodies were highly efficacious in preventing acute allograft rejections in nonhuman primates, but the effect was less prominent in recall responses; this is consistent with the model in which the CD154-CD40 pathway, interlinked with CD80/86-CD28 co-stimulation, is of central importance in primary T-cell responses. Treatment studies in human SLE, however, identified the unexpected side effect of frequent thromboembolic events [55]. It is now clear that platelets secrete a soluble form of CD154, which is important in thrombus formation and stability [56]. One but not the only action of soluble CD154 is its binding of CD40 on endothelial cells and promotion of their activation [57]. Studies employing direct targeting of CD40 rather than CD154 may in part circumvent the thromboembolic side effects, but they may be not without side effects, given the widespread distribution of CD40.
Co-stimulatory signals provided by activation-induced receptors
A set of co-stimulatory molecules is not constitutively expressed on T cells, but is rapidly induced upon activation and provides a second wave of co-stimulation (Figure 2) [20, 43]. Their major function is to sustain and to amplify the T-cell response, but they are also involved in T-cell differentiation and memory T-cell development. They have been proven to be important in autoimmune diseases in animal models, but their role in human disease is less well defined. Classical examples are CD30, 4-1BB, and Ox40 of the TNF receptor family and inducible co-stimulator (ICOS) of the immunoglobulin family.

Activation-induced co-stimulatory receptors and their ligands. Upon activation, T cells express a number of receptors that have co-stimulatory ability and are important for sustained activation, proliferation, and differentiation. Their ligands again are expressed on antigen-presenting cells. ICOS, inducible co-stimulator; MHC, major histocompatibility complex; TCR, T-cell receptor.
ICOS is a CD28 homolog that is upregulated on naïve T cells after activation and on memory and effector T cells [58, 59]. Similar to CD28, its cytoplasmic domain has a sequence motif that facilitates the binding of the p85 subunit of PI3-K [60]. Its potency to activate the PI3-K/AKT signaling pathway is greater than that of CD28; however, ICOS is also more selective in its activation mechanisms and, in contrast to CD28, it is unable to influence the c-Jun amino-terminal kinase pathway or promote the production of interleukin-2. Ligands for ICOS are constitutively expressed on APCs and, in contrast to CD80 and CD86, do not underlie the control of CD40 or signals involving the NF-κB pathway such as Toll-like receptors. mRNAs for ICOS ligands are also found in a variety of nonhematopoietic cells such as endothelial and epithelial cells and fibroblasts. In vitro, ICOS stimulation augments the production of various T-helper-1 and T-helper-2 cytokines, but does not contribute to clonal expansion. Experiments in transgenic and knockout animals, as well as in disease models, have generated a complex picture of its in vivo function. ICOS co-stimulation appears to be of particular importance for T-cell effector activity and for T-cell-dependent B-cell responses [61]. In most autoimmune disease models, ICOS blockade suppresses disease activity [62, 63], but it can also aggravate disease when it is given during the induction rather than the effector stage [64].
Ox40 (CD134), 4-1BB (CD137), and CD30 have similar functions in primary and secondary immune responses [43]. Their ligands are expressed on APCs, including B cells, but they may also be present on nonhematopoietic cells, such as endothelial cells. Ox40 is primarily inducible on CD4+ T cells, whereas 4-1BB expression is observed preferentially in CD8+ cells, but this subset specificity is not absolute. Both molecules are transiently expressed, and support the expansion of T cells and differentiation into effector-cell populations. Ox40-Ox40 ligand interaction has been shown to provide an important co-stimulatory signal in murine models of autoimmune encephalomyelitis, collagen-induced arthritis, and autoimmune colitis [65–69]. Antibody-mediated inhibition attenuated disease. In contrast, the role of 4-1BB in autoimmune disease appears to be more complex. Treatment with anti-4-1BB antibodies can have inhibitory as well as stimulatory effects, depending on the disease [70]. A possible explanation is that 4-1BB may be involved in the activation and differentiation of a regulatory cell population [71]. Expansion of CD8+ effector cells seen with agonistic 4-1BB-specific antibodies may have a regulatory impact on immune responses; also, cell populations other than T cells express 4-1BB and are influenced by agonistic antibodies. CD30, in many respects, behaves similarly to Ox40 and enhances proliferation and cytokine production induced by TCR stimulation. CD30 co-stimulation has primarily been explored in the NOD (nonobese diabetic) model of diabetes because it maps to the diabetes susceptibility locus idd9.2 and revealed several sequence differences in NOD mice compared with B10 mice [72].
In summary, a set of molecules is readily induced in T cells upon TCR signaling and provides a second wave of co-stimulation. Recognition of the ligands, mostly expressed on APCs, sustains the immune response and amplifies the clonal expansion or effector function. These co-stimulatory signals are equally relevant for the priming of naïve as well as the recall of memory T cells. The functional consequences of immune intervention targeting these receptor-ligand interactions can be complex, depending on the timing, the stage of the immune response, and the microenvironment; the results can be difficult to predict.
Regulatory control of T effector cells
For effector subpopulations, the context of the environment changes; in particular, CD8+ cells are no longer focused on APCs but on parenchymal cells. It is therefore not surprising that effector cells lose the expression of important co-stimulatory receptors that function in the initiation of the immune response, such as CD27 and CD28. Effector cell populations continue to be responsive to 4-1BB triggering; this receptor-ligand interaction may to some extent substitute for the loss of signals derived by CD27 and CD28. In addition, they gain the expression of regulatory molecules (Figure 3), frequently borrowed from the repertoire of natural killer (NK) cell receptors; these molecules include the killer immunoglobulin-like receptor (KIR) and the immunoglobulin-like transcript families, as well as natural-killer group 2, member D (NKG2D) [73, 74]. One characteristic feature of these families is the inclusion of negative and positive regulatory receptors that are co-expressed on individual cells; the balance between positive and negative signals determines the outcome. How and when T cells gain the expression of these molecules is unclear. Some of these molecules are expressed on a large percentage of effector cell populations; others are only present on small subsets. In general, the expression of all of these regulatory receptors favors CD8+ T cells, but they can also be found on CD4+ T cells [75]. One reason for this distribution pattern may be that the frequency of CD8+ effector subpopulations is higher than that of CD4+ T cells, in particular with increasing age. Negative regulatory receptors have ITIM (immunoreceptor tyrosine-based inhibitory motif) motifs in their cytoplasmic domain that recruit Src homology 2 (SH2)-containing tyrosine phosphatases (SHP) and exert inhibitory function when co-engaged with the TCR [10]. In contrast to NK cells, these inhibitory signals do not appear to be absolute, but rather modify the functional response pattern of T cells [76]. Many of these co-inhibitory receptors recognize common determinants on MHC class I molecules.

Co-stimulatory receptor-ligand pairs in the synovium. Effector T-cell populations frequently lose the expression of CD27 and CD28 and gain the expression of regulatory receptors that bind to ligands in the synovium. In general, these receptors as well as their ligands are constitutively expressed independent of cell activation. CD47 is expressed on all T cells irrespective of their differentiation status and receives a co-stimulatory signal from a trimolecular complex formation including CD36 on synoviocytes or endothelial cells and thrombospondin produced in the synovial tissue. KIR, killer immunoglobulin-like receptor; MHC, major histocompatibility complex; TCR, T-cell receptor.
The ligands for stimulatory KIR receptors are less well defined, but may represent complexes of MHC class I molecules and peptides. These stimulatory receptors are usually expressed in the minority on individual cells, but they can dominate selected effector cells in autoimmune diseases. The function of the stimulatory receptors is entirely dependent on adapter molecules that endow them with co-stimulatory or stimulatory function [77]. An adapter molecule conferring full stimulatory function is DAP12, which expresses an ITAM motif [78]. In the presence of DAP12, T cells expressing stimulatory KIR can be fully triggered just by the stimulation of the activating KIR receptors in the absence of antigenic stimulation [79]. In contrast, stimulatory KIR receptors provide co-stimulatory signals in T cells that lack the expression of DAP12 [80]. It is possible that in these T cells DAP10 plays a role as an adapter molecule. DAP10 has a PI3-K binding domain that resembles CD28 or ICOS and initiates activation of the PI3-K pathway. Preferential expression of co-stimulatory rather than co-inhibitory receptors appears to contribute to unopposed effector cell activity in chronic inflammatory diseases [81]. Stimulatory KIR polymorphisms have been found to be associated with psoriasis and extra-articular manifestations of RA [82].
Co-stimulatory ligands in the synovial tissue
It is the premise of co-stimulation to set the recognition of antigen into the context of the immediate environment. For naïve and central memory T cells, co-stimulatory signals are provided by ligands expressed on DCs and other APCs, which are plentiful in the inflamed synovium. Effector T cells frequently receive regulatory signals from nonpolymorphic determinants on MHC class I molecules that are expressed on all somatic cells. In addition, synovial fibroblasts express ligands that regulate T-cell responses specifically in the synovial environment.
CD47 is a cell membrane protein of the immunoglobulin superfamily with wide tissue distribution that has been implicated in both integrin-dependent and integrin-independent signaling cascades. It is expressed on all subsets of T cells, including effector cell populations that have lost the classical co-stimulatory molecules CD28 and CD27. Engagement in T cells leads to F-actin polymerization, PKCθ recruitment, phos-pholipase-γ activation, and calcium influx, amplifying TCR stimulation and enhancing proliferation as well as cytokine production [83]. In addition to the classical SIRP (signal regulatory protein) ligands, CD47 binds to thrombospondin, which is highly expressed in the synovial environment, in particular in diffuse synovitis that is poorly vascularized [84]. CD47 and thrombospondin can form a trimolecular complex with CD36 expressed on synovial fibroblasts. This complex formation is efficient in triggering CD47 activity in T cells [85, 86].
NKG2D is a cell surface molecule that can fully activate NK cells, but it has co-stimulatory function if found on T cells. Its ligands include seven members of the MIC and the ULBP/RAE families, which are induced as a stress response in epithelial cells [87]. Several of these members are also found to be expressed in synovial fibroblasts in RA [88, 89]. NKG2D is constitutively expressed on human CD8+ T cells and a subset of CD4+effector cells, which is in contrast to the rodent system, where its expression is activation dependent. NKG2D signals through DAP10, activating the PI3-K pathway [90]. The synovial microenvironment is therefore poised to provide co-stimulatory signals to NKG2D-expressing CD4+ and CD8+ T cells.
Similar to NKG2D, the fractalkine receptor CX3CR1 (CX3C chemokine receptor 1) is found on CD28- CD4+ T cells and on the CD8+effector T-cell population. CX3CR1 is a G-protein-coupled receptor that provides a co-stimulatory signal to complement TCR stimulation. Engagement of this receptor enhances production of effector cytokines such as TNF-α and interferon-γ [91]. Its ligand fractalkine is produced by synovial fibroblasts and exists in a soluble and a cell-bound form. Cell-bound fractalkine provides a co-stimulatory signal to T cells. The increased production of TNF-α by the effector T-cell population stimulates the synovial fibroblast to produce soluble fractalkine, which is a growth factor for fibroblast proliferation [92].
Conclusion
The concept of co-stimulation, initially originating from the two-signal hypothesis, has developed into the broader notion that T-cell responses are context dependent and are influenced by signals from their environment through a variety of receptor-ligand interactions. These signals amplify and modify the original TCR signal received by antigenic stimulation in a resting naïve or memory T cell, regulate T-cell expansion and differentiation in recently activated T cells, or control effector functions in a particular somatic environment. Co-stimulation blockade attenuates or modifies T-cell responses and therefore represents a powerful treatment tool in autoimmune diseases. By its very nature, co-stimulation is not antigen specific and therefore will not be specific for autoimmune T-cell responses, but it also inhibits or modifies responses to infectious or tumorigenic antigens. However, immune responses in the various autoimmune diseases and in normal responses may differ in their context dependency. Co-stimulation blockade will be unlikely to restore tolerance as originally envisioned, but selective pathway blockade or defined temporal administration may have the potential to be applied as a targeted intervention in an autoimmune disease.
Key messages
-
T-cell activation requires a combination of TCR-mediated and co-stimulatory signals.
-
Although antigen recognition is the determining factor for TCR stimulation, co-stimulatory stimuli provide information on the cellular context where recognition occurs.
-
Interfering with co-stimulatory pathways has the promise to modulate immune responses without knowledge of the antigen.
-
Co-stimulatory signals modify TCR signaling. T-cell responses to high-affinity antigens can occur in the absence of co-stimulatory signals. Conversely, co-stimulatory signals can overcome unresponsiveness to low-affinity antigens.
-
Many co-stimulatory receptor-ligand pairs have been described. Their roles depend on the differentiation state of the T cell and the environment in which the T-cell response occurs (Figures 1 to 3).
-
The CD28-CD80/CD86 pathway is the classical co-stimulatory pathway and has unique importance for the initiation of naïve CD4+ T-cell responses that are triggered by antigen presented by DCs (Figure 1).
-
Activation-induced co-stimulatory molecules such as Ox40, 4-1BB, and ICOS function to sustain and to amplify the T-cell response and to regulate T-cell differentiation into memory and effector cells (Figure 2).
-
Co-stimulatory as well as co-inhibitory receptors on effector T cells regulate the T-cell response in the synovium by virtue of their ligands, which are expressed on synoviocytes and parenchymal cells (Figure 3).
Abbreviations
- AP1:
-
activator protein-1
- APC:
-
antigen-presenting cell
- CTLA:
-
cytotoxic T-lymphocyte antigen
- DC:
-
dendritic cell
- Ig:
-
immunoglobulin
- ICOS:
-
inducible co-stimulator
- KIR:
-
killer immunoglobulin-like receptor
- MHC:
-
major histocompatibility complex
- NFAT:
-
nuclear factor of activated T cells
- NF-κB:
-
nuclear factor-κB
- NK:
-
natural killer
- NKG2D:
-
natural-killer group 2, member D
- PI3-K:
-
phosphoinositide-3 kinase
- PKC:
-
protein kinase C
- RA:
-
rheumatoid arthritis
- SLE:
-
systemic lupus erythematosus
- TCR:
-
T-cell receptor
- TNF:
-
tumor necrosis factor
- TRAF:
-
TNF receptor-associated factor.
References
Bretscher P, Cohn M: A theory of self-nonself discrimination. Science (New York). 1970, 169: 1042-1049.
Lafferty KJ, Misko IS, Cooley MA: Allogeneic stimulation modulates the in vitro response of T cells to transplantation antigen. Nature. 1974, 249: 275-276.
Jenkins MK, Schwartz RH: Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Exp Med. 1987, 165: 302-319.
Matzinger P: Tolerance, danger, and the extended family. Annu Rev Immunol. 1994, 12: 991-1045.
Janeway CA, Medzhitov R: Innate immune recognition. Annu Rev Immunol. 2002, 20: 197-216.
Lenschow DJ, Walunas TL, Bluestone JA: CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996, 14: 233-258.
Sharpe AH, Freeman GJ: The B7-CD28 superfamily. Nat Rev. 2002, 2: 116-126.
Kremer JM, Genant HK, Moreland LW, Russell AS, Emery P, Abud-Mendoza C, Szechinski J, Li T, Ge Z, Becker JC, Westhovens R: Effects of abatacept in patients with methotrexate-resistant active rheumatoid arthritis: a randomized trial. Ann Intern Med. 2006, 144: 865-876.
Weaver TA, Charafeddine AH, Kirk AD: Costimulation blockade: towards clinical application. Front Biosci. 2008, 13: 2120-2139.
Leibson PJ: The regulation of lymphocyte activation by inhibitory receptors. Curr Opin Immunol. 2004, 16: 328-336.
Keir ME, Francisco LM, Sharpe AH: PD-1 and its ligands in T-cell immunity. Curr Opin Immunol. 2007, 19: 309-314.
Murphy KM, Nelson CA, Sedy JR: Balancing co-stimulation and inhibition with BTLA and HVEM. Nat Rev. 2006, 6: 671-681.
Lucas PJ, Negishi I, Nakayama K, Fields LE, Loh DY: Naive CD28-deficient T cells can initiate but not sustain an in vitro antigen-specific immune response. J Immunol. 1995, 154: 5757-5768.
O'Neill SK, Cao Y, Hamel KM, Doodes PD, Hutas G, Finnegan A: Expression of CD80/86 on B cells is essential for autoreactive T cell activation and the development of arthritis. J Immunol. 2007, 179: 5109-5116.
Ferguson SE, Han S, Kelsoe G, Thompson CB: CD28 is required for germinal center formation. J Immunol. 1996, 156: 4576-4581.
Green JM, Noel PJ, Sperling AI, Walunas TL, Gray GS, Bluestone JA, Thompson CB: Absence of B7-dependent responses in CD28-deficient mice. Immunity. 1994, 1: 501-508.
Shahinian A, Pfeffer K, Lee KP, Kundig TM, Kishihara K, Wakeham A, Kawai K, Ohashi PS, Thompson CB, Mak TW: Differential T cell costimulatory requirements in CD28-deficient mice. Science (New York). 1993, 261: 609-612.
Vallejo AN: CD28 extinction in human T cells: altered functions and the program of T-cell senescence. Immunol Rev. 2005, 205: 158-169.
Bryl E, Vallejo AN, Weyand CM, Goronzy JJ: Down-regulation of CD28 expression by TNF-alpha. J Immunol. 2001, 167: 3231-3238.
Greenwald RJ, Freeman GJ, Sharpe AH: The B7 family revisited. Annu Rev Immunol. 2005, 23: 515-548.
McAdam AJ, Schweitzer AN, Sharpe AH: The role of B7 co-stimulation in activation and differentiation of CD4+ and CD8+T cells. Immunol Rev. 1998, 165: 231-247.
van Essen D, Kikutani H, Gray D: CD40 ligand-transduced co-stimulation of T cells in the development of helper function. Nature. 1995, 378: 620-623.
Acuto O, Michel F: CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev. 2003, 3: 939-951.
Kane LP, Lin J, Weiss A: It's all Relative: NF-kappaB and CD28 costimulation of T-cell activation. Trends Immunol. 2002, 23: 413-420.
Michel F, Mangino G, Attal-Bonnefoy G, Tuosto L, Alcover A, Roumier A, Olive D, Acuto O: CD28 utilizes Vav-1 to enhance TCR-proximal signaling and NF-AT activation. J Immunol. 2000, 165: 3820-3829.
Rincon M, Flavell RA: AP-1 transcriptional activity requires both T-cell receptor-mediated and co-stimulatory signals in primary T lymphocytes. EMBO J. 1994, 13: 4370-4381.
Michel F, Attal-Bonnefoy G, Mangino G, Mise-Omata S, Acuto O: CD28 as a molecular amplifier extending TCR ligation and signaling capabilities. Immunity. 2001, 15: 935-945.
Viola A, Lanzavecchia A: T cell activation determined by T cell receptor number and tunable thresholds. Science (New York). 1996, 273: 104-106.
Riley JL, Mao M, Kobayashi S, Biery M, Burchard J, Cavet G, Gregson BP, June CH, Linsley PS: Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc Natl Acad Sci USA. 2002, 99: 11790-11795.
Lühder F, Huang Y, Dennehy KM, Guntermann C, Müller I, Winkler E, Kerkau T, Ikemizu S, Davis SJ, Hanke T, Hünig T: Topological requirements and signaling properties of T cell-activating, anti-CD28 antibody superagonists. J Exp Med. 2003, 197: 955-966.
Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N: Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006, 355: 1018-1028.
Kane LP, Weiss A: The PI-3 kinase/Akt pathway and T cell activation: pleiotropic pathways downstream of PIP3. Immunol Rev. 2003, 192: 7-20.
Parry RV, Riley JL, Ward SG: Signalling to suit function: tailoring phosphoinositide 3-kinase during T-cell activation. Trends Immunol. 2007, 28: 161-168.
Berg LJ, Finkelstein LD, Lucas JA, Schwartzberg PL: Tec family kinases in T lymphocyte development and function. Annu Rev Immunol. 2005, 23: 549-600.
Appleman LJ, Berezovskaya A, Grass I, Boussiotis VA: CD28 costimulation mediates T cell expansion via IL-2-independent and IL-2-dependent regulation of cell cycle progression. J Immunol. 2000, 164: 144-151.
Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, Thompson CB: CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 1995, 3: 87-98.
Borowski AB, Boesteanu AC, Mueller YM, Carafides C, Topham DJ, Altman JD, Jennings SR, Katsikis PD: Memory CD8+ T cells require CD28 costimulation. J Immunol. 2007, 179: 6494-6503.
Fuse S, Zhang W, Usherwood EJ: Control of memory CD8+T cell differentiation by CD80/CD86-CD28 costimulation and restoration by IL-2 during the recall response. J Immunol. 2008, 180: 1148-1157.
Tay L, Leon F, Vratsanos G, Raymond R, Corbo M: Vaccination response to tetanus toxoid and 23-valent pneumococcal vaccines following administration of a single dose of abatacept: a randomized, open-label, parallel group study in healthy subjects. Arthritis Res Ther. 2007, 9: R38-
Donahue JG, Choo PW, Manson JE, Platt R: The incidence of herpes zoster. Arch Intern Med. 1995, 155: 1605-1609.
Vanderlugt CL, Miller SD: Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev. 2002, 2: 85-95.
Goronzy JJ, Weyand CM: Rheumatoid arthritis. Immunol Rev. 2005, 204: 55-73.
Watts TH: TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005, 23: 23-68.
Hendriks J, Xiao Y, Borst J: CD27 promotes survival of activated T cells and complements CD28 in generation and establishment of the effector T cell pool. J Exp Med. 2003, 198: 1369-1380.
Lens SM, Tesselaar K, van Oers MH, van Lier RA: Control of lymphocyte function through CD27-CD70 interactions. Semin Immunol. 1998, 10: 491-499.
Arens R, Tesselaar K, Baars PA, van Schijndel GM, Hendriks J, Pals ST, Krimpenfort P, Borst J, van Oers MH, van Lier RA: Constitutive CD27/CD70 interaction induces expansion of effector-type T cells and results in IFNgamma-mediated B cell depletion. Immunity. 2001, 15: 801-812.
Nakajima A, Oshima H, Nohara C, Morimoto S, Yoshino S, Kobata T, Yagita H, Okumura K: Involvement of CD70-CD27 interactions in the induction of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2000, 109: 188-196.
Tesselaar K, Arens R, van Schijndel GM, Baars PA, Valk van der MA, Borst J, van Oers MH, van Lier RA: Lethal T cell immunodeficiency induced by chronic costimulation via CD27-CD70 interactions. Nat Immunol. 2003, 4: 49-54.
Lu Q, Wu A, Richardson BC: Demethylation of the same promoter sequence increases CD70 expression in lupus T cells and T cells treated with lupus-inducing drugs. J Immunol. 2005, 174: 6212-6219.
Lee WW, Yang ZZ, Li G, Weyand CM, Goronzy JJ: Unchecked CD70 expression on T cells lowers threshold for T cell activation in rheumatoid arthritis. J Immunol. 2007, 179: 2609-2615.
Grewal IS, Flavell RA: CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. 1998, 16: 111-135.
Kang YM, Zhang X, Wagner UG, Yang H, Beckenbaugh RD, Kurtin PJ, Goronzy JJ, Weyand CM: CD8 T cells are required for the formation of ectopic germinal centers in rheumatoid synovitis. J Exp Med. 2002, 195: 1325-1336.
Ridge JP, Di Rosa F, Matzinger P: A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998, 393: 474-478.
Schoenberger SP, Toes RE, Voort van der EI, Offringa R, Melief CJ: T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998, 393: 480-483.
Yazdany J, Davis J: The role of CD40 ligand in systemic lupus erythematosus. Lupus. 2004, 13: 377-380.
Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, Kroczek RA: CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 1998, 391: 591-594.
Andre P, Prasad KS, Denis CV, He M, Papalia JM, Hynes RO, Phillips DR, Wagner DD: CD40L stabilizes arterial thrombi by a beta3 integrin-dependent mechanism. Nat Med. 2002, 8: 247-252.
Kroczek RA, Mages HW, Hutloff A: Emerging paradigms of T-cell co-stimulation. Curr Opin Immunol. 2004, 16: 321-327.
Suh WK, Tafuri A, Berg-Brown NN, Shahinian A, Plyte S, Duncan GS, Okada H, Wakeham A, Odermatt B, Ohashi PS, Mak TW: The inducible costimulator plays the major costimulatory role in humoral immune responses in the absence of CD28. J Immunol. 2004, 172: 5917-5923.
Coyle AJ, Lehar S, Lloyd C, Tian J, Delaney T, Manning S, Nguyen T, Burwell T, Schneider H, Gonzalo JA, Gosselin M, Owen LR, Rudd CE, Gutierrez-Ramos JC: The CD28-related molecule ICOS is required for effective T cell-dependent immune responses. Immunity. 2000, 13: 95-105.
Loke P, Allison JP: Emerging mechanisms of immune regulation: the extended B7 family and regulatory T cells. Arthritis Res Ther. 2004, 6: 208-214.
Nurieva RI, Treuting P, Duong J, Flavell RA, Dong C: Inducible costimulator is essential for collagen-induced arthritis. J Clin Invest. 2003, 111: 701-706.
Rottman JB, Smith T, Tonra JR, Ganley K, Bloom T, Silva R, Pierce B, Gutierrez-Ramos JC, Ozkaynak E, Coyle AJ: The costimulatory molecule ICOS plays an important role in the immunopathogenesis of EAE. Nat Immunol. 2001, 2: 605-611.
Chapoval AI, Ni J, Lau JS, Wilcox RA, Flies DB, Liu D, Dong H, Sica GL, Zhu G, Tamada K, Chen L: B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat Immunol. 2001, 2: 269-274.
Higgins LM, McDonald SA, Whittle N, Crockett N, Shields JG, MacDonald TT: Regulation of T cell activation in vitro and in vivo by targeting the OX40-OX40 ligand interaction: amelioration of ongoing inflammatory bowel disease with an OX40-IgG fusion protein, but not with an OX40 ligand-IgG fusion protein. J Immunol. 1999, 162: 486-493.
Malmstrom V, Shipton D, Singh B, Al-Shamkhani A, Puklavec MJ, Barclay AN, Powrie F: CD134L expression on dendritic cells in the mesenteric lymph nodes drives colitis in T cell-restored SCID mice. J Immunol. 2001, 166: 6972-6981.
Ndhlovu LC, Ishii N, Murata K, Sato T, Sugamura K: Critical involvement of OX40 ligand signals in the T cell priming events during experimental autoimmune encephalomyelitis. J Immunol. 2001, 167: 2991-2999.
Weinberg AD, Wegmann KW, Funatake C, Whitham RH: Blocking OX-40/OX-40 ligand interaction in vitro and in vivo leads to decreased T cell function and amelioration of experimental allergic encephalomyelitis. J Immunol. 1999, 162: 1818-1826.
Yoshioka T, Nakajima A, Akiba H, Ishiwata T, Asano G, Yoshino S, Yagita H, Okumura K: Contribution of OX40/OX40 ligand interaction to the pathogenesis of rheumatoid arthritis. Eur J Immunol. 2000, 30: 2815-2823.
Mittler RS, Foell J, McCausland M, Strahotin S, Niu L, Bapat A, Hewes LB: Anti-CD137 antibodies in the treatment of autoimmune disease and cancer. Immunol Res. 2004, 29: 197-208.
Myers LM, Vella AT: Interfacing T-cell effector and regulatory function through CD137 (4-1BB) co-stimulation. Trends Immunol. 2005, 26: 440-446.
Lyons PA, Hancock WW, Denny P, Lord CJ, Hill NJ, Armitage N, Siegmund T, Todd JA, Phillips MS, Hess JF, Chen SL, Fischer PA, Peterson LB, Wicker LS: The NOD Idd9 genetic interval influences the pathogenicity of insulitis and contains molecular variants of Cd30, Tnfr2, and Cd137. Immunity. 2000, 13: 107-115.
Vivier E, Anfossi N: Inhibitory NK-cell receptors on T cells: witness of the past, actors of the future. Nat Rev. 2004, 4: 190-198.
Young NT, Uhrberg M, Phillips JH, Lanier LL, Parham P: Differential expression of leukocyte receptor complex-encoded Ig-like receptors correlates with the transition from effector to memory CTL. J Immunol. 2001, 166: 3933-3941.
Czesnikiewicz-Guzik M, Lee WW, Cui D, Hiruma Y, Lamar DL, Yang ZZ, Ouslander JG, Weyand CM, Goronzy JJ: T cell subset-specific susceptibility to aging. Clin Immunol (Orlando, FL). 2008, 127: 107-118.
Henel G, Singh K, Cui D, Pryshchep S, Lee WW, Weyand CM, Goronzy JJ: Uncoupling of T-cell effector functions by inhibitory killer immunoglobulin-like receptors. Blood. 2006, 107: 4449-4457.
Snyder MR, Weyand CM, Goronzy JJ: The double life of NK receptors: stimulation or co-stimulation?. Trends Immunol. 2004, 25: 25-32.
Humphrey MB, Lanier LL, Nakamura MC: Role of ITAM-containing adapter proteins and their receptors in the immune system and bone. Immunol Rev. 2005, 208: 50-65.
Snyder MR, Nakajima T, Leibson PJ, Weyand CM, Goronzy JJ: Stimulatory killer Ig-like receptors modulate T cell activation through DAP12-dependent and DAP12-independent mechanisms. J Immunol. 2004, 173: 3725-3731.
Snyder MR, Lucas M, Vivier E, Weyand CM, Goronzy JJ: Selective activation of the c-Jun NH2-terminal protein kinase signaling pathway by stimulatory KIR in the absence of KARAP/DAP12 in CD4+ T cells. J Exp Med. 2003, 197: 437-449.
Rajagopalan S, Long EO: Understanding how combinations of HLA and KIR genes influence disease. J Exp Med. 2005, 201: 1025-1029.
Yen JH, Moore BE, Nakajima T, Scholl D, Schaid DJ, Weyand CM, Goronzy JJ: Major histocompatibility complex class I-recognizing receptors are disease risk genes in rheumatoid arthritis. J Exp Med. 2001, 193: 1159-1167.
Reinhold MI, Lindberg FP, Kersh GJ, Allen PM, Brown EJ: Costimulation of T cell activation by integrin-associated protein (CD47) is an adhesion-dependent, CD28-independent signaling pathway. J Exp Med. 1997, 185: 1-11.
Park YW, Kang YM, Butterfield J, Detmar M, Goronzy JJ, Weyand CM: Thrombospondin 2 functions as an endogenous regulator of angiogenesis and inflammation in rheumatoid arthritis. Am J Pathol. 2004, 165: 2087-2098.
Vallejo AN, Mugge LO, Klimiuk PA, Weyand CM, Goronzy JJ: Central role of thrombospondin-1 in the activation and clonal expansion of inflammatory T cells. J Immunol. 2000, 164: 2947-2954.
Vallejo AN, Yang H, Klimiuk PA, Weyand CM, Goronzy JJ: Synoviocyte-mediated expansion of inflammatory T cells in rheumatoid synovitis is dependent on CD47-thrombospondin 1 interaction. J Immunol. 2003, 171: 1732-1740.
Gonzalez S, Groh V, Spies T: Immunobiology of human NKG2D and its ligands. Curr Topics Microbiol Immunol. 2006, 298: 121-138.
Goronzy JJ, Henel G, Sawai H, Singh K, Lee EB, Pryshchep S, Weyand CM: Costimulatory pathways in rheumatoid synovitis and T-cell senescence. Ann N Y Acad Sci. 2005, 1062: 182-194.
Groh V, Bruhl A, El-Gabalawy H, Nelson JL, Spies T: Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc Natl Acad Sci USA. 2003, 100: 9452-9457.
Upshaw JL, Leibson PJ: NKG2D-mediated activation of cytotoxic lymphocytes: unique signaling pathways and distinct functional outcomes. Semin Immunol. 2006, 18: 167-175.
Sawai H, Park YW, Roberson J, Imai T, Goronzy JJ, Weyand CM: T cell costimulation by fractalkine-expressing synoviocytes in rheumatoid arthritis. Arthritis Rheum. 2005, 52: 1392-1401.
Sawai H, Park YW, He X, Goronzy JJ, Weyand CM: Fractalkine mediates T cell-dependent proliferation of synovial fibroblasts in rheumatoid arthritis. Arthritis Rheum. 2007, 56: 3215-3225.
Acknowledgements
The authors thank Tamela Yeargin for editing the manuscript.
This work was funded in part by grants from the National Institutes of Health (R01 AR41974, R01 AR42527, RO1 AG15043, RO1 AI44142, and RO1 EY11916).
This article is published as part of Arthritis Research & Therapy Volume 10 Supplement 1, 2008: Co-stimulation blockade: from bench to bedside. The full contents of the supplement are available online at http://arthritis-research.com/supplements/10/S1.
Publication of this supplement has been sponsored by Bristol-Myers Squibb Company.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
About this article
Cite this article
Goronzy, J.J., Weyand, C.M. T-cell co-stimulatory pathways in autoimmunity. Arthritis Res Ther 10 (Suppl 1), S3 (2008). https://doi.org/10.1186/ar2414
Published:
DOI: https://doi.org/10.1186/ar2414