
Abstract
Proteinases are involved in essential steps in cartilage and bone homeostasis. Consequently, efforts have been made to establish their potential role in the pathology of rheumatic conditions such as rheumatoid arthritis, osteoarthritis and spondyloarthritis. Matrix metalloproteinases (MMPs) are sensitive markers of disease severity and response to treatment, and therefore they have potential in the assessment of rheumatic diseases. Despite disappointing early results with synthetic inhibitors of MMPs, there is still much scope for developing effective and safe MMPs inhibitors, and consequently to deliver new options to inhibit joint destruction.
Similar content being viewed by others

Introduction
Proteases are responsible for enzymatic cleavage of peptide bonds [1, 2], which is a basic requirement for completion of diverse biological processes. Examples of contributions made by proteases can be found in digestion, blood coagulation and fibrinolysis. They are also involved in the processing of precursors related to the synthesis of collagen, immune functions, development and apoptosis [3]. The proteolytic activity of proteases must be rigorously controlled to avoid inappropriate degradation of proteins. Imbalance in regulation of proteolytic activity can be found in a wide range of diseases, including cancer, rheumatoid arthritis (RA) and osteoarthritis (OA) [4].
Of particular importance is that proteases have been found to play diverse and strategic roles in cartilage and bone remodelling, which in recent years has engendered increased interest in these enzymes in the field of rheumatology. To highlight the clinical relevance of proteinases to joint destruction, we discuss their contribution to cartilage and bone homeostasis in health and give particular emphasis to their crucial role in diseases such as RA, OA and spondyloarthritis.
General features of proteinases
Proteases selectively hydrolyze a peptide bond in a polypeptide chain of a target molecule. Depending on the position of the peptide bond, proteases are referred to as exopeptidases or endopeptidases. Exopeptidases specifically cleave substrates at the amino-terminal or carboxyl-terminal positions of polypeptides, and therefore can be subdivided into aminopeptidases and carboxypeptidases [5, 6]. Endopeptidases (also called proteinases) break peptide bonds in the middle of the molecule. They can be subclassified based on their mechanism of catalysis, which is related to the chemical group involved in the process of hydrolysis. As a consequence, endopeptidases are described as aspartate, cysteine and threonine types, which act intracellularly in an acid pH, or as serine and metallo catalytic types, which act extracellularly in a neutral pH environment [6]. Each of these catalytic types is described in the following discussion (a summary is provided in Figure 1).

Summary of proteases. MMP, matrix metalloproteinase; MT, membrane-type; tPA, tissue-type plasminogen activator; uPA, urokinase-type plasminogen activator.
Aspartate proteinases
A well known representative aspartic proteinase is cathepsin D. The major function of cathepsin D is to digest proteins and peptides within the acidic compartment of the lysosome [7]. It apparently is also involved in the processing of hormones, neuropeptides and antigens [7–9]. Therefore, cathepsin D has been proposed to be a potential target that could allow modulation of autoimmune diseases [8].
Cysteine proteinases
Cysteine proteinases are generally known as cathepsins (types B, K, L, S, H, F, C, X and O) [10]. Cathepsin S is the major processing enzyme of the major histocompatibility complex class II invariant chain. Cathepsins L and F participate in the same process, primarily in tissues or cells that do not express cathepsin S. Finally, cathepsin K was found to be crucial in bone remodelling and is predominantly expressed in osteoclasts. Interestingly, it has also been described in synovial fibroblasts and macrophages of RA joints [11].
Threonine proteinases
This class of proteinases represents a crucial element in the proteosome. Along with lysosomal proteolysis, the ubiquitinproteosome pathway is the main intracellular cascade for controlled degradation of proteins [12]. It plays an important role in a variety of fundamental cellular processes, including cell cycle progression, cell division, development, differentiation and apoptosis. Furthermore, it influences cell trafficking and modulates immune and inflammatory responses [13].
Serine proteinases
This is a family of enzymes that contain a serine residue in their active site [14]. They are of particular interest because they have been implicated in a variety of physiological and pathological processes. For example, the urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator play a critical role in several processes, including clot dissolution, extracellular matrix (ECM) remodelling, angiogenesis and wound healing, as well as tumour invasion and metastasis [15]. uPA converts plasminogen to plasmin, which is a broad-spectrum enzyme that can degrade not only fibrin but also proteins of the joint ECM and cartilage. By single proteolytic cleavage, both uPA and plasmin produce active forms of matrix metalloproteinases (MMPs) [16].
Metalloproteinases
This group of proteases is divided into five families: the serralysins, the astacins, the adamalysins, the MMPs and the pappalysins [5]. The family of MMPs is best known for its ability to cleave components of the ECM, but they also cleave other proteinases and proteinase inhibitors, latent growth factors and growth factor binding proteins, chemotactic molecules, cell surface receptors and cell-cell adhesion molecules. MMPs regulate many biological processes and consequently they are precisely controlled at various critical steps, including synthesis and secretion, activation of proenzymes and inhibition of active enzymes. However, the localization and clearance of MMPs is also tightly controlled [17].
Matrix metalloproteinases and adamalysins: key characteristics
The MMPs and adamalysins are considered to be major mediators of cartilage destruction in arthritic diseases and have attracted particular research interest.
Matrix metalloproteinases
In general, MMPs are composed of three distinct domains [18]: a pre-domain, which is required for enzyme maturation and release from the cell; a prodomain, which maintains the enzyme in an inactive state; and the catalytic domain, which characteristically contains a zinc atom and is responsible for enzyme activity. Because of their rather diverse structures and biological functions, they are classified into at least five main groups (Figure 1) according to their substrate specificity, primary structure and cellular localization (Table 1): collagenases, gelatinases, stromelysins, the matrilysins and membrane-type (MT) MMPs. Apart from those included in these main groups, other MMPs have been described including MMP-12 (metalloelastase), MMP-19, MMP-20 (enamelysin), MMP-20 and MMP-23, as well as XMMP (Xenopus) and CMMP (chicken) [19] (for review, see [20]).
As mentioned above, various mechanisms are involved in the regulation of MMPs [18], including transcription control, pro-enzyme activation, and inhibition of active enzymes by natural inhibitors. MMP gene expression is regulated at the transcriptional level, controlled by the stimulating effects of cytokines (such as IL-1β and tumour necrosis factor [TNF]-α) and growth factors (such as epidermal growth factor, platelet-derived growth factor, basic fibroblast growth factor and transforming growth factor-β). After binding to their membrane receptor these cytokines and growth factor generate a signalling cascade, which involves activator protein-1 (AP-1) transcription factors and finally leads to the transcription of MMPs [21].
The production of MMPs as pro-enzymes is another important mechanism of regulation. They are produced as inactive forms and require further cleavage by other proteinases to become active. MMPs can be activated by MT1-MMP, MT2-MMP and MT5-MMP [19], or by plasmin, uPA and tissue-type plasminogen activator [21]. The initial proteolytic activation of MMPs occurs at an exposed region of the pro-domain. First, the pro-domain is removed, which leads to destabilization of the molecule. The next step includes participation of the cysteine switch-zinc mechanism [22]. This mechanism involves the dissociation of a cysteine residue from the zinc atom in the catalytic domain to expose the active site. Finally, the active form can be autocatalytically cleaved by the activated metalloprotease [21].
MMPs are also regulated by tissue inhibitors of metalloproteinases (TIMPs) [19]. TIMPs are produced by the same cells that produce the MMPs and bind to them at a ratio of 1:1 in order to induce their inactivation. Changes in levels of TIMPs are particularly important because they directly affect MMP activity [22]. Thus far, four TIMPS have been described; TIMP-1, TIMP-2 and TIMP-4 are present in soluble forms [23], whereas TIMP-3 is tightly attached to the matrix by binding to proteoglycans [24].
Adamalysins
ADAM
The adamalysin family includes the adamalysins (ADAM [a disintegrin and metalloproteinase]) and ADAMTS (a disintegrin and metalloproteinase with thrombospondin motif). There are more than 30 members of the ADAM family, with a prominent sheddase activity described. Sheddases proteolytically cleave cellular membrane proteins by detachment of their extracellular region [25]. Most relevant to bone and cartilage remodelling is ADAM-17, because it sheds TNF-α and TNF-α receptors from the membrane. After the shedding and releasing of TNF-α, it can function in a paracrine and endocrine manner [25].
ADAMTS
Aggrecanases are the main proteinases responsible for aggrecan cleavage in the early events of cartilage remodelling. Later, MMPs participate in this process and continue with the degradation of collagen [26]. In the cartilage, two different aggrecanases have been isolated, aggrecanase-1 (ADAMTS-4) and aggrecanase-2 (ADAMTS-5) [26]. Like all metalloproteinases, both ADAMTS-4 and ADAMTS-5 rely on the cysteine switch mechanism for activation. In addition, they can be activated by furin-like proprotein convertases [27, 28]. Like MMPs, ADAMTS-4 and ADAMTS-5 are inhibited by TIMP-3 [29].
Proteinases in the joint
It is generally accepted that proteolytic enzymes are involved in the catabolic aspect of normal tissue remodelling and that altered activity of these enzymes is responsible for the cartilage destruction and bone erosion associated with disorders such as OA and RA [30].
Articular cartilage in adults is a comparatively acellular tissue, with a cell volume approximating 2% of the total cartilage volume. The remainder is occupied by an extensive ECM [31]. The structural backbone of this matrix is the collagen fibril. Articular cartilage is mainly composed by type II collagen, but it also contains types IX and XI collagen, both on the surface and within. Many other matrix molecules are found in association with the collagen fibrils, the most common and largest of which is the large proteoglycan aggrecan. It forms molecular aggregates with hyaluronic acid, which in turn interacts with the collagen fibrillar network [32]. Further elements of the cartilage matrix are leucine-rich proteoglycans, including decorin, fibromodulin and biglycan [32].
Cartilage and bone turnover is a complex process in which proteinases play a prominent role in health and disease. The cartilage remodelling process is conducted entirely by a single cell type, namely the chondrocyte. This cell is not only responsible for the synthesis of the complex ECM of the articular cartilage, but it is also the source of proteinases and other mediators that degrade the damaged matrix to permit repair [33]. The production of these proteolytic enzymes is regulated by various local mediators, such as cytokines, growth factors, prostaglandins, matrix breakdown products, complement, oxygen species and neuropeptides [34].
Similar to cartilage, the bone tissue undergoes continuous remodelling. Osteoclasts play a prominent role in the resorption of bone in this remodelling cycle. By secreting H+ ions and proteinases, osteoclasts dissolve bone mineral and degrade organic bone matrix [35]. Osteoclast-mediated bone resorption is a multistep process that is initiated by the attachment of osteoclasts to the bone surface. After they have attached to the bone surface, a tightly sealed resorption lacuna is created. Then proteolytic enzymes expressed in osteoclasts, such as cathepsin K and MMPs (MMP-13, MMP-2 and MMP-9), are secreted into the lacuna for removal of bone mineral and degradation of organic matrix protein [32]. MMPs are essential for the initiation of the osteoclastic resorption process by removing the collagenous layer from the bone surface, which must be achieved before the demineralization process can be initiated. MMPs have also been implicated in the cleansing of bone lining cells from resorption pits of remaining collagen fibrils before the pits are refilled with new bone matrix components produced by osteoblasts [11]. Additionally, MMP-12 produced by osteoclasts or periosteoclastic cells contributes to bone matrix solubilization and osteoclast migration, thereby controlling the cell-matrix interactions that are required for cell attachment/detachment [36]. In summary, proteinases participate importantly in normal bone and cartilage turnover, whereas deregulation of proteinases is relevant to several joint diseases.
Proteinases in the diseased joint
The cellular and molecular mechanisms that underlie pathological bone destruction have partially been identified. In particular, molecular insight into osteoclast biology has revealed that even though inflammation and destruction are independent processes, they are linked to each other [30]. The link is established through the network of cytokines and growth factors that are produced by cells in the inflamed joint [35]. Proinflammatory cytokines such as TNF-α and IL-1 are abundant in the synovium of patients with various types of chronic arthritis, and they disrupt normal tissue homeostasis in cartilage and bone [30]. The influence of cytokines in osteoclast differentiation provides a link between inflammation and the process of bone destruction. Some cytokines such as IL-1 directly induce osteoclast differentiation, whereas others such as TNF-α act indirectly by upregulating RANKL (receptor activator of nuclear factor-κB ligand), which is the major factor involved in osteoclast differentiation and activation [37].
One of the strongest predictors of long-term outcome in RA and OA is progressive joint damage. In RA and OA, this progressive cartilage and bone destruction are considered to be driven by excess MMP enzymes [38]. The profile of MMPs expressed by activated cells in arthritic joints is sufficient to destroy completely the structural collagens that build up the articular cartilage, the adjacent bones and tendons, as well as the noncollagen matrix molecules [7].
Proteinases in rheumatoid arthritis
In RA functional disability is a multifactorial process, and damage to the joint structures contributes significantly to the overall functional status of the patient [31]. Degradation of the cartilage, tendon and bone ECM proteins by MMPs is the hallmark of synovial joint destruction [39]. In this process loss of aggrecans, considered a critical event in arthritis, initially occurs at the surface of the cartilage and then progresses to deeper zones. This is followed by degradation of collagen fibrils and mechanical failure of the tissue [39].
There are two principal mechanisms by which RA synovial tissue contributes to loss of cartilage. The first and direct mechanism involves the production of MMPs and cathepsins by the RA synovium [33]. The second mechanism indirectly induces cartilage remodelling by deregulation of chondrocyte function through the release of cytokines and other mediators from the synovium [33]. As part of the inflammatory process in RA, macrophages are recruited to the joints, where they release inflammatory cytokines such as IL-1β, TNF-α and IL-6. These cytokines induce expression of proteinases in synovial fibroblasts and chondrocytes [40].
It has been demonstrated that synovial cells stimulated by TNF-α or IL-1β increase the transcription of cathepsin B [40–42]. The location of cathepsin B appeared to be restricted to synovial cells attached to cartilage and bone at sites of RA joint erosion [43]. Accumulation of active cathepsin B in the synovial fluid of RA patients is probably related to destruction of subchondral bone [44]. Interestingly, cathepsin B, along with MMP-1 and cathepsin L, has been detected in the synovium soon after onset of symptoms, implying that the potential for joint destruction exists even at very early stages of the disease [45, 46].
Also, cathepsin K is highly expressed in synovial fibroblasts and macrophages in joints of RA patients [11, 47, 48]. In addition to its major role in bone resorption, it has been demonstrated that cathepsin K plays a critical role in cartilage degradation as well. Co-cultures of synovial fibroblasts on cartilage disks revealed the ability of fibroblasts to phagocytose collagen fibrils [49]. This degenerative property of fibroblasts was prevented by a potent cathepsin K inhibitor.
The important role of MMPs in cartilage and bone destruction in RA has been demonstrated using various approaches. High levels of MMP-1, MMP-3, MMP-8 and MMP-9 are found in the synovial fluid of patients with RA [50]. Moreover, the synovial tissue exhibits high constitutive expression of MMP-2, MMP-11 and MMP-19 [51]. The expression of MMP-3 is particularly high in synovial tissue from RA patients, suggesting that MMP-3 plays a significant role in the progression of erosions of the cartilage [52]. Interestingly, a high MMP/TIMP ratio was identified in the serum of RA patients [52], implying an imbalance in the proteolytic system in favour of the MMPs.
Finally, it has been shown that RA synovial fibroblasts exhibit increased production of MMPs (MMP-1, MMP-3, MMP-13, MMP-14 and MMP-15) and contribute significantly to the joint destruction observed in RA [53, 54]. This high expression of MMPs by RA synovial fibroblasts is not only upregulated by elevated levels of IL-1β and TNF-α [40] but is also sustained intrinsically by RA synovial fibroblasts, which display a transformed phenotype [55].
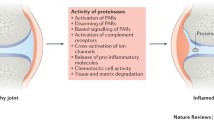
In summary, the inflammatory environment observed in the synovial tissue allows the production and secretion of cytokines and growth factors by infiltrating cells and resident synovial cells. This leads to increased production of ADAMTSs and MMPs by synovial fibroblasts and by chondrocytes, favouring cartilage and bone destruction (Figure 2b).

Proteinases in joint destruction. Shown are the proteinases that are involved in the joint destruction that occurs in (a) osteoarthritis and (b) rheumatoid arthritis. ADAM, a disintegrin and metalloproteinase-like; ADAMTS, ADAM-thrombospondin; IL, interleukin; MMP, matrix metalloproteinase; MT, membrane-type; RANKL, receptor activator of nuclear factor-κB ligand; TNF, tumour necrosis factor; tPA, tissue-type plasminogen activator; uPA, urokinase-type plasminogen activator.
Because of the significant roles played by MMPs in joint destruction, they have been regarded as useful biomarkers and therapeutic targets. Levels of MMP-1 and MMP-3 in the serum of RA patients correlate with disease activity [56]. For that reason, it has been suggested that MMP-3 could be a useful marker for predicting of bone and cartilage damage in early untreated RA [57]. Successful treatment with leflunomide [58], methotrexate [53], or anti-TNF-α antibodies efficiently downregulates serum levels of MMPs [59, 60]. Although these recent advances in RA treatment arrest radiological joint destruction for some time, none of the disease-modifying antirheumatic dugs, including the biological agents, have yet provided long-term, problem-free protection against joint destruction [54]. Therefore, there remains a need to develop novel therapeutic strategies.
Proteinases in osteoarthritis
The aetiology of OA is not completely understood, but it appears to result from mechanical, biochemical and enzymatic factors. The final common pathway of these interactions is the failure of the chondrocytes to maintain a homeostatic balance between matrix synthesis and degradation [19, 61]. Therefore, excessive digestion of cartilage collagen is considered a critical issue in loss of articular cartilage in OA [62]. However, the chondrocytes not only secrete the proteinases responsible of cartilage destruction, but they also produce proinflammatory cytokines such as IL-1 and TNF-α, creating an inflammatory environment that also favours increased synthesis of proteinases [17] (Figure 2a).
The destruction of the ECM in OA results from several events that take place in sequence. The first critical step is loss of cartilage aggrecans mediated by ADAMTS-4 and ADAMTS-5. Then, diverse MMPs continue to degrade the major components of the ECM [63]. Secondary cartilage breakdown products, such as fibronectin fragments, are released into the joint fluid and irritate the synovial membrane lining in the joint space. The resulting synovitis provokes release of inflammatory mediators from synovial tissue and initiates recruitment of mononuclear inflammatory cells into the joint space. These arriving cells secrete IL-1 and TNF-α, which further upregulate the production of proteinases by chondrocytes and synovial fibroblasts [64, 65] (Figure 2).
In OA, expression of MMPs, ADAMTS and TIMPs exhibits a particular pattern [66]. The coexistence of multiple collagenases, plus their distinct localization and distribution in the cartilage, points to a specific role for each of them. For example, it is proposed that MMP-1 is involved in tissue destruction initiated in the superficial zone of the cartilage during inflammation, whereas MMP-13 plays a role in the remodelling phase of the disease [19]. Furthermore, it has been shown that MMP-2 and MMP-9 are increased in OA joints and that their expression is enhanced by IL-1 and TNF-α [19]. In OA, TIMPs do not increase to the same extent as proteinases do, producing a disproportionate ratio of MMP to TIMP. The result is an excess of proteinases, which aggravates cartilage breakdown and promotes joint destruction [66, 67].
Proteinases in spondyloarthritis
The spondyloarthropathies (SpAs) represent a group of related arthritides that include ankylosing spondylitis, psoriatic arthritis, reactive arthritis, and enteropathic and undifferentiated SpA. They are characterized by their association with HLA-B27 and development of sacroilitis and enthesitis. The observed functional impairment, disability and impaired quality of life resemble those in RA [68].
MMPs and TIMPs are expressed in the synovial compartment of SpA patients [67]. In particular, high expression of MMP-3 has been demonstrated in serum, synovial tissue and synovial fluids of SpA patients [69, 70]. TNF-α blockade can induce a downregulation of MMPs and TIMPs in the synovium of affected joints and decreases levels of MMP-3 in the serum [70]. Consequently, some authors have predicted a possible role for MMPs as biomarkers of disease activity or response to treatment [71].
Most recently, our group showed that new small erosions appear in the bony processes of the vertebrae even in late stages of the disease (at the time of surgical correction), and that these sites of destruction appear to be caused by osteoclasts producing MMPs, in particular cathepsin K [72]. These findings might explain why patients still experience significant pain at late stages of the disease.
Proteinases as therapeutic targets
Because of the prominent roles of metalloproteinases observed in degenerative diseases, they are attractive as therapeutic targets. Several approaches to block the deleterious effects of proteinases in pathological conditions have been evaluated. These approaches include use of synthetic metalloproteinase inhibitors, inhibition of signal transduction pathways and gene transfer technology.
Synthetic metalloproteinase inhibitors
The synthetic inhibitors of MMPs can be divided into three pharmacological categories [73]: collagen peptidomimetics and nonpeptidomimetics; tetracycline derivatives; and bisphosphonates.
Collagen peptidomimetics and nonpeptidomimetics
Peptidomimetic MMP inhibitors are pseudopeptide derivatives that have been synthesized to mimic the structure of collagen at the site where MMPs bind to cleave it. The compounds batismastat and marismastat are examples of peptidomimetic MMP inhibitors [73, 74]. The clinical development of batismastat was hampered by poor oral bioavailability and solubility [75]. The preclinical studies of marimastat have demonstrated inhibition of progression of lung and breast tumours. However, several published clinical trials have been unable to demonstrate benefit from using marimastat as a single agent in diverse cancers. Nevertheless, the combination of marimastat with other chemotherapeutic agents has proven to be well tolerated and to have synergistic action in inhibiting tumour growth [75].
Prinomastat, BMS-275291 and BAY 12–9566 (tanomastat) are examples of nonpeptidomimetic inhibitors. Their use in cancer therapy has been disappointing. Prinomastat failed to improve the outcome in advanced non-small-cell lung cancer [76]. A clinical trial of prinomastat in patients with adenocarcinoma of the oesophagus required early closure because of unexpected thromboembolic events [77]. Also, BMS-275291 increases toxicity to chemotherapy and does not improve survival in advanced non-small-cell lung cancer [78]. Finally, BAY 12–9566 was well tolerated but there was no evidence of an impact on outcome in patients with advanced ovarian cancer [79].
With regard to joint diseases, Ro 32–3555 protected cartilage from degradative changes in a mouse model of OA [80] and it was well tolerated in a trial including RA patients [81]. In general, peptidomimetic and nonpeptidomimetic metalloproteinase inhibitors have been tested in bone diseases but, although they inhibited bone destruction in animal models, they failed to confer benefit in human trials.
Tetracycline derivatives
Use of tetracycline derivatives is another possible therapeutic strategy. They can inhibit the activity and production of MMPs (MMP-1, MMP-3 and MMP-13, and MMP-2 and MMP-9). This family of agents includes tetracycline, doxycycline, minocycline and the tetracycline analogues that have been chemically modified to eliminate their antimicrobial activity.
Some of these tetracycline derivatives have been evaluated in preclinical cancer models and have entered early clinical trials in patients with malignant diseases [73]. Thus far, doxycycline has been effective in reducing the rate of joint-space narrowing in patients with established knee OA [82].
Bisphosphonates
Published data regarding the role of bisphosphonates as MMPs inhibitors include those from preclinical studies only. These agents inhibited transforming growth factor-β1 induced MMP-2 secretion in PC-3 prostate cancer cell lines. Clodronate also inhibited the expression of the MT1-MMP protein and mRNA in the HT1080 fibrosarcoma cell line, and decreased the invasion of C8161 melanoma and HT1080 fibrosarcoma cell lines [73].
Inhibition of signal transduction pathways
The development of selective protein kinase inhibitors that can block or modulate diseases caused by abnormalities in these signalling pathways is widely considered a promising approach to drug development [22]. The signal transduction pathways activated when IL-1β and TNF-α bind to their cognate receptors on synovial cells and chondrocytes are potential drug targets. Chemical blockade of mitogenactivated protein kinase pathways inhibits expression of MMP genes in tissue culture experiments and blocks the progression of arthritis in animal models. SB203580, a p38 mitogen-activated protein kinase inhibitor, blocks both MMP-13 gene expression in cultured chondrocytes and IL-1 mediated collagen degradation in cartilage explants. However, further clinical research is needed [17].
Gene transfer
Gene transfer technology has also been applied to treatment of RA. A transgene is a gene that is artificially introduced into an organism or cell to modify their genome and function [83]. Gene transfer improves the delivery of therapeutic proteins and allows stable and high concentrations of therapeutic peptides to be achieved [83]. Gene transfer studies in the severe combined immunodeficient mouse model of cartilage invasion made it possible to evaluate selective inhibition of specific enzymes and their individual contributions to joint destruction. In this regard, inhibition of cathepsin L by a specific ribozyme was able to reduce expression of cathepsin L mRNA to 44% [84]. A ribozyme was also utilized to target MMP-1 in the same model [85]. In other work, an anti-sense construct against MT1-MMP was designed and transferred into RA synovial fibroblasts for studies in the severe combined immunodeficient mouse model [84], but invasiveness of RA synovial fibroblasts into the co-transplanted cartilage could only be moderately reduced. Similarly, transfer of a cell surface targeted plasmin inhibitor was not as effective in vivo as it was in vitro [86]. In contrast, transferring TIMPs into RA synovial fibroblasts has resulted in a remarked inhibition of RA synovial fibroblast mediated cartilage destruction [87]. Because TIMP-3, for instance, both inhibits MT1-MMP (which can activate other MMPs) and inhibits the shedding of TNF-α converting enzyme and IL-6, therapeutic application of a TIMP-3-like molecule could potentially prevent both the cytokine-driven activation of synovial cells and cytokine-independent activation of RA synovial fibroblasts [87].
Conclusion
Tight control of proteinases in the joints allows the integrity of the bone and cartilage to be conserved. Failure to regulate the synthesis, activation and inhibition of the proteinases favours the deleterious effects of MMPs, including MMP-1, MMP-3 and others, which finally lead to degradation. Despite early disappointing results with synthetic inhibitors of MMPs in human trials, there remains much scope for developing effective but highly selective and safe MMP inhibitors. This novel group of drugs could provide new therapeutic options to inhibit joint destruction, which is the main reason for the disability observed in RA, OA and the SpAs.
Abbreviations
- ADAM:
-
= a disintegrin and metalloproteinase
- ADAMTS:
-
= a disintegrin and metalloproteinase with thrombospondin motif
- ECM:
-
= extracellular matrix
- IL:
-
= interleukin
- MMP:
-
= matrix metalloproteinase
- MT:
-
= membrane-type
- OA:
-
= osteoarthritis
- RA:
-
= rheumatoid arthritis
- SpA:
-
= spondy-loarthropathy
- TIMP:
-
= tissue inhibitor of metalloproteinases
- TNF:
-
= tumour necrosis factor
- uPA:
-
= urokinase-type plasminogen activator.
References
Turk B: Targeting proteases: successes, failures and future prospects. Nat Rev Drug Discov. 2006, 5: 785-799. 10.1038/nrd2092.
Lah TT, Duran Alonso MB, Van Noorden CJ: Antiprotease therapy in cancer: hot or not?. Expert Opin Biol Ther. 2006, 6: 257-79. 10.1517/14712598.6.3.257.
Dickinson DP: Cysteine peptidases of mammals: their biological roles and potential effects in the oral cavity and other tissues in health and disease. Crit Rev Oral Biol Med. 2002, 13: 238-275.
Turk D, Guncar G: Lysosomal cysteine proteases (cathepsins): promising drug targets. Acta Crystallogr D Biol Crystal-logr. 2003, 59: 203-213. 10.1107/S0907444902021479.
Barrett AJ: The many forms and functions of cellular proteinases. Fed Proc. 1980, 39: 9-14.
Cawston TE, Wilson AJ: Understanding the role of tissue degrading enzymes and their inhibitors in development and disease. Best Pract Res Clin Rheumatol. 2006, 20: 983-1002. 10.1016/j.berh.2006.06.007.
Fusek M, Vetvicka V: Dual role of cathepsin D: ligand and protease. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2005, 149: 43-50.
Chapman HA: Endosomal proteases in antigen presentation. Curr Opin Immunol. 2006, 18: 78-84. 10.1016/j.coi.2005.11.011.
Lkhider M, Castino R, Bouguyon E, Isidoro C, Ollivier-Bousquet M: Cathepsin D released by lactating rat mammary epithelial cells is involved in prolactin cleavage under physiological conditions. J Cell Sci. 2004, 117: 5155-5164. 10.1242/jcs.01396.
Turk V, Turk B, Turk D: Lysosomal cysteine proteases: facts and opportunities. EMBO J. 2001, 3: 4629-4633. 10.1093/emboj/20.17.4629.
Yasuda Y, Kaleta J, Bromme D: The role of cathepsins in osteoporosis and arthritis: rationale for the design of new therapeutics. Adv Drug Deliv Rev. 2005, 57: 973-993. 10.1016/j.addr.2004.12.013.
Mitsiades CS, Mitsiades N, Hideshima T, Richardson PG, Anderson KC: Proteasome inhibition as a new therapeutic principle in hematological malignancies. Curr Drug Targets. 2006, 7: 1341-1347. 10.2174/138945006778559247.
Wang J, Maldonado MA: The ubiquitin-proteasome system and its role in inflammatory and autoimmune diseases. Cell Mol Immunol. 2006, 3: 255-261.
Pham CT: Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol. 2006, 6: 541-550. 10.1038/nri1841.
Walker B, Lynas JF: Strategies for the inhibition of serine proteases. Cell Mol Life Sci. 2001, 58: 596-624. 10.1007/PL00000884.
Jin T, Tarkowski A, Carmeliet P, Bokarewa M: Urokinase, a constitutive component of the inflamed synovial fluid, induces arthritis. Arthritis Res Ther. 2003, 5: R9-R17. 10.1186/ar606.
Burrage PS, Mix KS, Brinckerhoff CE: Matrix metalloproteinases: role in arthritis. Front Biosci. 2006, 11: 529-543. 10.2741/1817.
Rannou F, Francois M, Corvol MT, Berenbaum F: Cartilage breakdown in rheumatoid arthritis. Joint Bone Spine. 2006, 73: 29-36. 10.1016/j.jbspin.2004.12.013.
Martel-Pelletier J, Welsch DJ, Pelletier JP: Metalloproteases and inhibitors in arthritic diseases. Best Pract Res Clin Rheumatol. 2001, 15: 805-829. 10.1053/berh.2001.0195.
Murphy G, Knauper V, Atkinson S, Butler G, English W, Hutton M, Stracke J, Clark I: Matrix metalloproteinases in arthritic disease. Arthritis Res. 2002, 4 (Suppl 3): S39-S49. 10.1186/ar572.
Chakraborti S, Mandal M, Das S, Mandal A, Chakraborti T: Regulation of matrix metalloproteinases: an overview. Mol Cell Biochem. 2003, 253: 269-285. 10.1023/A:1026028303196.
Visse R, Nagase H: Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003, 92: 827-839. 10.1161/01.RES.0000070112.80711.3D.
Lambert E, Dasse E, Haye B, Petitfrere E: TIMPs as multifacial proteins. Crit Rev Oncol Hematol. 2004, 49: 187-198. 10.1016/j.critrevonc.2003.09.008.
Baker AH, Edwards DR, Murphy G: Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J Cell Sci. 2002, 115: 3719-3727. 10.1242/jcs.00063.
Huovila AP, Turner AJ, Pelto-Huikko M, Karkkainen I, Ortiz RM: Shedding light on ADAM metalloproteinases. Trends Biochem Sci. 2005, 30: 413-422. 10.1016/j.tibs.2005.05.006.
Nagase H, Kashiwagi M: Aggrecanases and cartilage matrix degradation. Arthritis Res Ther. 2003, 5: 94-103. 10.1186/ar630.
Arner EC: Aggrecanase-mediated cartilage degradation. Curr Opin Pharmacol. 2002, 2: 322-329. 10.1016/S1471-4892(02)00148-0.
Longpre JM, Leduc R: Identification of prodomain determinants involved in ADAMTS-1 biosynthesis. J Biol Chem. 2004, 279: 33237-33245. 10.1074/jbc.M313151200.
Milner JM, Rowan AD, Cawston TE, Young DA: Metalloproteinase and inhibitor expression profiling of resorbing cartilage reveals pro-collagenase activation as a critical step for collagenolysis. Arthritis Res Ther. 2006, 8: R142-10.1186/ar2034.
Luyten FP, Lories RJ, Verschueren P, de Vlam K, Westhovens R: Contemporary concepts of inflammation, damage and repair in rheumatic diseases. Best Pract Res Clin Rheumatol. 2006, 20: 829-848. 10.1016/j.berh.2006.06.009.
Poole AR, Kojima T, Yasuda T, Mwale F, Kobayashi M, Laverty S: Composition and structure of articular cartilage: a template for tissue repair. Clin Orthop Relat Res. 2001, 391 (Suppl): S26-S33. 10.1097/00003086-200110001-00004.
Poole AR, Kobayashi M, Yasuda T, Laverty S, Mwale F, Kojima T, Sakai T, Wahl C, El-Maadawy S, Webb G, et al: Type II collagen degradation and its regulation in articular cartilage in osteoarthritis. Ann Rheum Dis. 2002, 61 (Suppl 2): ii78-ii81.
Goldring SR: Pathogenesis of bone and cartilage destruction in rheumatoid arthritis. Rheumatology (Oxford). 2003, 42 (Suppl 2): ii11-ii16.
Mandelin J, Hukkanen M, Li TF, Korhonen M, Liljestrom M, Sillat T, Hanemaaijer R, Salo J, Santavirta S, Konttinen YT: Human osteoblasts produce cathepsin K. Bone. 2006, 38: 769-777. 10.1016/j.bone.2005.10.017.
Walsh NC, Crotti TN, Goldring SR, Gravallese EM: Rheumatic diseases: the effects of inflammation on bone. Immunol Rev. 2005, 208: 228-251. 10.1111/j.0105-2896.2005.00338.x.
Delaisse JM, Andersen TL, Engsig MT, Henriksen K, Troen T, Blavier L: Matrix metalloproteinases (MMP) and cathepsin K contribute differently to osteoclastic activities. Microsc Res Tech. 2003, 61: 504-513. 10.1002/jemt.10374.
Anandarajah AP, Schwarz EM: Anti-RANKL therapy for inflammatory bone disorders: mechanisms and potential clinical applications. J Cell Biochem. 2006, 97: 226-232. 10.1002/jcb.20674.
Mandal M, Mandal A, Das S, Chakraborti T, Sajal C: Clinical implications of matrix metalloproteinases. Mol Cell Biochem. 2003, 252: 305-329. 10.1023/A:1025526424637.
Malemud CJ: Matrix metalloproteinases (MMPs) in health and disease: an overview. Front Biosci. 2006, 11: 1696-1701. 10.2741/1915.
Huet G, Flipo RM, Colin C, Janin A, Hemon B, Collyn-d'Hooghe M, Lafyatis R, Duquesnoy B, Degand P: Stimulation of the secretion of latent cysteine proteinase activity by tumor necrosis factor alpha and interleukin-1. Arthritis Rheum. 1993, 36: 772-780. 10.1002/art.1780360606.
Lemaire R, Huet G, Zerimech F, Grard G, Fontaine C, Duquesnoy B, Flipo RM: Selective induction of the secretion of cathepsins B and L by cytokines in synovial fibroblast-like cells. Br J Rheumatol. 1997, 36: 735-743. 10.1093/rheumatology/36.7.735.
Kaneko M, Tomita T, Nakase T, Ohsawa Y, Seki H, Takeuchi E, Takano H, Shi K, Takahi K, Kominami E, et al: Expression of proteinases and inflammatory cytokines in subchondral bone regions in the destructive joint of rheumatoid arthritis. Rheumatology (Oxford). 2001, 40: 247-255. 10.1093/rheumatology/40.3.247.
Trabandt A, Gay RE, Fassbender HG, Gay S: Cathepsin B in synovial cells at the site of joint destruction in rheumatoid arthritis. Arthritis Rheum. 1991, 34: 1444-1451.
Hashimoto Y, Kakegawa H, Narita Y, Hachiya Y, Hayakawa T: Significance of cathepsin B accumulation in synovial fluid of rheumatoid arthritis. Biochem Biophys Res Commun. 2001, 283: 334-339. 10.1006/bbrc.2001.4787.
Cunnane G, FitzGerald O, Hummel KM, Gay RE, Gay S, Bresnihan B: Collagenase, cathepsin B and cathepsin L gene expression in the synovial membrane of patients with early inflammatory arthritis. Rheumatology (Oxford). 1999, 38: 34-42. 10.1093/rheumatology/38.1.34.
Cunnane G, FitzGerald O, Hummel KM, Youssef PP, Gay RE, Gay S, Bresnihan B: Synovial tissue protease gene expression and joint erosions in early rheumatoid arthritis. Arthritis Rheum. 2001, 44: 1744-1753. 10.1002/1529-0131(200108)44:8<1744::AID-ART309>3.0.CO;2-K.
Hou WS, Li W, Keyszer G, Weber E, Levy R, Klein MJ, Gravallese EM, Goldring SR, Bromme D: Comparison of cathepsins K and S expression within the rheumatoid and osteoarthritic synovium. Arthritis Rheum. 2002, 46: 663-674. 10.1002/art.10114.
Hummel KM, Petrow PK, Franz JK, Muller-Ladner U, Aicher WK, Gay RE, Bromme D, Gay S: Cysteine proteinase cathepsin K mRNA is expressed in synovium of patients with rheumatoid arthritis and is detected at sites of synovial bone destruction. J Rheumatol. 1998, 25: 1887-1894.
Hou WS, Li Z, Gordon RE, Chan K, Klein MJ, Levy R, Keysser M, Keyszer G, Bromme D: Cathepsin k is a critical protease in synovial fibroblast-mediated collagen degradation. Am J Pathol. 2001, 159: 2167-2177.
Tchetverikov I, Ronday HK, Van El B, Kiers GH, Verzijl N, TeKoppele JM, Huizinga TW, DeGroot J, Hanemaaijer R: MMP profile in paired serum and synovial fluid samples of patients with rheumatoid arthritis. Ann Rheum Dis. 2004, 63: 881-883. 10.1136/ard.2003.013243.
Konttinen YT, Ainola M, Valleala H, Ma J, Ida H, Mandelin J, Kinne RW, Santavirta S, Sorsa T, Lopez-Otin C, et al: Analysis of 16 different matrix metalloproteinases (MMP-1 to MMP-20) in the synovial membrane: different profiles in trauma and rheumatoid arthritis. Ann Rheum Dis. 1999, 58: 691-697.
Ainola MM, Mandelin JA, Liljestrom MP, Li TF, Hukkanen MV, Konttinen YT: Pannus invasion and cartilage degradation in rheumatoid arthritis: involvement of MMP-3 and interleukin-1beta. Clin Exp Rheumatol. 2005, 23: 644-650.
Fiedorczyk M, Klimiuk PA, Sierakowski S, Gindzienska-Sieskiewicz E, Chwiecko J: Serum matrix metalloproteinases and tissue inhibitors of metalloproteinases in patients with early rheumatoid arthritis. J Rheumatol. 2006, 33: 1523-1529.
Pap T, Meinecke I, Muller-Ladner U, Gay S: Are fibroblasts involved in joint destruction?. Ann Rheum Dis. 2005, 64 (Suppl 4): iv52-iv54. 10.1136/ard.2005.042424.
Karouzakis E, Neidhart M, Gay RE, Gay S: Molecular and cellular basis of rheumatoid joint destruction. Immunol Lett. 2006, 106: 8-13. 10.1016/j.imlet.2006.04.011.
Muller-Ladner U, Gay S: MMPs and rheumatoid synovial fibroblasts : Siamese twins in joint destruction?. Ann Rheum Dis. 2002, 61: 957-959. 10.1136/ard.61.11.957.
Green MJ, Gough AK, Devlin J, Smith J, Astin P, Taylor D, Emery P: Serum MMP-3 and MMP-1 and progression of joint damage in early rheumatoid arthritis. Rheumatology (Oxford). 2003, 42: 83-88. 10.1093/rheumatology/keg037.
Yamanaka H, Matsuda Y, Tanaka M, Sendo W, Nakajima H, Taniguchi A, Kamatani N: Serum matrix metalloproteinase 3 as a predictor of the degree of joint destruction during the six months after measurement, in patients with early rheumatoid arthritis. Arthritis Rheum. 2000, 43: 852-858. 10.1002/1529-0131(200004)43:4<852::AID-ANR16>3.0.CO;2-7.
Litinsky I, Paran D, Levartovsky D, Wigler I, Kaufman I, Yaron I, Yaron M, Caspi D, Elkayam O: The effects of leflunomide on clinical parameters and serum levels of IL-6, IL-10, MMP-1 and MMP-3 in patients with resistant rheumatoid arthritis. Cytokine. 2006, 33: 106-110. 10.1016/j.cyto.2005.12.009.
Catrina AI, Lampa J, Ernestam S, af Klint E, Bratt J, Klareskog L, Ulfgren AK: Anti-tumour necrosis factor (TNF)-alpha therapy (etanercept) down-regulates serum matrix metalloproteinase (MMP)-3 and MMP-1 in rheumatoid arthritis. Rheumatology (Oxford). 2002, 41: 484-489. 10.1093/rheumatology/41.5.484.
Klimiuk PA, Sierakowski S, Domyslawska I, Chwiecko J: Effect of repeated infliximab therapy on serum matrix metallopro-teinases and tissue inhibitors of metalloproteinases in patients with rheumatoid arthritis [abstract]. J Rheumatol. 2004, 31: 238-242.
Kurz B, Lemke AK, Fay J, Pufe T, Grodzinsky AJ, Schunke M: Pathomechanisms of cartilage destruction by mechanical injury. Ann Anat. 2005, 187: 473-485.
Smith GN: The role of collagenolytic matrix metalloproteinases in the loss of articular cartilage in osteoarthritis. Front Biosci. 2006, 11: 3081-3095. 10.2741/2034.
Yasuda T: Cartilage destruction by matrix degradation products. Mod Rheumatol. 2006, 16: 197-205. 10.1007/s10165-006-0490-6.
Wu CW, Tchetina EV, Mwale F, Hasty K, Pidoux I, Reiner A, Chen J, Van Wart HE, Poole AR: Proteolysis involving matrix metalloproteinase 13 (collagenase-3) is required for chondrocyte differentiation that is associated with matrix mineralization. J Bone Miner Res. 2002, 17: 639-651. 10.1359/jbmr.2002.17.4.639.
Kevorkian L, Young DA, Darrah C, Donell ST, Shepstone L, Porter S, Brockbank SM, Edwards DR, Parker AE, Clark IM: Expression profiling of metalloproteinases and their inhibitors in cartilage. Arthritis Rheum. 2004, 50: 131-141. 10.1002/art.11433.
Malemud CJ, Islam N, Haqqi TM: Pathophysiological mechanisms in osteoarthritis lead to novel therapeutic strategies. Cells Tissues Organs. 2003, 174: 34-48. 10.1159/000070573.
Dean DD, Martel-Pelletier J, Pelletier JP, Howell DS, Woessner JF: Evidence for metalloproteinase and metalloproteinase inhibitor imbalance in human osteoarthritic cartilage [abstract]. J Clin Invest. 1989, 84: 678-685.
Huang W, Schwarz EM: Mechanisms of bone resorption and new bone formation in spondyloarthropathies. Curr Rheumatol Rep. 2002, 4: 513-517. 10.1007/s11926-002-0059-0.
Chen CH, Lin KC, Yu DT, Yang C, Huang F, Chen HA, Liang TH, Liao HT, Tsai CY, Wei JC, et al: Serum matrix metalloproteinases and tissue inhibitors of metalloproteinases in ankylosing spondylitis: MMP-3 is a reproducibly sensitive and specific biomarker of disease activity. Rheumatology (Oxford). 2006, 45: 414-420. 10.1093/rheumatology/kei208.
Vandooren B, Kruithof E, Yu DT, Rihl M, Gu J, De Rycke L, Van Den Bosch F, Veys EM, De Keyser F, Baeten D: Involvement of matrix metalloproteinases and their inhibitors in peripheral synovitis and down-regulation by tumor necrosis factor alpha blockade in spondylarthropathy. Arthritis Rheum. 2004, 50: 2942-2953. 10.1002/art.20477.
Braun J, Baraliakos X, Yelder C, Seemeyer C, Gay R, Boehm H, Gay S, Neidhart M: Clinical and histopathological findings in patients with ankylosing spondylitis before and after surgical treatment for axis correction and erection of the spine [abstract]. Arthritis Rheum. 2006, 54 (Suppl): S466-
Hidalgo M, Eckhardt SG: Development of matrix metalloproteinase inhibitors in cancer therapy. J Natl Cancer Inst. 2001, 93: 178-193. 10.1093/jnci/93.3.178.
Murphy G, Lee MH: What are the roles of metalloproteinases in cartilage and bone damage?. Ann Rheum Dis. 2005, 64 (Suppl 4): iv44-iv47. 10.1136/ard.2005.042465.
Yip D, Ahmad A, Karapetis CS, Hawkins CA, Harper PG: Matrix metalloproteinase inhibitors: applications in oncology. Invest New Drugs. 1999, 17: 387-399. 10.1023/A:1006386406584.
Bissett D, O'Byrne KJ, von Pawel J, Gatzemeier U, Price A, Nicolson M, Mercier R, Mazabel E, Penning C, Zhang MH, et al: Phase III study of matrix metalloproteinase inhibitor prinomastat in non-small-cell lung cancer. J Clin Oncol. 2005, 23: 842-849. 10.1200/JCO.2005.03.170.
Heath EI, Burtness BA, Kleinberg L, Salem RR, Yang SC, et al: Phase II, parallel-design study of preoperative combined modality therapy and the matrix metalloprotease (MMP) inhibitor prinomastat in patients with esophageal adenocarcinoma. Invest New Drugs. 2006, 24: 135-140. 10.1007/s10637-006-5934-5.
Leighl NB, Paz-Ares L, Douillard JY, Peschel C, Arnold A, Depierre A, Santoro A, Betticher DC, Gatzemeier U, Jassem J, et al: Randomized phase III study of matrix metalloproteinase inhibitor BMS-275291 in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: National Cancer Institute of Canada-Clinical Trials Group Study BR.18. J Clin Oncol. 2005, 23: 2831-2839. 10.1200/JCO.2005.04.044.
Hirte H, Vergote IB, Jeffrey JR, Grimshaw RN, Coppieters S, Schwartz B, Tu D, Sadura A, Brundage M, Seymour L: A phase III randomized trial of BAY 12–9566 (tanomastat) as maintenance therapy in patients with advanced ovarian cancer responsive to primary surgery and paclitaxel/platinum containing chemotherapy: a National Cancer Institute of Canada Clinical Trials Group Study. Gynecol Oncol. 2006, 102: 300-308. 10.1016/j.ygyno.2005.12.020.
Brewster M, Lewis EJ, Wilson KL, Greenham AK, Bottomley KM: Ro 32-an orally active collagenase selective inhibitor, prevents structural damage in the STR/ORT mouse model of osteoarthritis [abstract]. Arthritis Rheum. 1998, 41: 1639-1644. 10.1002/1529-0131(199809)41:9<1639::AID-ART15>3.0.CO;2-0.
Hemmings FJ, Farhan M, Rowland J, Banken L, Jain R: Tolerability and pharmacokinetics of the collagenase-selective inhibitor Trocade in patients with rheumatoid arthritis. Rheumatology (Oxford). 2001, 40: 537-543. 10.1093/rheumatology/40.5.537.
Brandt KD, Mazzuca SA, Katz BP, Lane KA, Buckwalter KA, Yocum DE, Wolfe F, Schnitzer TJ, Moreland LW, Manzi S, et al: Effects of doxycycline on progression of osteoarthritis: results of a randomized, placebo-controlled, double-blind trial. Arthritis Rheum. 2005, 52: 2015-2025. 10.1002/art.21122.
Moritz F, Distler O, Ospelt C, Gay RE, Gay S: Technology insight: gene transfer and the design of novel treatments for rheumatoid arthritis. Nat Clin Pract Rheumatol. 2006, 2: 153-162. 10.1038/ncprheum0117.
Schedel J, Seemayer CA, Pap T, Neidhart M, Kuchen S, Michel BA, Gay RE, Muller-Ladner U, Gay S, Zacharias W: Targeting cathepsin L (CL) by specific ribozymes decreases CL protein synthesis and cartilage destruction in rheumatoid arthritis. Gene Ther. 2004, 11: 1040-1047. 10.1038/sj.gt.3302265.
Rutkauskaite E, Zacharias W, Schedel J, Muller-Ladner U, Mawrin C, Seemayer CA, Alexander D, Gay RE, Aicher WK, Michel BA: Ribozymes that inhibit the production of matrix metalloproteinase 1 reduce the invasiveness of rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2004, 50: 1448-1456. 10.1002/art.20186.
van der Laan WH, Pap T, Ronday HK, Grimbergen JM, Huisman LG, TeKoppele JM, Breedveld FC, Gay RE, Gay S, Huizinga TW, et al: Cartilage degradation and invasion by rheumatoid synovial fibroblasts is inhibited by gene transfer of a cell surface-targeted plasmin inhibitor. Arthritis Rheum. 2000, 43: 1710-1718. 10.1002/1529-0131(200008)43:8<1710::AID-ANR6>3.0.CO;2-Y.
van der Laan WH, Quax PH, Seemayer CA, Huisman LG, Pieterman EJ, Grimbergen JM, Verheijen JH, Breedveld FC, Gay S: Cartilage degradation and invasion by rheumatoid synovial fibroblasts is inhibited by gene transfer of TIMP-1 and TIMP-3. Gene Ther. 2003, 10: 234-242. 10.1038/sj.gt.3301871.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have received a research grant to test different compounds from Novartis, which are not mentioned in this review.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Rengel, Y., Ospelt, C. & Gay, S. Proteinases in the joint: clinical relevance of proteinases in joint destruction. Arthritis Res Ther 9, 221 (2007). https://doi.org/10.1186/ar2304
Published:
DOI: https://doi.org/10.1186/ar2304