
Abstract
There is significant evidence arising from experimental models that autoantibodies play a key role in the pathogenesis of inflammatory arthritis. In addition to autoantibody production, B cells efficiently present antigen to T cells, produce soluble factors, including cytokines and chemokines, and form B cell aggregates in the target organ of rheumatoid arthritis. In this review we analyze the multifaceted role that B cells play in the pathogenesis of rheumatoid arthritis and discuss how this information can be used to guide more specific targeting of B cells for the therapy of this disease.
Similar content being viewed by others

Introduction
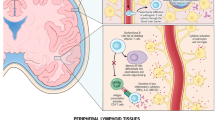
The advent of biological therapy has provided a powerful tool to improve our understanding of the pathogenesis of disease. As well as benefiting patients, the success of targeted biological therapies demonstrates the importance of particular molecules or cells in disease pathogenesis. The potent efficacy of rituximab, a B-cell depleting agent, in the treatment of patients with rheumatoid arthritis (RA) has revitalized interest in the central role played by B cells in disease pathogenesis [1] (Fig. 1).

B cell participation in RA. Illustrated is the potential role of B cells in the regulation of immune responses in RA. Mature B cells, upon antigen encounter and TLR stimulation, expand and differentiate into short-lived plasma cells or can enter into a GC reaction, which is necessary for the generation of both memory B cells, and long-lived plasma cells that can produce autoantibodies. Autoantibodies form immune complexes that further activate the immune system via Fc and complement receptors expressed on target cells. Antigen-activated mature B cells provide help to T cells and induce differentiation of effector T cells that produce proinflammatory cytokines (known to be directly/indirectly involved in cartilage and bone destruction). Mature B cells, via mechanisms yet to be elucidated, can also differentiate into IL-10 producing B cells that can dampen the autoreactive T-cell response. GC, germinal centre; IFN, interferon; IL, interleukin; RA, rheumatoid arthritis; TLR, Toll-like receptor ligand; TNF, tumour necrosis factor.
Accumulation of B cells in the synovium is driven by a variety of signals
RA is one of only a few diseases in which ectopic germinal centre-like structures can be observed at the site of inflammation [2]. These structures, which range from loose aggregates of T and B cells to distinct follicle-like structures, are often observed in close contact with the inflamed synovial membrane of RA patients. A variety of cells, including fibroblast-like synoviocytes and dendritic cells, that are present in the synovium of patients with RA produce factors that affect B-cell survival, organization and trafficking, such as B cell-activating factor of the TNF family (BAFF), CXC chemokine ligand (CXCL)13, CXCL12 and lymphotoxin beta (Table 1) [2–4]. Based on their immunological function and location, each of these factors could contribute to the recruitment and maintenance of B cells in arthritic joints, thus representing potential therapeutic targets. For example, blockade of surface lymphotoxin using a decoy lymphotoxin receptor-immunoglobulin and BAFF, as discussed below, are currently in clinical trials. Interestingly, the efficacy observed in RA patients treated with etanercept, which binds to lymphotoxin-α as well as tumour necrosis factor (TNF)-α, may partly be related to blockade of the former cytokine [5]. Simultaneous blockade of more than one factor that drives B-cell accumulation may be a more efficient therapeutic approach than targeting a single cytokine or chemokine.
The role played by B cells in the maintenance of ectopic germinal centre-like structures, as well as in the immune response in RA synovium, has been addressed by using a humanized experimental model in which synovial tissue derived from patients was implanted into severe combined immunodeficient (SCID) mice [6]. B cells were then depleted by administration of anti-CD20 (rituximab), and T-cell responses were measured. The removal of B cells led to disruption of the lymphoid-like structures and to a reduction in T-helper (Th)1 interferon-γ producing cells, which are known to be involved in the induction and maintenance of the proinflammatory cytokine cascade.
Role played by B cells as antigen-presenting cells in rheumatoid arthritis
B cells actively participate in an autoimmune process through interaction with T cells by a variety of mechanisms, including antigen presentation and cytokine production. B cells process antigens, which are presented to T cells via major histocompatibility complex class II. Inherited susceptibility to RA has been associated with DRB1 genes that encode the HLA-DR4 and HLA-DR1 molecules [7]. These findings suggest a pathogenic role for antigen presentation in RA. Although dendritic cells are believed to be important in priming naïve T cells, B cells represent the predominant population of antigen-presenting cells in later phases of the immune response [8]. Rheumatoid factor (RF)-producing B cells are particularly effective in presenting immune complexes to T cells, irrespective of the antigen contained in the antigen-antibody complex [9]. Thus, T cells of other specificities could easily be activated if immune complexes in RA contain other antigens.
T-cell priming by B cells has been shown to be important in the pathogenesis of a murine model of arthritis. Specifically, the involvement of B cells in priming T cells was dissected within the context of proteoglycan (PG)-induced arthritis by using a mouse deficient in a secretory antibody (mIgM) [10]. These mice express a membrane-bound heavy chain transgene, which pairs with an endogenous light chain specific for hapten 4-hydroxy-3-nitro-phenyl acetyl (NP). T cells isolated from PG-immunized mIgM mice failed to induce arthritis in SCID mice, even if they were co-transferred with wild-type B cells, suggesting that T cells do not become properly primed in this experimental setting. However, targeting PG to B cells using NP coupled with PG led to differentiation of arthritogenic T cells that are able to transfer disease. Other antigen-presenting cells could not substitute for B cells in this T-cell priming, supporting a central role for B cells in driving autoreactive T cells. Autoantibody production was also essential for development of severe disease, indicating that B cells play two complementary roles in the pathogenesis of arthritis.
Immune complexes can activate B cells via Toll-like receptor ligands
It was recently shown that chromatin-containing immune complexes can activate B cells through Toll-like receptor ligand (TLR)9. These immune complexes activate B cells to produce RF by synergistic engagement of B-cell receptor and TLR9 [11]. TLRs were originally described as a family of pattern-recognition receptors that can differentiate between microbial molecular patterns and host components [12]. Their engagement results in rapid activation of the innate and adaptive immune systems to effect clearance of pathogens.
There is mounting evidence suggesting involvement of TLR signalling in the pathogenesis of experimental arthritis. Mice deficient for MyD88, the essential adaptor molecule involved in signalling by TLR family members, failed to develop streptococcal cell wall induced arthritis, and TLR2-deficient mice exhibited reduced disease [13]. Furthermore, direct injection of CpG DNA or double-stranded RNA into joints of susceptible mice results in the development of transient arthritis [14]. Heat shock protein, fibrinogen and hyaluronan, which are known to bind to TLR4, have all been detected in the inflamed joint [15]. In the KB × N model of murine antibody transferred arthritis, TLR4-deficient mice exhibit reduced disease [16]. Although there is enough evidence from experimental arthritis implicating TLRs in the development of arthritis, whether TLR activation is involved in human RA remains to be formally demonstrated.
Autoantibodies as effector molecules in rheumatoid arthritis
The pathological involvement of antibodies in inflammatory arthritis was first proved using DBA/1 mice immunized with collagen type II in complete Freund's adjuvant (CFA). These mice develop a severe arthritis that shares some pathological features with human RA. It has been demonstrated that SCID mice (which lack T and B cells), when treated with serum isolated from arthritic DBA/1 mice, develop an inflammatory arthritis [17, 18]. However, the disease was transient and less severe than collagen-induced arthritis (CIA). A more severe arthritis can be induced in recipient mice if serum is cotransferred with T cells presensitized with heat denatured collagen [19].
The pathological relevance of B cells in arthritis was further demonstrated by Holdhmal and colleagues [20], using μMT mice (which lack B cells) immunized with type II collagen in CFA. Lack of B cells completely prevented induction of arthritis and resulted in an impaired T-cell response to type II collagen (Mauri C, unpublished data); this suggests that although autoantibodies can initiate disease, other components of the immune system are needed to fuel the pathogenic response.
Compared with evidence from experimental models of arthritis, proof that antibodies are also pathogenic in human RA is more difficult to obtain. The presence of RF in serum, which binds to the constant region of IgG, was first identified in 1957 [21] and has long been recognized as a marker in the majority of patients with RA. The severity of RA has been correlated with RF levels, and patients who are seropositive for RF have more aggressive disease and worse prognosis [22, 23]. Important studies have indicated that the presence of RF can be detected many years before arthritis begins [24, 25].
The possible pathogenic role of RF-positive B cells has been revisited with the increased use of rituximab in RA therapy. RF can cause tissue damage through formation of immune complexes, by activation of complement, thereby recruiting cells into the synovium. Although preliminary data suggested that patients who are RF negative appear less likely to respond to B-cell depletion therapy [26], a larger trial [27] did not identify substantial differences in the response between RF-positive and RF-negative patients with RA. However, RF titres fell by 55% in those patients who were RF positive, which corroborates previous findings [28]. Moreover, routine assays for RF are not particularly sensitive and do not exclude their presence. Thus, patients identified as being RF-negative may still have low titres of RF. It is also possible that the many other autoantibody specificities present in patients with RA may distinguish those patients who respond to rituximab. These other autoantibody specificities include those directed toward the nuclear antigen RA-33 and heavy chain binding protein, both of which are found in early RA and pre-disease sera [29]. Antibody and T-cell reponses to heavy chain binding protein have been identified both in patients with RA and in animal models, suggesting that this may be an important autoantigen. Although antibodies to type II collagen have been shown to induce disease in animal models, it is unlikely that anti-collagen antibodies are relavant to human disease.
Recently, antibodies to citrulline-modified peptides (anti-cyclic citrullinated peptide [CCP] antibodies) have attracted considerable attention, and their measurement has now entered into routine clinical use. From a clinical perspective, anti-CCP antibodies represent a useful test for predicting which patients with early arthritis will go on to develop RA. The presence of both anti-CCP antibodies and RF predicts the development of RA in patients with early arthritis with high sensitivity and specificity [25]. The importance of anti-CCP antibodies is further emphasized by their link with HLA-DRB1 shared epitope alleles, the most important genetic risk factor for RA. Recent work has suggested that this HLA genetic risk factor is linked to the development of anti-CCP antibodies rather than to the disease itself [30]. Thus, the presence of these autoantibodies in RA, often preceding disease by many years, may indicate a breakdown in central and/or peripheral tolerance.
The pathological role played by anti-CCP antibodies was recently confirmed in the CIA arthritis model. As in human RA, anti-CCP antibodies can be detected before the onset of disease and are present in inflamed synovium of mice in the acute phase of CIA. Although the amount of anti-CCP antibodies measured in serum of mice with acute inflammation is similar to levels of antibodies to collagen type II, transfer of anti-CCP antibodies alone failed to induce disease in recipient mice. However, transfer of anti-CCP antibodies to SCID mice significantly reduced the amount of anti-collagen type II antibodies necessary to induce disease, demonstrating a contributing role in the development of arthritis [31]. The relevant targets of these antibodies in joints or in peripheral tissue remain unknown.
Are both Fc receptors and complement component C5a required for autoantibodies to drive the effector phase in arthritis?
Antibodies can act directly on target organs and induce disease through Fc-mediated activation of the complement system or through the formation of immune complexes. In addition, antibodies can directly activate Fcγ receptors (FcγRs) expressed on both myeloid and lymphoid cells. A clear insight into how antibodies work in arthritis has been gleaned from the K/B × N model of RA. In these mice glucose-6-phosphate isomerase (GPI) is the target autoantigen, and T-cell reactivity to this ubiquitous antigen results in recruitment of anti-GPI B cells and subsequent immune complex-mediated arthritis [32, 33]. Arthritis can be induced in non-autoimmune recipients, or in RAG2-/- mice (which lack both T and B cells), by transfer of sera or purified antibodies. Similar to the SCID model mentioned above, inflammation begins to subside between 15 and 30 days after antibody transfer. Histologically, analysis of joints 30 days after transfer revealed less inflammation than in the K/B × N model itself, and little cartilage damage, supporting the notion that other abnormalities in cellular types and soluble factors are needed for full expression of disease [32]. FcγRs are intimately involved in the pathogenesis of this arthritis. In particular, much milder arthritis was observed in mice lacking the FcγRIII receptor [34], whereas FcγRII-deficient mice exhibited accelerated disease.
The involvement of FcγRIIB in mediating antibody damage has also been investigated in the CIA model of arthritis. FcγRIIB is an inhibitory receptor that suppresses B cells, mast cells and macrophages, and transmits its inhibitory signal via its immunoreceptor tyrosine-based inhibitory motif. Deletion of FcγRIIB renders DBA/1 mice more susceptible to disease [35]. Recently, the pathogenicity of human RA-associated antibodies was also tested in a passive transfer model using FcγRIIB deficient mice. Transfer of serum from active RA patients, or an immunoglobulin-rich fraction, to 8- to 12-week-old B6.FcγRIIB-/- mice induced a mild transient arthritis [36], indicating that serum from patients with RA can induce an inflammatory arthritis. The administration of a large amount of intravenous IgG (IVIG) is a common treatment for a number of autoimmune conditions and is thought to modulate Fc receptor function [37]. IVIG has been shown to have a protective effect in the K/B × N mouse model of arthritis discussed above through induction of FcγRIIB [38]. This property of IVIG has been linked to sialylation of the Fc portion of IgG. The proportion of sialylated IgG molecules in commercial IVIG may account for the very mixed results obtained when patients with RA were treated with IVIG [39].
Involvement of the complement system in the development of mouse models of arthritis caused by autoantibodies has been demonstrated using C5-deficient mice. Both the K/B × N and collagen-induced model of arthritis are dependent on C5a for disease expression, and antibodies to C5 ameliorated disease in the K/B × N model [40, 41]. This has led to clinical trials of C5a receptor based peptides in RA, with mixed results. Other components of the complement system such as C4 do not participate in disease pathogenesis [40]. Therefore, the effector function of arthritogenic antibodies rely both on Fc receptors and C5a.
Immunoregulation by B cells
Although the pathogenic role played by mature B cells in RA has been extensively studied, new data have demonstrated that a distinct subset of B cells, namely those that produce IL-10, are involved in the downregulation of the immune system. It was originally demonstrated that B-cell-deficient mice developed an exacerbated experimental autoimmune encephalomyelitis as compared with wild-type animals, suggesting a protective role for B cells in the development of autoimmune disease [42]. B cells that produce cytokines, and in particular IL-10, have been reported to play an immunoregulatory role in autoimmunity, chronic inflammatory bowel disorders, asthma and infectious diseases [43, 44]. In the context of arthritis, we previously showed that stimulation of splenic B cells isolated during the acute phase of disease, with an agonistic anti-CD40 antibody, induces differentiation of IL-10 producing B cells. Transfer of anti-CD40 stimulated B cells to DBA/1 mice immunized with collagen type II in CFA prevented or ameliorated arthritis [45]. The mechanisms by which this subset of B cell regulates the immune response against autoantigens are not fully understood. However, we showed that mice treated with anti-CD40 challenged B cells exhibit an impaired Th1 response [45]. Therefore one plausible explanation is that production of IL-10 might restore the dysregulated Th1/Th2 balance, or it could directly modulate effector cells, including macrophages and dendritic cells, thus downmodulating inflammatory responses. IL-10 producing B cells could also act as secondary antigen-presenting cells, leading to an abortive response and induction of anergic CD4+ T cells, or they could recruit regulatory T cells or induce their differentiation. If an equivalent population exists in humans, then removal of these B cells by rituximab might be detrimental.
Targeting bad B cells
An understanding of which B cells are relevant to the pathogenesis of disease is important in designing therapeutic strategies to target B cells. The vast majority of B cells found in peripheral blood are removed by rituximab, but the extent and nature of B cell removal in other tissues in RA patients remain to be established. For example, experiments conducted in monkeys revealed that B cells residing in tissues are less effectively removed, and that memory B cells are more resistant to depletion than naïve ones [46]. Similar observations have been made in murine studies using anti-CD20, where marginal zone B cells, B1 cells and germinal centre B cells are more resistant to depletion [47–49]. As mentioned above, it is likely that plasma cells that producing RF are likely to be important in disease pathogenesis, but their depletion by rituximab is hampered by lack of CD20 expression. However, those plasma cells that have a short life span rely on CD20-expressing B cell precursors for continued renewal. Examination of peripheral blood indicates that CD19+CD20- plasmablasts decrease following rituximab therapy [50]. The observation that RF titres decrease following rituximab treatment suggests that short-lived, rather than long-lived, plasma cells are at least partly responsible for their formation. A number of factors are known to be important in plasma cell survival, including cytokines such as TNF-α and the cell adhesion molecule CD44 [51]. Perhaps use of anti-TNF-α together with rituximab may have synergistic benefit through their combined targeting of B cells and plasma cells, although infection-related side effects may prohibit use of this combination.
Antagonists to BAFF also lead to incomplete removal of peripheral and lymphoid B cells in monkeys, with marginal zone-like B cells being particularly susceptible to depletion [52]. Trials have begun to evaluate the anti-BAFF agent belimumab (LymphoStat-B; Human Genome Sciences, Rockville, MD, USA) in RA and have demonstrated limited efficacy, perhaps because of incomplete blockade or because other related B-cell survival factors such as a proliferation-inducing ligand (APRIL) would not be affected.
Conclusion
Renewed interest in B cells in RA has been initiated by a global B-cell-depleting agent, but it is likely that only a small proportion of B cells contribute to disease pathogenesis whereas others may actually be protective. It is hoped that research in patients with RA using these new agents will reveal correlations between pathogenic B cell subsets and improvement in clinical disease activity, thereby enhancing our understanding of the role played by B cells in human disease.
Note
This review is part of a series on Cells of the synovium in rheumatoid arthritis edited by Gary Firestein.
Other articles in this series can be found at http://arthritis-research.com/articles/review-series.asp?series=ar_Cells
Abbreviations
- RA:
-
= rheumatoid arthritis
- BAFF:
-
= B cell-activating factor of the TNF family
- NP:
-
= hapten 4-hydroxy-3-nitro-phenyl acetyl
- CCP:
-
= cyclic citrullinated peptide
- CFA:
-
= complete Freund's adjuvant
- CIA:
-
= collagen-induced arthritis
- CXCL:
-
= CXC chemokine ligand
- FcγR:
-
= Fcγ receptor
- IL:
-
= interleukin
- IVIG:
-
= intravenous IgG
- PG:
-
= prostaglandin
- RF:
-
= rheumatoid factor
- SCID:
-
= severe combined immunodeficient
- Th:
-
= T-helper (cell)
- TLR:
-
= Toll-like receptor ligand
- TNF:
-
= tumour necrosis factor
References
Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T: Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004, 350: 2572-2581. 10.1056/NEJMoa032534.
Takemura S, Braun A, Crowson C, Kurtin PJ, Cofield RH, O'Fallon WM, Goronzy JJ, Weyand CM: Lymphoid neogenesis in rheumatoid synovitis. J Immunol. 2001, 167: 1072-1080.
Tan SM, Xu D, Roschke V, Perry JW, Arkfeld DG, Ehresmann GR, Migone TS, Hilbert DM, Stohl W: Local production of B lymphocyte stimulator protein and APRIL in arthritic joints of patients with inflammatory arthritis. Arthritis Rheum. 2003, 48: 982-992. 10.1002/art.10860.
Seki T, Selby J, Haupl T, Winchester R: Use of differential subtraction method to identify genes that characterize the phenotype of cultured rheumatoid arthritis synoviocytes. Arthritis Rheum. 1998, 41: 1356-1364. 10.1002/1529-0131(199808)41:8<1356::AID-ART4>3.0.CO;2-X.
Buch MH, Conaghan PG, Quinn MA, Bingham SJ, Veale D, Emery P: True infliximab resistance in rheumatoid arthritis: a role for lymphotoxin alpha?. Ann Rheum Dis. 2004, 63: 1344-1346. 10.1136/ard.2003.014878.
Goronzy JJ, Weyand CM: Rheumatoid arthritis. Immunol Rev. 2005, 204: 55-73. 10.1111/j.0105-2896.2005.00245.x.
Newton JL, Harney SM, Wordsworth BP, Brown MA: A review of the MHC genetics of rheumatoid arthritis. Genes Immun. 2004, 5: 151-157. 10.1038/sj.gene.6364045.
MacLennan IC, Gulbranson-Judge A, Toellner KM, Casamayor-Palleja M, Chan E, Sze DM, Luther SA, Orbea HA: The changing preference of T and B cells for partners as T-dependent antibody responses develop. Immunol Rev. 1997, 156: 53-66. 10.1111/j.1600-065X.1997.tb00958.x.
Roosnek E, Lanzavecchia A: Efficient and selective presentation of antigen-antibody complexes by rheumatoid factor B cells. J Exp Med. 1991, 173: 487-489. 10.1084/jem.173.2.487.
O'Neill SK, Shlomchik MJ, Glant TT, Cao Y, Doodes PD, Finnegan A: Antigen-specific B cells are required as APCs and autoanti-body-producing cells for induction of severe autoimmune arthritis. J Immunol. 2005, 174: 3781-3788.
Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A: Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002, 416: 603-607. 10.1038/416603a.
Takeda K, Kaisho T, Akira S: Toll-like receptors. Annu Rev Immunol. 2003, 21: 335-376. 10.1146/annurev.immunol.21.120601.141126.
Joosten LA, Koenders MI, Smeets RL, Heuvelmans-Jacobs M, Helsen MM, Takeda K, Akira S, Lubberts E, van de Loo FA, van den Berg WB: Toll-like receptor 2 pathway drives streptococcal cell wall-induced joint inflammation: critical role of myeloid differentiation factor 88. J Immunol. 2003, 171: 6145-6153.
Deng GM, Nilsson IM, Verdrengh M, Collins LV, Tarkowski A: Intra-articularly localized bacterial DNA containing CpG motifs induces arthritis. Nat Med. 1999, 5: 702-705. 10.1038/9554.
van der Heijden IM, Wilbrink B, Tchetverikov I, Schrijver IA, Schouls LM, Hazenberg MP, Breedveld FC, Tak PP: Presence of bacterial DNA and bacterial peptidoglycans in joints of patients with rheumatoid arthritis and other arthritides. Arthritis Rheum. 2000, 43: 593-598. 10.1002/1529-0131(200003)43:3<593::AID-ANR16>3.0.CO;2-1.
Choe JY, Crain B, Wu SR, Corr M: Interleukin 1 receptor dependence of serum transferred arthritis can be circumvented by toll-like receptor 4 signaling. J Exp Med. 2003, 197: 537-542. 10.1084/jem.20021850.
Stuart JM, Tomoda K, Yoo TJ, Townes AS, Kang AH: Serum transfer of collagen-induced arthritis. II. Identification and localization of autoantibody to type II collagen in donor and recipient rats. Arthritis Rheum. 1983, 26: 1237-1244. 10.1002/art.1780261011.
Taylor PC, Plater-Zyberk C, Maini RN: The role of the B cells in the adoptive transfer of collagen-induced arthritis from DBA/1 (H-2q) to SCID (H-2d) mice. Eur J Immunol. 1995, 25: 763-769. 10.1002/eji.1830250321.
Nandakumar KS, Backlund J, Vestberg M, Holmdahl R: Collagen type II (CII)-specific antibodies induce arthritis in the absence of T or B cells but the arthritis progression is enhanced by CII-reactive T cells. Arthritis Res Ther. 2004, 6: R544-R550. 10.1186/ar1217.
Svensson L, Jirholt J, Holmdahl R, Jansson L: B cell-deficient mice do not develop type II collagen-induced arthritis (CIA). Clin Exp Immunol. 1998, 111: 521-526. 10.1046/j.1365-2249.1998.00529.x.
Franklin EC, Holman HR, Muller-Eberhard HJ, Kunkel HG: An unusual protein component of high molecular weight in the serum of certain patients with rheumatoid arthritis. J Exp Med. 1957, 105: 425-438. 10.1084/jem.105.5.425.
van Zeben D, Hazes JM, Zwinderman AH, Cats A, van der Voort EA, Breedveld FC: Clinical significance of rheumatoid factors in early rheumatoid arthritis: results of a follow up study. Ann Rheum Dis. 1992, 51: 1029-1035.
Symmons DP, Silman AJ: Aspects of early arthritis. What determines the evolution of early undifferentiated arthritis and rheumatoid arthritis? An update from the Norfolk Arthritis Register. Arthritis Res Ther. 2006, 8: 214-
Aho K, Palosuo T, Raunio V, Puska P, Aromaa A, Salonen JT: When does rheumatoid disease start?. Arthritis Rheum. 1985, 28: 485-489. 10.1002/art.1780280503.
Rantapaa-Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, Sundin U, van Venrooij WJ: Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003, 48: 2741-2749. 10.1002/art.11223.
De Vita S, Zaja F, Sacco S, De Candia A, Fanin R, Ferraccioli G: Efficacy of selective B cell blockade in the treatment of rheumatoid arthritis: evidence for a pathogenetic role of B cells. Arthritis Rheum. 2002, 46: 2029-2033. 10.1002/art.10467.
Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, Keystone EC, Loveless JE, Burmester GR, Cravets MW, et al: Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multi-center, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006, 54: 2793-2806. 10.1002/art.22025.
Cambridge G, Leandro MJ, Edwards JC, Ehrenstein MR, Salden M, Bodman-Smith M, Webster AD: Serologic changes following B lymphocyte depletion therapy for rheumatoid arthritis. Arthritis Rheum. 2003, 48: 2146-2154. 10.1002/art.11181.
Mewar D, Wilson AG: Autoantibodies in rheumatoid arthritis: a review. Biomed Pharmacother. 2006, 60: 648-655. 10.1016/j.biopha.2006.09.002.
van der Helm-van Mil AH, Verpoort KN, Breedveld FC, Huizinga TW, Toes RE, de Vries RR: The HLA-DRB1 shared epitope alleles are primarily a risk factor for anti-cyclic citrullinated peptide antibodies and are not an independent risk factor for development of rheumatoid arthritis. Arthritis Rheum. 2006, 54: 1117-1121. 10.1002/art.21739.
Kuhn KA, Kulik L, Tomooka B, Braschler KJ, Arend WP, Robinson WH, Holers VM: Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J Clin Invest. 2006, 116: 961-973. 10.1172/JCI25422.
Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D: A new mouse model of rheumatoid arthritis: organ-specific disease provoked by systemic autoimmunity. Ryumachi. 1997, 37: 147-
Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D: Organ-specific disease provoked by systemic autoimmunity. Cell. 1996, 87: 811-822. 10.1016/S0092-8674(00)81989-3.
Corr M, Crain B: The role of FcgammaR signaling in the K/B × N serum transfer model of arthritis. J Immunol. 2002, 169: 6604-6609.
Kleinau S, Martinsson P, Heyman B: Induction and suppression of collagen-induced arthritis is dependent on distinct fcgamma receptors. J Exp Med. 2000, 191: 1611-1616. 10.1084/jem.191.9.1611.
Petkova SB, Konstantinov KN, Sproule TJ, Lyons BL, Awwami MA, Roopenian DC: Human antibodies induce arthritis in mice deficient in the low-affinity inhibitory IgG receptor Fc gamma RIIB. J Exp Med. 2006, 203: 275-280. 10.1084/jem.20051951.
Samuelsson A, Towers TL, Ravetch JV: Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science. 2001, 291: 484-486. 10.1126/science.291.5503.484.
Kaneko Y, Nimmerjahn F, Ravetch JV: Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006, 313: 670-673. 10.1126/science.1129594.
Braun-Moscovici Y, Furst DE: Immunoglobulin for rheumatic diseases in the twenty-first century: take it or leave it?. Curr Opin Rheumatol. 2003, 15: 237-245. 10.1097/00002281-200305000-00010.
Ji H, Ohmura K, Mahmood U, Lee DM, Hofhuis FM, Boackle SA, Takahashi K, Holers VM, Walport M, Gerard C, et al: Arthritis critically dependent on innate immune system players. Immunity. 2002, 16: 157-168. 10.1016/S1074-7613(02)00275-3.
Grant EP, Picarella D, Burwell T, Delaney T, Croci A, Avitahl N, Humbles AA, Gutierrez-Ramos JC, Briskin M, Gerard C, et al: Essential role for the C5a receptor in regulating the effector phase of synovial infiltration and joint destruction in experimental arthritis. J Exp Med. 2002, 196: 1461-1471. 10.1084/jem.20020205.
Wolf SD, Dittel BN, Hardardottir F, Janeway CA: Experimental autoimmune encephalomyelitis induction in genetically B cell-deficient mice. J Exp Med. 1996, 184: 2271-2278. 10.1084/jem.184.6.2271.
Pistoia V: Production of cytokines by human B cells in health and disease. Immunol Today. 1997, 18: 343-350. 10.1016/S0167-5699(97)01080-3.
Mizoguchi A, Bhan AK: A case for regulatory B cells. J Immunol. 2006, 176: 705-710.
Mauri C, Gray D, Mushtaq N, Londei M: Prevention of arthritis by interleukin 10-producing B cells. J Exp Med. 2003, 197: 489-501. 10.1084/jem.20021293.
Vugmeyster Y, Beyer J, Howell K, Combs D, Fielder P, Yang J, Qureshi F, Sandlund B, Kawaguchi L, Dummer W, et al: Depletion of B cells by a humanized anti-CD20 antibody PRO70769 in Macaca fascicularis. J Immunother. 2005, 28: 212-219. 10.1097/01.cji.0000155050.03916.04.
Uchida J, Hamaguchi Y, Oliver JA, Ravetch JV, Poe JC, Haas KM, Tedder TF: The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J Exp Med. 2004, 199: 1659-1669. 10.1084/jem.20040119.
Gong Q, Ou Q, Ye S, Lee WP, Cornelius J, Diehl L, Lin WY, Hu Z, Lu Y, Chen Y, et al: Importance of cellular microenvironment and circulatory dynamics in B cell immunotherapy. J Immunol. 2005, 174: 817-826.
Hamaguchi Y, Uchida J, Cain DW, Venturi GM, Poe JC, Haas KM, Tedder TF: The peritoneal cavity provides a protective niche for B1 and conventional B lymphocytes during anti-CD20 immunotherapy in mice. J Immunol. 2005, 174: 4389-4399.
Leandro MJ, Cooper N, Cambridge G, Ehrenstein MR, Edwards JC: Bone marrow B-lineage cells in patients with rheumatoid arthritis following rituximab therapy. Rheumatology (Oxford). 2007, 46: 29-36. 10.1093/rheumatology/kel148.
Cassese G, Arce S, Hauser AE, Lehnert K, Moewes B, Mostarac M, Muehlinghaus G, Szyska M, Radbruch A, Manz RA: Plasma cell survival is mediated by synergistic effects of cytokines and adhesion-dependent signals. J Immunol. 2003, 171: 1684-1690.
Vugmeyster Y, Seshasayee D, Chang W, Storn A, Howell K, Sa S, Nelson T, Martin F, Grewal I, Gilkerson E, et al: A soluble BAFF antagonist, BR3-Fc, decreases peripheral blood B cells and lymphoid tissue marginal zone and follicular B cells in cynomolgus monkeys. Am J Pathol. 2006, 168: 476-489.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Mauri, C., Ehrenstein, M.R. Cells of the synovium in rheumatoid arthritis. B cells . Arthritis Res Ther 9, 205 (2007). https://doi.org/10.1186/ar2125
Published:
DOI: https://doi.org/10.1186/ar2125