
Abstract
Periarticular osteolysis, a crippling complication of rheumatoid arthritis, is the product of enhanced osteoclast recruitment and activation. The osteoclast, which is a member of the monocyte/macrophage family, is the exclusive bone resorptive cell, and its differentiation and activation are under the aegis of a variety of cytokines. Receptor activator of NF-κB ligand (RANKL) and macrophage colony-stimulating factor are the essential osteoclastogenic cytokines and are increased in inflammatory joint disease. Tumor necrosis factor-α, which perpetrates arthritic bone loss, exerts its osteoclastogenic effect in the context of RANKL with which it synergizes. Achieving an understanding of the mechanisms by which the three cytokines affect the osteoclast has resulted in a number of active and candidate therapeutic targets.
Similar content being viewed by others
Introduction
Recent years have witnessed a revolution in the treatment of inflammatory arthritis largely as a result of insights made into the role of cytokines in the pathogenesis of this family of diseases. Thus, inhibition of cytokines, such as members of the tumor necrosis factor (TNF) superfamily, that broadly impact the osteoclast, has proven a successful strategy for prevention of pathological bone loss [1].
The osteoclast is the principal and probably exclusive resorptive cell of bone and is therefore central to the pathogenesis of inflammatory osteolysis. It is abundant in affected joints of patients with rheumatoid [2] or psoriatic [3] arthritis as well as in implant particle-induced inflammation prompting prosthetic loosening [4]. Thus, understanding the mechanisms by which osteoclasts resorb bone, and the cytokines that regulate their differentiation and activity, provides mechanism-based candidate therapeutic targets to prevent periarticular osteolysis.
Much of what is known about the osteoclast comes from the study of the osteopetroses [5]. This family of disorders is characterized by enhanced bone mass caused by a failure of osteoclast recruitment or function. The fact that an osteopetrotic child was cured by marrow transplantation in the early 1980s established that the human osteoclast is of hematopoietic origin [6]. Subsequent studies document that the resorptive cell is a member of the monocyte/macrophage family [7] and provide the tools for generating the cell in culture and therefore the performance of meaningful biochemical and molecular experiments. As a result of these efforts, the past two decades have witnessed major insights into osteoclast biology.
How do osteoclasts resorb bone?
The osteoclast precursor arises principally in the marrow as an early mononuclear macrophage; it circulates and binds to the bone surface [8]. Whether the site to which the osteoclast precursor attaches, and which the differentiated osteoclast will ultimately resorb, is a selective or stochastic process is unknown. The process of bone remodeling must, however, replace effete bone with new to prevent brittleness and tendency to fracture, a condition that may be compromising long-term anti-bone resorptive therapy [9].
Once attached to bone, the mononuclear osteoclast precursor fuses with its sister cells to form a terminally differentiated polykaryon, which no longer has the capacity to replicate. Indirect evidence indicates that the life span of the osteoclast, in vivo, is about 2 weeks.
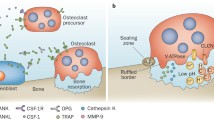
Although the osteoclast, like the foreign body giant cell, is multinucleate and the product of macrophage fusion, the two are distinct. The osteoclast, upon contact with bone, uniquely polarizes, which endows it with the capacity to degrade both the organic and the inorganic components of the skeleton [8]. This polarization process involves reorganization of the osteoclast cytoskeleton. Thus, under the influence of the Rho family of GTPases [10], the osteoclast's fibrillar actin forms a novel circular anchoring structure at the cell/bone interface, known as the 'actin ring' or 'sealing zone', that isolates the resorptive microenvironment from the general extracellular space [11]. At the same time, cytosol-residing acidified vesicles track to the resorptive surface of the cell [12], where they fuse with the bone-apposed plasma membrane under the aegis of Rab3D [13]. This insertion of large numbers of acidifiying vesicles into the plasma membrane results in the formation of a complex villous structure unique to, and diagnostic of, the resorbing osteoclast: the 'ruffled membrane' [14]. Once it has accomplished its resorptive mission at a particular location in bone, the osteoclast disassembles its actin ring and ruffled membrane, and migrates to its next site of activity, where it once again reorganizes its cytoskeleton to the resorptive phenotype [11]. Thus, changes in the osteoclast cytoskeleton are diagnostic of, and essential to, various steps in its bone degradative cycle (Fig. 1).

The osteoclast's bone resorptive cycle. (a) The osteoclast, when unattached to bone, is a non-polarized polykaryon with fibrillar actin (red material) diffusely distributed throughout the cell. (b) Upon attachment to bone the actin cytoskeleton forms a ring, or sealing zone, which isolates the resorptive microenvironment from the general extracellular space. (c) At the same time, acidifying vesicles polarize and insert into the plasma membrane juxtaposed to bone to generate the cell's resorptive organelle, the ruffled membrane. (d) The polarized osteoclast secretes hydrochloric acid (HCL), which acidifies the resorptive microenvironment, leading to mobilization of the mineral phase of bone. The exposed organic matrix is then degraded by cathepsin K (Cath K). Having resorbed the underlying bone to a depth of about 50 μm, the osteoclast detaches, disassembles its actin ring and ruffled membrane, and migrates to its next site of resorption.
The study of murine and human models of osteopetrosis established a paradigm by which the osteoclast first mobilizes the mineralized and, then, the organic phase of bone. Having generated the isolated extracellular microenvironment at its interface with bone, the osteoclast acidifies it by means of an electrogenic H+-ATPase that has been inserted into the ruffled membrane by polarized cytosolic vesicles [14]. This proton pump, which is similar to that residing in clathrin-coated vesicles [15], is essential to the resorptive process, and its dysfunction is the principal known cause of human osteopetrosis [16]. The massive extracellular transport of protons by the osteoclast has the potential for intracellular alkalization, which the cell prevents by a chloride–bicarbonate exchange mechanism located in the anti-resorptive plasma membrane [17]. The Cl- entering the cell moves transcellularly to the ruffled membrane and is transported into the resorptive microenvironment by an anion channel, which is charge-coupled to the H+-ATPase [18]. Interestingly, mutation of this Cl- channel also prompts osteopetrosis in humans [19]. Thus, by the generation of HCl, the osteoclast creates a pH of about 4.5 in the isolated microenvironment, the initial impact of which is to degrade the mineralized component of bone, thereby exposing its organic matrix consisting largely of type 1 collagen [20]. After mobilization of its mineral phase, the collagenous component of bone is degraded by the lysosomal enzyme cathepsin K, whose loss of function is responsible for the sclerosing skeletal disease pyknodysostosis [21].
The fact that contact with bone organizes the osteoclast cytoskeleton, and endows the cell with its resorptive capacity, indicates that molecules that mediate bone–cell recognition must be central to osteoclast formation and function. Integrins are heterodimeric transmembrane matrix receptors whose intracellular domains interact with signaling molecules and cytoskeletal proteins. In fact, integrins transmit extracellular matrix-derived signals that organize the osteoclast's fibrillar actin and prompt acidifying vesicles to migrate towards the ruffled membrane [12].
αvβ3 is the principal integrin mediating osteoclast function; it is specifically expressed when macrophage precursors commit to the bone resorptive but not the host defense phenotype [22]. This heterodimeric receptor, in osteoclasts, is localized within mobile matrix recognition structures known as podosomes, which also contain actin and other cytoskeletal proteins [23]. The location of podosomes within the osteoclast varies with the phase of the resorptive cycle, because these structures participate in the cell's migratory and bone degradative activities [23]. The fact that osteoclasts derived from mice lacking the integrin are dysfunctional, largely because of failure to organize their actin cytoskeleton and generate a normal ruffled membrane, establishes that αvβ3 transmits essential signals to the cell's interior [24]. These observations indicate that the αvβ3 integrin is a candidate anti-bone resorptive target, and small-molecule drugs that compete for the matrix receptor are in clinical trial [9, 25, 26]. Whether, as proposed, they arrest the bone loss of inflammatory arthritis [27] is yet to be determined.
αvβ3 occupancy organizes the osteoclast cytoskeleton by activating a series of signaling pathways. These include prolonged induction of the mitogen-activated protein (MAP) kinase Erk1/2 leading to enhanced expression of the activator protein-1 (AP-1) transcription factor, c-Fos [28]. c-Fos is essential for osteoclast generation [29], and mice deleted of the molecule are resistant to the bone loss of inflammatory arthritis. Interestingly, c-Fos overexpression in αvβ3-deficient osteoclasts substantially rescues the cells' capacity to organize their cytoskeleton [28]. In contrast, the integrin is itself necessary for the cell to adequately degrade bone [28].
The best characterized method by which αvβ3 mediates the resorptive process is through the Rho GTPase, Rac [30]. In this paradigm, αvβ3 occupancy recruits the proto-oncogene c-Src, which in turn phosphorylates the tyrosine kinase Syk. Activated Syk stimulates the guanine nucleotide exchange factor Vav3, the dominant isoform in osteoclasts, which transits Rac from its inactive GDP-bound form to active Rac-GTP [31]. Deletion of any of the above-mentioned signaling molecules results in a disturbance of the osteoclast cytoskeleton and the cell's capacity to resorb bone [24, 31–33]. Like αvβ3, c-Src appears coincidentally with osteoclast differentiation [34, 35] and is currently an anti-resorptive therapeutic target [36].
How do cytokines regulate osteoclast formation?
RANK ligand
Osteoclast precursors, like other members of the monocyte/macrophage family, are both the source and target of a variety of cytokines. Identification of the key cytokines regulating basal osteoclast formation and function followed the observation that generation of osteoclasts in culture requires contact of their precursors with marrow stromal cells, including osteoblasts [7]. Thus, the two essential cytokines promoting osteoclastogenesis are receptor activator of NF-κB ligand (RANKL) [37] and macrophage colony-stimulating factor (M-CSF) [38] (also known as CSF-1), each of which is produced by the marrow stromal cell family.
RANKL is a homotrimeric member of the TNF superfamily [39] and the essential osteoclastogenic cytokine. It is expressed as a transmembrane protein by osteoblasts and their precursors and its production is enhanced by osteoclast-stimulating agents such as parathyroid hormone [40] and TNF-α [41, 42]. In physiological circumstances cell-surface-residing RANKL interacts with its receptor, RANK, on osteoclast progenitors, explaining the requirement for contact between the two cells during osteoclastogenesis. In pathological conditions, such as inflammatory arthritis, RANKL is also expressed by activated T lymphocytes and in this circumstance is cleaved from the membrane and functions as a soluble ligand. In fact, T cell-produced RANKL is a major contributor to inflammation-mediated periarticular bone loss [43].
The unique osteoclastogenic properties of RANKL are due to specific structural features of loop components of its external domain, absent from other members of the TNF superfamily, that enable it to recognize its receptor [39]. RANK activation, in turn, recruits a number of TNF-receptor-associated proteins (TRAFs). However, it is TRAF6 that endows RANK with its unique osteoclastogenic potential. Although TRAF6 also associates with CD40 and the IL-1 and Toll-like receptors, it does not do so as abundantly as with RANK, probably accounting for at least a significant component of their lack of osteoclastogenic capacity [44, 45].
Osteoclast recruitment and function are also regulated by the LIM domain-only protein, FHL2, which binds TRAF6 and thus inhibits its association with RANK [46]. FHL2 is not detectable in naive osteoclasts in vivo but appears under the influence of RANKL or in animals with inflammatory arthritis. Establishing functional relevance, mice lacking FHL2 have hyper-resorptive osteoclasts and enhanced bone loss stimulated by RANKL and inflammatory arthritis. The accelerated resorption in this circumstance is due to aggressive organization of the osteoclast cytoskeleton, reflecting the capacity of RANKL to activate the mature resorptive cell in addition to promoting osteoclast differentiation [37, 47, 48].
The osteoclast-activating properties of RANKL are mediated via a complex composed of its receptor, TRAF6 and c-Src, which the cytokine specifically recruits to lipid rafts. Reflecting the cytoskeletal impact of c-Src, this event involves the organization of fibrillar actin and is mediated via the phosphoinositide 3-kinase (PI-3K)/Akt pathway, which also exerts an anti-apoptotic effect on the cells [49].
The discovery of the pivotal role of RANKL in the osteoclastogenic process actually followed on that of the secreted protein, osteoprotegerin (OPG) [50]. OPG, like RANKL, is synthesized by osteoblasts and their precursors and is also a member of the TNF superfamily [51]. It recognizes RANKL and thus functions as a decoy receptor, competing with RANK for its ligand. As would be predicted, OPG overexpression results in the arrest of osteoclastogenesis and hence leads to osteopetrosis [50]. Alternatively, deletion of the OPG gene, Tnfrsf11b, results in severe osteoporosis due to increased osteoclast number and activity [52]. Importantly, many of the same resorptive agents that enhance RANKL secretion suppress OPG production, and the ratio of the two molecules dictates the rate of bone loss in a variety of pathological conditions [53].
Activation of the RANK/TRAF6 composite induces a series of intracellular signaling pathways, each of which participates in the osteoclast phenotype. Activation of calcinurin by RANKL-enhanced intracellular calcium is among the most important of these events. Activated calcinurin dephosphorylates nuclear factor of activated T cells 1 (NFAT1), which translocates to the nucleus where, in association with c-Fos and c-Jun, it induces NFAT2 gene expression [54]. NFAT2, also in the context of the same AP-1 proteins, has the central role in the transactivation of osteoclastic genes such as tartrate-resistant acid phosphatase, the β3 integrin subunit and the calcitonin receptor [55]. Thus, whereas RANKL is the key osteoclastogenic cytokine, NFAT2 seems to be a key osteoclastogenic transcription factor.
The NF-κB family of transcription factors is also downstream of RANKL and central to osteoclast differentiation. In fact, deletion of the p50 and p52 NF-κB subunits, in concert, completely arrests osteoclastogenesis, resulting in severe osteopetrosis [56]. This realization prompted exploration of the NF-κB signaling pathway in the context of the osteoclast, and several intermediary signaling molecules have been identified as crucial to the event.
NF-κB activation occurs via both classical (canonical) and alternative signaling pathways. In both circumstances the IκB kinase (IKK) complex initiates the activation of NF-κB. This complex consists of three subunits, namely IKKα and β, which are catalytic, and IKKγ, which is regulatory. There is little question that IKKγ (also known as NEMO) is essential to the osteoclastogenic process because inhibition of its association with the α and β subunits, by cell-permeable peptides, arrests RANKL-induced osteoclastogenesis and prevents both the inflammatory and bone-destructive components of antigen-induced [57] and serum-transfer arthritis [58].
IKKβ activates the classical pathway by phosphorylating the cytosolic NF-κB binding proteins, IκBs, thereby targeting them for ubiquitin-mediated degradation. Most NF-κB subunits, particularly p65 and p50, are thus liberated and free to translocate to the nucleus and to function as transcriptional regulators. Importantly, the direct administration of non-degradable IκB peptides to mice prevents the development of inflammatory arthritis and its attendant bone destruction [59, 60].
The IKKβ-activated classical pathway generates osteoclasts in response to RANKL and participates in the bone-destructive components of inflammatory arthritis by promoting the differentiation of osteoclasts and prolonging their lifespan [61, 62]. There is, however, disagreement about the role of IKKα in basal and pathological osteoclastogenesis. IKKα modulates the alternative pathway leading to the generation of p52 NF-κB subunits [63]. On the one hand, mice lacking NF-κB-inducing kinase (NIK), which activates IKKα but not IKKβ, are resistant to RANKL-induced osteoclastogenesis and the bone destruction attending a variety of forms of inflammatory arthritis [64]. The fact that IKKα-/- mice exhibit defective osteoclast formation in vivo is in keeping with these NIK-based observations [65]. On the other hand, mice bearing an IKKα-inactivating mutation mirror wild-type animals as regards lipopolysaccharide-induced osteoclastogenesis and periarticular osteolysis [61]. Although specifics remain to be resolved, the NF-κB signaling pathway is clearly central to physiological and pathological bone resorption and its various components represent potential therapeutic targets.
M-CSF
M-CSF promotes the survival, proliferation and maturation of monocyte/macrophage precursors. It recognizes only one receptor, the tyrosine kinase c-Fms. The central role of the cytokine and its receptor in osteoclastogenesis is established by the fact that op/op mice, with a loss of function mutation in the Csf1 gene [38], and those deleted of c-Fms [66], lack osteoclasts and develop osteopetrosis. Interestingly, the osteopetrotic lesion of op/op mice resolves with age, reflecting a progressively increasing expression of granulocyte/macrophage colony-stimulating factor [67] and vascular endothelial growth factor [68], which compensate for the absence of M-CSF.
Like RANKL, M-CSF production by osteoblasts and their precursors, or by T cells, is stimulated by a variety of osteoclastogenic molecules, often with pathological consequences. For example, c-Fms activation participates in the bone loss attending inflammatory arthritis [69]. In this circumstance, inflammation-enhanced IL-1 and TNF-α stimulate the release of IL-7 from stromal cells, which in turn prompts T cells to produce M-CSF. Similarly, increased levels of parathyroid hormone promote the release of M-CSF from osteoblasts and stromal cells in the bone microenvironment [70]. An analogous scenario may hold for estrogen deprivation, perhaps participating in the pathogenesis of post-menopausal osteoporosis [71].
Activation of c-Fms involves its dimerization and auto-phosphorylation on specific tyrosine residues. The occupied receptor transmits a variety of signals affecting a broad array of events within the osteoclast and its precursor. For example, M-CSF-induced osteoclast precursor proliferation is mediated by both Erk1/2 and PI-3K/Akt. The latter also prolongs longevity of the mature cell [28]. Prolonged Erk activation stimulates osteoclast differentiation via the induction of c-Fos and, probably, NFAT2 [28]. M-CSF also regulates macrophage and osteoclast migration via cytoskeletal organization mediated by PI-3K and c-Src [72, 73]. The guanine nucleotide exchange factor Vav is phosphorated in response to M-CSF, leading to Rac-stimulated motility [31, 74].
SHIP1 is a 5' lipid phosphatase that dephosphorylates phosphatidylinositol 3,4,5-trisphosphate and thus inactivates Akt. SHIP1-deficient osteoclasts and their precursors are also hypersensitive to M-CSF [75]. A lack of SHIP1 therefore accelerates macrophage proliferation and dampens osteoclast apoptosis. These distinct effects of SHIP1 deletion on osteoclasts and their precursors result in increased numbers of enlarged, hypernucleated cells that aggressively resorb bone and produce an osteoporotic phenotype.
As demonstrated by the above, both M-CSF and the αvβ3 integrin activate several of the same signaling pathways in the osteoclast. In fact, they collaborate in osteoclast regulation. For example, the capacity of M-CSF to organize the cell's cytoskeleton depends on αvβ3-mediated matrix adhesion [76]. Furthermore, the retarded differentiation and cytoskeletal function of β3-/- osteoclasts are rescued by a high dose of M-CSF [28]. These findings reflect at least one common signaling pathway emanating from the integrin and c-Fms, involving prolonged activation of Erk leading to increased c-Fos expression. The essential role of c-Fos in αvβ3-mediated osteoclast cytoskeletal organization is confirmed by rescue of β3-/- osteoclasts by overexpression of the AP1 transcription factor [28]. M-CSF and αvβ3 also share Rac as a common downstream target in osteoclast cytoskeletal organization, an event mediated in both circumstances by activation of Vav3 [31].
TNF-α
Rheumatoid arthritis is a complicated condition because a host of cytokines, produced by a variety of cells, contributes to its pathogenesis. Although RANKL and IL-1 are important participants in the development of focal bone erosions that result in joint collapse, TNF-α is the principal and rate-limiting culprit in that its blockade dampens both the inflammatory and osteoclastogenic components of the disease.
TNF-α binds to two distinct receptors, each of which is expressed by osteoclast precursors. However, the osteoclastogenic properties of TNF-α are mediated via its p55 receptor (p55r). Marrow derived from mice expressing only this receptor generate substantially more osteoclasts in response to the cytokine than do the wild type, whereas those bearing only the other TNF receptor, p75r, produce fewer [77]. In keeping with this observation, soluble TNF-α, which preferentially activates p55r, has potent osteoclastogenic properties whereas those of its membrane-residing precursor, which recognizes p75r, are negligible. Similarly, whereas lipopolysaccharide seems to mediate osteoclast formation via its Toll-like receptors, it also stimulates the process via p55r [34]. TNF-α and RANKL are synergistic, and minimal levels of one markedly enhance the osteoclastogenic capacity of the other [41]. Alternatively, TNF-α recruits osteoclasts when precursors are exposed to, or primed by, permissive (that is, constitutive) levels of RANKL [41]. This observation in vitro is in keeping with the fact that OPG-treated mice fail to generate an osteoclastogenic response when subjected to inflammatory arthritis [43]. Thus, in the presence of M-CSF, RANKL – but not TNF-α – is necessary and sufficient to generate osteoclasts.
Many of the signaling pathways induced by p55 TNF receptor mirror those emanating from activated RANK, calling into question the reason that TNF-α on its own is incapable of promoting osteoclast differentiation. The most compelling evidence in this regard relates to the association of TRAF6 with the RANKL but not the TNF receptor.
TNF-α is a promiscuous cytokine, produced and recognized by a host of cells that participate in inflammatory osteolysis. Marrow stromal cells and macrophages are particular targets of TNF-α in this condition, but the greater contribution to osteoclast recruitment is made by the stromal cells [42]. In the presence of relatively mild inflammatory conditions, such as particle-induced implant loosening, TNF-α exerts its effect by stimulating stromal-cell production of cytokines, including RANKL, IL-1 and M-CSF, which in turn target macrophages to promote osteoclast differentiation. As the magnitude of inflammation and TNF-α production increases, substantial osteoclastogenesis is achieved by direct targeting of the cytokine to the osteoclast precursor even in the absence of TNF-α-responsive stromal cells.
M-CSF produced by stromal cells is particularly important in the pathogenesis of TNF-α-induced osteolysis. M-CSF stimulates RANK expression by osteoclast precursors and mediates the capacity of TNF-α to increase the number of these mononuclear cells. Most importantly, M-CSF inhibition selectively and completely arrests the profound osteoclastogenesis attending this condition or after TNF-α administration.
TNF-α enjoys an intimate relationship with IL-1 in pathological bone loss, including that attending loss of ovarian function [78, 79]. In this circumstance, decreased estrogen levels promote interferon-γ expression by T cells. The interferon enhances MHC class II expression by antigen-presenting cells, which in turn promotes T cell proliferation and their production of TNF-α and IL-1. These two cytokines stimulate RANKL expression by stromal cells, thereby increasing osteoclast number, which characterizes the accelerated bone loss of post-menopausal osteoporosis.
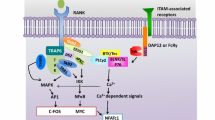
IL-1 mediates a substantial portion, but not all, of TNF-α-induced osteoclast recruitment in inflammatory osteoclastogenesis [80] (Fig. 2). Under the aegis of the p38 MAP kinase, IL-1 stimulates RANKL production by marrow stromal cells and, in the context of constitutive RANKL, directly promotes osteoclast precursor differentiation. Like TNF-α, IL-1 on its own is incapable of osteoclast recruitment despite a single TRAF6-binding site on the IL-1 receptor-associated kinase, IRAK. Attesting to their interdependence, blockade of either TNF-α or IL-1 does not completely arrest the periarticular damage of inflammatory arthritis, whereas inhibition of the two cytokines in combination is substantially more effective [81]. Thus, TNF-α signaling through p38 MAP kinase induces stromal-cell expression of IL-1, which upregulates its own receptor. Occupancy of the now abundant IL-1 receptor similarly activates p38, which promotes RANKL production. In macrophages, TNF-α enhances RANK expression and the synthesis of IL-1 whose functional receptor is in turn upregulated by the same three cytokines. Thus, the interdependence of TNF-α, RANKL and IL-1 in the generation of osteoclasts lends credence to the observation that combined blockade is most effective in preventing pathological bone loss.

Mechanisms of osteoclastogenesis induced by tumour necrosis factor (TNF)-α/IL-1. TNF-α interacts with its p55 receptor (TNFR) on both marrow stromal cells and osteoclast precursors in the form of marrow macrophages. Activation of the TNFR stimulates the expression of macrophage colony-stimulating factor (M-CSF) by stromal cells, which occupies its receptor, c-Fms, on osteoclast precursors. Signaling through p38 mitogen-activated protein kinase, TNF-α also induces stromal-cell synthesis of IL-1, which upregulates its own functional receptor, IL-1RI. Occupancy of now abundant IL-1RI similarly activates p38, which promotes RANKL production. In macrophages, TNF-α enhances RANK expression and the synthesis of IL-1, whose functional receptor is upregulated by the same three cytokines, also in a p38-dependent manner. Coincidentally, RANKL suppresses the IL-1 decoy receptor IL-1RII. TNF-α-induced IL-1RI upregulation in macrophages occurs by a combination of IL-1-dependent and IL-1-independent mechanisms. IL-1 interacting with its receptor on osteoclast precursors, in conjunction with RANKL and M-CSF, directly induces these cells to commit to the osteoclast phenotype. IL-1 mediates about 50% of the osteoclastogenic effect of TNF-α. (Modified from [80].)
Conclusion
Patients with rheumatoid arthritis face complications of the bony skeleton that result in joint destruction. Periarticular osteolysis, which may be particularly draconian, reflects accelerated osteoclast differentiation and function under the aegis of cytokines produced within the inflammatory environment. These cytokines, such as RANKL, M-CSF and TNF-α, induce the expression of molecules, like the αvβ3 integrin, necessary for osteoclasts to accomplish their bone-destructive mission. Delineating the means by which osteoclasts differentiate and resorb bone in an inflammatory environment has provided new therapeutic targets that are now being assessed in clinical trials.
Abbreviations
- AP-1:
-
activator protein-1
- M-CSF:
-
macrophage colony-stimulating factor
- IKK:
-
IκB kinase
- IL:
-
interleukin
- NF:
-
nuclear factor
- NFAT:
-
nuclear factor of activated T cells
- OPG:
-
osteoprotegerin
- PI-3K:
-
phosphoinositide 3-kinase
- RANKL:
-
receptor activator of NF-κB ligand
- TNF-α:
-
tumor necrosis factor-α
- TRAF:
-
TNF-receptor-associated protein.
References
Smolen JS, Steiner G: Therapeutic strategies for rheumatoid arthritis. Nat Rev Drug Discov. 2003, 2: 473-488. 10.1038/nrd1109.
Scott DL, Pugner K, Kaarela K, Doyle DV, Woolf A, Holmes J, Hieke K: The links between joint damage and disability in rheumatoid arthritis. Rheumatology. 2000, 39: 122-132. 10.1093/rheumatology/39.2.122.
Ritchlin CT, Haas-Smith SA, Li P, Hicks DG, Schwarz EM: Mechanisms of TNF-α- and RANKL-mediated osteoclastogenesis and bone resorption in psoriatic arthritis. J Clin Invest. 2003, 111: 821-831. 10.1172/JCI200316069.
Goldring SR, Clark CR, Wright TM: The problem in total joint arthroplasty: aseptic loosening. Journal of Bone & Joint Surgery – American Volume. 1993, 75: 799-801.
Tolar J, Teitelbaum SL, Orchard PJ: Osteopetrosis. N Engl J Med. 2004, 351: 2839-2849. 10.1056/NEJMra040952.
Coccia PF, Krivit W, Cervenka J, Clawson C, Kersey JH, Kim TH, Nesbit ME, Ramsay NK, Warkentin PI, Teitelbaum SL, et al: Successful bone-marrow transplantation for infantile malignant osteopetrosis. N Engl J Med. 1980, 302: 701-708.
Udagawa N, Takahashi N, Akatsu T, Tanaka H, Sasaki T, Nishihara T, Suda T: Origin of osteoclasts: mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc Natl Acad Sci USA. 1990, 87: 7260-7264. 10.1073/pnas.87.18.7260.
Teitelbaum SL: Bone resorption by osteoclasts. Science. 2000, 289: 1504-1508. 10.1126/science.289.5484.1504.
Teitelbaum SL: Osteoporosis and Integrins. J Clin Endocrinol Metab. 2005, 90: 2466-2468. 10.1210/jc.2005-0338.
Chellaiah MA, Soga N, Swanson S, McAllister S, Alvarez U, Wang D, Dowdy SF, Hruska KA: Rho-A is critical for osteoclast podosome organization, motility, and bone resorption. J Biol Chem. 2000, 275: 11993-12002. 10.1074/jbc.275.16.11993.
Vaananen HK, Horton M: The osteoclast clear zone is a specialized cell-extracellular matrix adhesion structure. J Cell Sci. 1995, 108: 2729-2732.
Abu-Amer Y, Ross FP, Schlesinger P, Tondravi MM, Teitelbaum SL: Substrate recognition by osteoclast precursors induces s-crc/microtubule association. J Cell Biol. 1997, 137: 247-258. 10.1083/jcb.137.1.247.
Pavlos NJ, Xu J, Riedel D, Yeoh JSG, Teitelbaum SL, Papadimitriou JM, Jahn R, Ross FP, Zheng MH: Rab3D regulates a novel vesicular trafficking pathway that is required for osteoclastic bone resorption. Mol Cell Biol. 2005, 25: 5253-5269. 10.1128/MCB.25.12.5253-5269.2005.
Blair HC, Teitelbaum SL, Ghiselli R, Gluck S: Osteoclastic bone resorption by a polarized vacuolar proton pump. Science. 1989, 245: 855-857. 10.1126/science.2528207.
Mattsson JP, Schlesinger PH, Keeling DJ, Teitelbaum SL, Stone DK, Xie X-S: Isolation and reconstitution of a vacuolar-type proton pump of osteoclast membranes. J Biol Chem. 1994, 269: 24979-24982.
Frattini A, Orchard PJ, Sobacchi C, Giliani S, Abinun M, Mattsson JP, Keeling DJ, Andersson AK, Wallbrandt P, Zecca L, et al: Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat Genet. 2000, 25: 343-346. 10.1038/77131.
Teti A, Blair HC, Teitelbaum SL, Kahn AJ, Carano A, Grano M, Santacroce G, Schlesinger P, Zambonin-Zallone A: Cytoplasmic pH is regulated in isolated avian osteoclasts by a Cl-/HCO3 exchanger. Boll Soc Ital Biol Sper. 1989, 65: 589-595.
Schlesinger PH, Blair HC, Teitelbaum SL, Edwards JC: Characterization of the osteoclast ruffled border chloride channel and its role in bone resorption. J Biol Chem. 1997, 272: 18636-18643. 10.1074/jbc.272.30.18636.
Kornak U, Kasper D, Bosl MR, Kaiser E, Schweizer M, Schulz A, Friedrich W, Delling G, Jentsch TJ: Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell. 2001, 104: 205-215. 10.1016/S0092-8674(01)00206-9.
Blair HC, Kahn AJ, Crouch EC, Jeffrey JJ, Teitelbaum SL: Isolated osteoclasts resorb the organic and inorganic components of bone. J Cell Biol. 1986, 102: 1164-1172. 10.1083/jcb.102.4.1164.
Gelb BD, Shi GP, Chapman HA, Desnick RJ: Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science. 1996, 273: 1236-1238. 10.1126/science.273.5279.1236.
Teitelbaum SL, Ross FP: Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003, 4: 638-649. 10.1038/nrg1122.
Faccio R, Novack DV, Zallone A, Ross FP, Teitelbaum SL: Dynamic changes in the osteoclast cytoskeleton in response to growth factors and cell attachment are controlled by β3 integrin. J Cell Biol. 2003, 162: 499-509. 10.1083/jcb.200212082.
McHugh KP, Hodivala-Dilke K, Zheng MH, Namba N, Lam J, Novack D, Feng X, Ross FP, Hynes RO, Teitelbaum SL: Mice lacking β3 integrins are osteosclerotic because of dysfunctional osteoclasts. J Clin Invest. 2000, 105: 433-440.
Engleman VW, Nickols GA, Ross FP, Horton MA, Settle SL, Ruminski PG, Teitelbaum SL: A peptidomimetic antagonist of the αvβ3 integrin inhibits bone resorption in vitro and prevents osteoporosis in vivo. J Clin Invest. 1997, 99: 2284-2292.
Murphy MG, Cerchio K, Stoch SA, Gottesdiener K, Wu M, Recker R, for the L-000845704 Study Group: Effect of L-000845704, an αVβ3 integrin antagonist, on markers of bone turnover and bone mineral density in postmenopausal osteoporotic women. J Clin Endocrinol Metab. 2005, 90: 2022-2028. 10.1210/jc.2004-2126.
Wilder RL: Integrin αVβ3 as a target for treatment of rheumatoid arthritis and related rheumatic diseases. Ann Rheum Dis. 2002, 61: 96ii-99.
Faccio R, Zallone A, Ross FP, Teitelbaum SL: c-Fms and the αvβ3 integrin collaborate during osteoclast differentiation. J Clin Invest. 2003, 111: 749-758. 10.1172/JCI200316924.
Grigoriadis AE, Wang Z-Q, Cecchini MG, Hofstetter W, Felix R, Fleisch HA, Wagner EF: c-Fos: A key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science. 1994, 266: 443-448. 10.1126/science.7939685.
Razzouk S, Lieberherr M, Cournot G: Rac-GTPase, osteoclast cytoskeleton and bone resorption. Eur J Cell Biol. 1999, 78: 249-255.
Faccio R, Teitelbaum SL, Fujikawa K, Chappel JC, Zallone A, Tybulewicz VL, Ross FP, Swat W: Vav3 regulates osteoclast function and bone mass. Nat Med. 2005, 11: 284-290. 10.1038/nm1194.
Faccio R, Zou W, Colaianni G, Teitelbaum SL, Ross FP: High dose M-CSF partially rescues the Dap12-/- osteoclast phenotype. J Cell Biochem. 2003, 90: 871-883. 10.1002/jcb.10694.
Boyce BF, Yoneda T, Lowe C, Soriano P, Mundy GR: Requirement of pp60c-src expression for osteoclasts to form ruffled borders and resorb bone in mice. J Clin Invest. 1992, 90: 1622-1627.
Abu-Amer Y, Ross FP, Edwards J, Teitelbaum SL: Lipopolysaccharide-stimulated osteoclastogenesis is mediated by tumor necrosis factor via its p55 receptor. J Clin Invest. 1997, 100: 1557-1565.
Merkel KD, Erdmann JM, McHugh KP, Abu-Amer Y, Ross FP, Teitelbaum SL: Tumor necrosis factor-α mediates orthopedic implant osteolysis. Am J Pathol. 1999, 154: 203-210.
Shakespeare WC, Metcalf CAR, Wang Y, Sundaramoorthi R, Keenan T, Weigele M, Bohacek RS, Dalgarno DC, Sawyer TK: Novel bone-targeted Src tyrosine kinase inhibitor drug discovery. Curr Opin Drug Discov Devel. 2003, 6: 729-741.
Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, et al: Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998, 93: 165-176. 10.1016/S0092-8674(00)81569-X.
Yoshida H, Hayashi S-I, Kunisada T, Ogawa M, Nishikawa S, Okamura H, Sudo T, Shultz LD, Nishikawa S-I: The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990, 345: 442-444. 10.1038/345442a0.
Lam J, Nelson CA, Ross FP, Teitelbaum SL, Fremont DL: Crystal structure of TRANCE/RANKL cytokine reveals determinants of receptor-ligand specificity. J Clin Invest. 2001, 108: 971-980. 10.1172/JCI200113890.
Lee SK, Lorenzo JA: Parathyroid hormone stimulates TRANCE and inhibits osteoprotegerin messenger ribonucleic acid expression in murine bone marrow cultures: correlation with osteoclast-like cell formation. Endocrinol. 1999, 140: 3552-3561. 10.1210/en.140.8.3552.
Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL: TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000, 106: 1481-1488.
Kitaura H, Sands MS, Aya K, Zhou P, Hirayama T, Uthgenannt B, Wei S, Takeshita S, Novack DV, Silva MJ, et al: Marrow stromal cells and osteoclast precursors differentially contribute to TNF-α induced osteoclastogenesis in vivo. J Immunol. 2004, 173: 4838-4846.
Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe S, et al: Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999, 402: 304-309. 10.1038/46303.
Kadono Y, Okada F, Perchonock C, Jang HD, Lee SY, Kim N, Choi Y: Strength of TRAF6 signalling determines osteoclastogenesis. EMBO Reports. 2005, 6: 171-176. 10.1038/sj.embor.7400345.
Gohda J, Akiyama T, Koga T, Takayanagi H, Tanaka S, Inoue J-i: RANK-mediated amplification of TRAF6 signaling leads to NFATc1 induction during osteoclastogenesis. EMBO J. 2005, 24: 790-799. 10.1038/sj.emboj.7600564.
Bai S, Kitaura H, Zhao H, Chen J, Muller JM, Schule R, Darnay B, Novack DV, Ross FP, Teitelbaum SL: FHL2 inhibits the activated osteoclast in a TRAF6 dependent manner. J Clin Invest. 2005, 115: 2742-2751. 10.1172/JCI24921.
Nakamura I, Kadono Y, Takayanagi H, Jimi E, Miyazaki T, Oda H, Nakamura K, Tanaka S, Rodan GA, Duong LT: IL-1 regulates cytoskeletal organization in osteoclasts via TNF receptor-associated factor 6/c-Src complex. J Immunol. 2002, 168: 5103-5109.
Armstrong AP, Tometsko ME, Glaccum M, Sutherland CL, Cosman D, Dougall WC: A RANK/TRAF6-dependent signal transduction pathway is essential for osteoclast cytoskeletal organization and resorptive function. J Biol Chem. 2002, 277: 44347-44356. 10.1074/jbc.M202009200.
Wang MW-H, Wei S, Faccio R, Takeshita S, Tebas P, Powderly WG, Teitelbaum SL, Ross FP: The HIV protease inhibitor riton-avir blocks osteoclastogenesis and function by impairing RANKL-induced signaling. J Clin Invest. 2004, 114: 206-213. 10.1172/JCI200415797.
Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, et al: Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997, 89: 309-319. 10.1016/S0092-8674(00)80209-3.
Thomas GP, Baker SUK, Eisman JA, Gardiner EM: Changing RANKL/OPG mRNA expression in differentiating murine primary osteoblasts. J Endocrinol. 2001, 170: 451-460. 10.1677/joe.0.1700451.
Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, et al: Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12: 1260-1268.
Hofbauer LC, Heufelder AE: Role of receptor activator of nuclear factor-κB ligand and osteoprotegerin in bone cell biology. J Mol Med. 2001, 79: 243-253. 10.1007/s001090100226.
Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, et al: Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002, 3: 889-901. 10.1016/S1534-5807(02)00369-6.
Ikeda F, Nishimura R, Matsubara T, Tanaka S, Inoue J-i, Reddy SV, Hata K, Yamashita K, Hiraga T, Watanabe T, et al: Critical roles of c-Jun signaling in regulation of NFAT family and RANKL-regulated osteoclast differentiation. J Clin Invest. 2004, 114: 475-484. 10.1172/JCI200419657.
Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD, Leonardi A, Tran T, Boyce BF, Siebenlist U: Requirement for NF-κB in osteoclast and B-cell development. Genes Dev. 1997, 11: 3482-3496.
Jimi E, Aoki K, Saito H, D'Acquisto F, May JJ, Nakamura I, Sudo T, Kojima T, Okamoto F, Fukushima H, et al: Selective inhibition of NF-B blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nat Med. 2004, 10: 617-624. 10.1038/nm1054.
Dai S, Hirayama T, Abbas S, Abu-Amer Y: The IκB kinase (IKK) inhibitor, NEMO-binding domain peptide, blocks osteoclastogenesis and bone erosion in inflammatory arthritis. J Biol Chem. 2004, 279: 37219-37222. 10.1074/jbc.C400258200.
Clohisy JC, Roy BC, Biondo C, Frazier E, Willis D, Teitelbaum SL, Abu-Amer Y: Direct inhibition of NF-κB blocks bone erosion associated with inflammatory arthritis. J Immunol. 2003, 171: 5547-5553.
Abu-Amer Y, Dowdy SF, Ross FP, Clohisy JC, Teitelbaum SL: TAT fusion proteins containing tyrosine 42-deleted IκBα arrest osteoclastogenesis. J Biol Chem. 2001, 276: 30499-30503. 10.1074/jbc.M104725200.
Ruocco MG, Maeda S, Park JM, Lawrence T, Hsu L-C, Cao Y, Schett G, Wagner EF, Karin M: IκB kinase (IKK)β, but not IKKα, is a critical mediator of osteoclast survival and is required for inflammation-induced bone loss. J Exp Med. 2005, 201: 1677-1687. 10.1084/jem.20042081.
Abbas S, Abu-Amer Y: Dominant-negative IκB facilitates apoptosis of osteoclasts by tumor necrosis factor-α. J Biol Chem. 2003, 278: 20077-20082. 10.1074/jbc.M208619200.
Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun S-C, et al: Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science. 2001, 293: 1495-1499. 10.1126/science.1062677.
Novack DV, Yin L, Hagen-Stapleton A, Schreiber RD, Goeddel DV, Ross FP, Teitelbaum SL: The IκB function of NF-κB2 p100 controls stimulated osteoclastogenesis. J Exp Med. 2003, 198: 771-781. 10.1084/jem.20030116.
Chaisson ML, Branstetter DG, Derry JM, Armstrong AP, Tometsko ME, Takeda K, Akira S, Dougall WC: Osteoclast differentiation is impaired in the absence of inhibitor of κB kinase α. J Biol Chem. 2004, 279: 54841-54848. 10.1074/jbc.M406392200.
Dai X-M, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, Sylvestre V, Stanley ER: Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood. 2002, 99: 111-120. 10.1182/blood.V99.1.111.
Myint YY, Miyakawa K, Naito M, Shultz LD, Oike Y, Yamamura K, Takahashi K: Granulocyte/macrophage colony-stimulating factor and interleukin-3 correct osteopetrosis in mice with osteopetrosis mutation. Am J Pathol. 1999, 154: 553-566.
Niida S, Kaku M, Amano H, Yoshida H, Kataoka H, Nishikawa S, Tanne K, Maeda N, Nishikawa S, Kodama H: Vascular endothelial growth factor can substitute for macrophage colony-stimulating factor in the support of osteoclastic bone resorption. J Exp Med. 1999, 190: 293-298. 10.1084/jem.190.2.293.
Weitzmann MN, Cenci S, Rifas L, Brown C, Pacifici R: IL-7 stimulates osteoclast formation by upregulating the T-cell production of soluble osteoclastogenic cytokines. Blood. 2000, 96: 1873-1878.
Weir EC, Lowik CW, Paliwal I, Insogna KL: Colony stimulating factor-1 plays a role in osteoclast formation and function in bone resorption induced by parathyroid hormone and parathyroid hormone-related protein. J Bone Miner Res. 1996, 11: 1474-1481.
Srivastava S, Weitzmann MN, Kimble RB, Rizzo M, Zahner M, Milbrandt J, Ross FP, Pacifici R: Estrogen blocks M-CSF gene expression and osteoclast formation by regulating phosphorylation of Egr-1 and its interaction with Sp-1. J Clin Invest. 1998, 102: 1850-1859.
Fuller K, Owens JM, Jagger CJ, Wilson A, Moss R, Chambers TJ: Macrophage colony-stimulating factor stimulates survival and chemotactic behavior in isolated osteoclasts. J Exp Med. 1993, 178: 1733-1744. 10.1084/jem.178.5.1733.
Golden LH, Insogna KL: The expanding role of PI3-kinase in bone. Bone. 2004, 34: 3-12. 10.1016/j.bone.2003.09.005.
Vedham V, Phee H, Coggeshall KM: Vav activation and function as a Rac guanine nucleotide exchange factor in macrophage colony-stimulating factor-induced macrophage chemotaxis. Mol Cell Biol. 2005, 25: 4211-4220. 10.1128/MCB.25.10.4211-4220.2005.
Takeshita S, Namba N, Zhao JJ, Jiang Y, Genant HK, Silva MJ, Brodt MD, Helgason CD, Kalesnikoff J, Rauh MJ, et al: SHIP-deficient mice are severely osteoporotic due to increased numbers of hyper-resorptive osteoclasts. Nat Med. 2002, 8: 943-949. 10.1038/nm752.
Teti A, Taranta A, Migliaccio S, Degiorgi A, Santandrea E, Villanova I, Faraggiana T, Chellaiah M, Hruska KA: Colony stimulating factor-1-induced osteoclast spreading depends on substrate and requires the vitronectin receptor and the c-src proto-oncogene. J Bone Miner Res. 1998, 13: 50-58. 10.1359/jbmr.1998.13.1.50.
Abu-Amer Y, Erdmann J, Kollias G, Alexopoulou L, Ross FP, Teitelbaum SL: Tumor necrosis factor receptors types 1 and 2 differentially regulate osteoclastogenesis. J Biol Chem. 2000, 275: 27307-27310.
Gao Y, Qian W-P, Dark K, Toraldo G, Lin ASP, Guldberg RE, Flavell RA, Weitzmann MN, Pacifici R: Estrogen prevents bone loss through transforming growth factor β signaling in T cells. Proc Natl Acad Sci USA. 2004, 101: 16618-16623. 10.1073/pnas.0404888101.
Teitelbaum SL: Postmenopausal osteoporosis, T cells, and immune dysfunction. PNAS. 2004, 101: 16711-16712. 10.1073/pnas.0407335101.
Wei S, Kitaura H, Zhou P, Ross FP, Teitelbaum SL: IL-1 mediates TNF-induced osteoclastogenesis. J Clin Invest. 2005, 115: 282-290. 10.1172/JCI200523394.
Zwerina J, Hayer S, Tohidast-Akrad M, Bergmeister H, Redlich K, Feige U, Dunstan C, Kollias G, Steiner G, Smolen J, et al: Single and combined inhibition of tumor necrosis factor, interleukin-1, and RANKL pathways in tumor necrosis factor-induced arthritis: effects on synovial inflammation, bone erosion, and cartilage destruction. Arthritis Rheum. 2004, 50: 277-290. 10.1002/art.11487.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Teitelbaum, S.L. Osteoclasts; culprits in inflammatory osteolysis. Arthritis Res Ther 8, 201 (2005). https://doi.org/10.1186/ar1857
Published:
DOI: https://doi.org/10.1186/ar1857