
Abstract
Microsomal prostaglandin E synthase (mPGES)-1 is a newly identified inducible enzyme of the arachidonic acid cascade with a key function in prostaglandin (PG)E2 synthesis. We investigated the kinetics of inducible cyclo-oxygenase (COX)-2 and mPGES-1 expression with respect to the production of 6-keto-PGF1α and PGE2 in rat chondrocytes stimulated with 10 ng/ml IL-1β, and compared their modulation by peroxisome-proliferator-activated receptor (PPAR)γ agonists. Real-time PCR analysis showed that IL-1β induced COX-2 expression maximally (37-fold) at 12 hours and mPGES-1 expression maximally (68-fold) at 24 hours. Levels of 6-keto-PGF1α and PGE2 peaked 24 hours after stimulation with IL-1β; the induction of PGE2 was greater (11-fold versus 70-fold, respectively). The cyclopentenone 15-deoxy-Δ12,14prostaglandin J2 (15d-PGJ2) decreased prostaglandin synthesis in a dose-dependent manner (0.1 to 10 μM), with more potency on PGE2 level than on 6-keto-PGF1α level (-90% versus -66% at 10 μM). A high dose of 15d-PGJ2 partly decreased COX-2 expression but decreased mPGES-1 expression almost completely at both the mRNA and protein levels. Rosiglitazone was poorly effective on these parameters even at 10 μM. Inhibitory effects of 10 μM 15d-PGJ2 were neither reduced by PPARγ blockade with GW-9662 nor enhanced by PPARγ overexpression, supporting a PPARγ-independent mechanism. EMSA and TransAM® analyses demonstrated that mutated IκBα almost completely suppressed the stimulating effect of IL-1β on mPGES-1 expression and PGE2 production, whereas 15d-PGJ2 inhibited NF-κB transactivation. These data demonstrate the following in IL-1-stimulated rat chondrocytes: first, mPGES-1 is rate limiting for PGE2 synthesis; second, activation of the prostaglandin cascade requires NF-κB activation; third, 15d-PGJ2 strongly inhibits the synthesis of prostaglandins, in contrast with rosiglitazone; fourth, inhibition by 15d-PGJ2 occurs independently of PPARγ through inhibition of the NF-κB pathway; fifth, mPGES-1 is the main target of 15d-PGJ2.
Similar content being viewed by others


Introduction
Prostaglandins (PGs) are well-known lipid mediators that reproduce the cardinal signs of inflammation [1] but also contribute to tumorigenesis, gastrointestinal protection or osteogenesis [2–5]. Decreasing their biosynthesis by the inhibition of cyclo-oxygenases (COXs) is thought to account for most of the therapeutical properties of non-steroidal anti-inflammatory drugs. During inflammation, the pathophysiological contribution of prostaglandins is supported by PGE2, the major mediator produced by monocytes in response to inflammatory stimulus, and prostacyclin (PGI2). However, since the discovery of at least two COX isoenzymes, the pathophysiological relevance of PG must be considered from a different point of view. First, inflammation can be ascribed to inducible COX-2-derived PG rather than to basal COX-1-derived PG [6]. Second, PGE2 and PGI2 are now recognized as end-point products of a coordinate enzymatic cascade comprising phospholipases A2, cyclooxygenases and terminal PG synthases whose activities are coupled preferentially between constitutive and inducible isoforms [7]. Third, PG produced by COX-2 switches from PGE2 to 15-deoxy-Δ12,14prostaglandin J2 (15d-PGJ2) in the course of acute inflammation [8]. Because 15d-PGJ2, a cyclopentenone by-product of PGD2, has shown anti-inflammatory properties in various experimental models [9, 10], it has been proposed as an endogenous regulator of inflammation favouring the resolution of acute flares [11].
PGE synthase-1 (PGES-1), the enzyme converting the COX-derived PGH2 into PGE2, exists in multiple forms with distinct enzymatic properties, modes of expression, subcellular localizations and intracellular functions [12]. One of its isoforms, cPGES-1, is a cytosolic protein found as a complex with heat shock protein 90 (Hsp90) that is constitutively expressed in a wide variety of cells and tissues. Another isoform, microsomal PGE synthase-1 (mPGES-1), is a perinuclear membrane-associated protein belonging to the microsomal glutathione S-transferase family. In contrast with cPGES-1, its expression is induced by pro-inflammatory cytokines, growth factors, bacterial endotoxins and phorbol esters and is downregulated by anti-inflammatory corticosteroids [12]. As mentioned above, PGES-1 isoforms display distinct functional coupling with upstream COX in cells; cPGES-1 is predominantly coupled with constitutive COX-1, thereby contributing to basal PG synthesis, whereas mPGES-1 is preferentially linked with inducible COX-2 and contributes to stimulated PG synthesis [7]. Recently a novel PGES, mPGES-2 [13], was cloned and was shown to be highly expressed in heart and brain. Its role remains largely unknown, especially in inflammatory conditions.
Peroxisome-proliferator-activated receptor γ (PPARγ) is a ligand-activated nuclear transcription factor belonging to the nuclear hormone receptor superfamily. PPARγ binds, as a heterodimer with retinoid X receptor, to peroxisome-proliferator-response element (PPRE) located in the promoter of numerous target genes whose expression is regulated by PPARγ agonists. Agonists of PPARγ include synthetic ligands, as antidiabetic thiazolidinediones, and natural compounds, as fatty acids and 15d-PGJ2, which were shown initially to have a major function in adipocyte differentiation and glucose homeostasis [14–16]. However, PPARγ agonists were recently thought to contribute to the control of inflammation by inhibiting the transcriptional induction of pro-inflammatory cytokines (tumour necrosis factor-α, IL-1 and IL-6) or genes encoding inflammatory enzymes (inducible nitric oxide synthase and COX-2) in activated monocytic cells [17, 18]. Similar pharmacological potencies were reported in chondrocytes [19] and synoviocytes [20] exposed to an inflammatory stimulus, giving a rationale to the anti-inflammatory effect of PPARγ agonists in experimental arthritis [10, 21]. Because 15d-PGJ2 was thought to be a negative regulator of experimental inflammation [11], it is tempting to speculate that part of this effect could be supported by the regulation of PPARγ target genes, possibly through the control of transcription factors such as NF-κB or activator protein-1 [22, 23].
Chondrocytes express both COX isoenzymes [24] and produce large amounts of eicosanoids under inflammatory conditions [25]. However, COX-2 represents only the first inducible step in the stimulated synthesis of PG [12] and its inhibition by PPARγ ligands remains moderate in articular cells [19, 20]. We therefore investigated whether PPARγ agonists could reduce PG synthesis by inhibiting mPGES-1 in rat chondrocytes stimulated with IL-1β. Such a mechanism would be consistent with the ability of 15d-PGJ2 to inhibit PGE2 production and to downregulate mPGES-1 in microsomal fractions from CHO cells overexpressing mPGES [26].
The present study demonstrates an early induction of COX-2 and a delayed induction of mPGES-1 by IL-1β in rat chondrocytes, with the stimulated synthesis of prostaglandins fitting well the expression profile of mPGES-1 for PGE2 while remaining lower than the extent of COX-2 induction for 6-keto-PGF1α (the stable metabolite of PGI2). In our experimental system, 15d-PGJ2 lowered the 6-keto-PGF1α level and the expression of COX-2 but was much more potent towards the PGE2 level and the expression of mPGES-1, supporting the view that mPGES-1 is the rate-limiting step in PGE2 synthesis. The dose-dependent inhibitory potency of 15d-PGJ2 was not reproduced by the high-affinity PPARγ agonist rosiglitazone and was affected neither by blockade of PPARγ with the antagonist GW-9662 nor by PPARγ overexpression. Consistent with a PPARγ-independent mechanism was our final observation that 15d-PGJ2 decreased NF-κB transactivation, which is crucial for the induction of mPGES-1 and the stimulation of PGE2 synthesis by IL-1β in rat chondrocytes.
Materials and methods
Isolation and culture of rat chondrocytes
Chondrocytes were isolated from femoral heads of healthy Wistar male rats (130 to 150 g) (Charles River, Saint-Aubin-les-Elbeuf, France), killed under general anaesthesia (AErrane™; Baxter SA, Maurepas, France) in accordance with national animal care guidelines, after approval by our internal ethics committee. Cells were obtained by sequential digestion with pronase and collagenase [27], then washed twice in PBS and cultured to confluence in 75 cm2 flasks at 37°C in a humidified atmosphere containing 5% CO2. The medium used was DMEM/Ham's F-12 supplemented with L-glutamine (2 mM), penicillin (100 U/ml), streptomycin (100 μg/ml) and either 10% heat-inactivated FCS (Life Technologies) during subculturing or 1% FCS during experiments. Chondrocytes were used between passages 1 and 3 to prevent dedifferentiation.
Study design
Chondrocytes maintained in low (1%) FCS medium were stimulated with 10 ng/ml IL-1β (Sigma, St-Quentin-Fallavier, France) in the presence or absence (vehicle alone, 0.1% of final concentration in dimethylsulphoxide) of PPAR agonists added 4 hours before IL-1β. In a preliminary kinetic study, mRNA levels of COX-2 and mPGES-1 in cell layers were determined from 6 to 48 hours after challenge with IL-1β, whereas 6-keto-PGF1α and PGE2 levels were assayed from 6 to 36 hours in culture supernatants. Thereafter, COX-2 mRNA level was checked 12 hours after exposure to IL-1β, whereas the mPGES-1 mRNA level, the COX-2 and mPGES-1 protein levels, and the secreted 6-keto-PGF1α and PGE2 levels were evaluated at 24 hours. The PPARγ agonists rosiglitazone (Cayman, Ann Arbor, MI, USA) or 15d-PGJ2 (Calbiochem, Meudon, France) were used in the range 0.1 to 10 μM, whereas additional PPARγ agonist troglitazone (Cayman) and PPARγ antagonist GW-9662 (Cayman) were used at 10 μM.
Assay for chondrocyte viability
Cell viability was assessed by the mitochondrial-dependent reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT; Sigma) into formazan [28]. In brief, cells were incubated for 24 hours at 37°C in the presence or absence of IL-1β and/or PPARγ agonists (added 4 hours before IL-1β) in low-FCS (1%) DMEM/Ham's F-12 medium. Chondrocytes were incubated further with MTT (1 mg/ml final concentration) for 4 hours at 37°C before the addition of lysing buffer (20% w/v SDS in a 50% aqueous solution of dimethylformamide, pH 4.7). After 24 hours of incubation at 37°C, solubilization of formazan crystals was quantified by measuring A580 on a Multiskan® microplate reader (Labsystems, Montigny-le-Bretonneux, France).
RNA extraction and real-time PCR analysis
Total RNA was isolated from chondrocyte layers using Trizol® (Invitrogen, Cergy-Pontoise, France). Two micrograms of total RNA were reverse-transcribed for 90 minutes at 37°C with 200 U of Moloney Murine Leukaemia Virus reverse transcriptase (Invitrogen) and hexamer random primers. Expression of COX-2, mPGES-1 and adiponectin (chosen as a specific PPARγ target gene [29]) mRNAs were quantified by real-time PCR with the Lightcycler® (Roche) technology and the SYBRgreen master mix system® (Qiagen, Courtabœuf, France). After amplification, a melting curve was constructed to determine the melting temperature of each PCR product; their sizes were checked on a 2% agarose gel stained with ethidium bromide (0.5 μg/ml). Each run included standard dilutions and positive and negative reaction controls. The mRNA levels of each gene of interest and of the ribosomal protein S29, chosen as a housekeeping gene, were determined in parallel for each sample. Results are expressed as the normalized ratio of mRNA level of each gene of interest over the S29 gene.
The gene-specific primer pairs used were as follows: mPGES-1, sense 5'-TCGCCTGGATACATTTCCTC-3', antisense 5'-GTCCCCCATTGTGGTATCTG-3'; COX-2, sense 5'-TACAAGCAGTGGCAAAGGCC-3', antisense 5'-CAGTATTGAGGAGAACAGATGGG-3'; adiponectin, sense 5'-AATCCTGCCCAGTCATGAAG-3', antisense 5'-TCTCCAGGAGTGCCATCTCT-3'; S29, sense 5'-AAGATGGGTCACCAGCAGCTCTACG-3', antisense 5'-AGACGCGGCAAGAGCGAGAA-3'.
Transient transfection experiments
Chondrocytes were seeded in six-well plates at 5 × 105 cells per well and grown to 80% confluence. Cells were transfected with either 500 ng of a PPARγ expression vector (pcDNA3.1 PPARγ, a gift from Dr H. Fahmi, Centre Hospitalier de l'Université de Montréal, Montréal, Canada), or 500 ng of a dominant-negative vector of NF-κB (IκBαΔN (Ala32, Ala36) from Clontech). Transfections were performed for 2 hours with 10 μl of polyethyleneimine reagent (Euromedex, Souffelweyersheim, France) in 1 ml of culture medium. At 24 hours after transfection, cells were stimulated with IL-1β for 24 hours in the presence or absence of PPARγ agonists.
Preparation of nuclear extracts and electrophoretic mobility-shift assay (EMSA)
Nuclear proteins were isolated as described elsewhere [30] with minor modifications. In brief, cells were scraped in a lysis buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 1 mM dithiothreitol (DTT)) containing a protease-inhibitor cocktail and 0.5% Igepal®, then incubated for 15 min on ice. Nuclei were collected by centrifugation at 2,000 g for 5 min at 4°C and resuspended in 50 μl of HEPES buffer without Igepal® and KCl, but containing 420 mM NaCl. After a 30 min incubation on ice, nuclear debris were removed by centrifugation at 13,000 g for 10 min at 4°C; supernatants were collected and then stored at -80°C before use. The DNA sequences of the double-stranded oligonucleotides specific for NF-κB were 5'-GATCCAGTTGAGGGGACTTTCCCAGGCG-3' and 5'-GATCCGCCTGGGAAAGTCCCCTCAACTG-3'. Complementary strands were annealed and double-stranded oligonucleotides were labelled with [32P]dCTP by using the Klenow fragment of DNA polymerase (Invitrogen). Nuclear proteins (5 μg) were incubated for 10 min at 4°C in a binding buffer (20 mM Tris/HCl, pH 7.9, 5 mM MgCl2, 0.5 mM DTT, 0.5 mM EDTA and 20% glycerol) in the presence of 2 μg of poly(dIdC). The extracts were then incubated for 30 min at 4°C with 10,000 c.p.m. of 32P-labelled NF-κB probe. The samples were loaded on a 5% native polyacrylamide gel and run in 0.5 × Tris/borate/EDTA buffer. NF-κB-specific bands were confirmed by competition with a 100-fold excess of unlabelled probe, which resulted in no shifted band.
NF-κB transactivation analysis
Nuclear proteins were prepared with the TransAM® nuclear extract kit in accordance with the manufacturer's protocol (Active Motif Europe, Rixensart, Belgium). In brief, cells were scraped into PBS containing phosphatase and protease inhibitors, centrifuged, resuspended in 1 × hypotonic buffer and then kept on ice for 15 min. After the addition of detergent, lysates were centrifuged at 14,000 g for 30 s at 4°C. The pellets were resuspended in complete lysis buffer (20 mM HEPES, pH 7.5, 350 mM NaCl, 20% glycerol, 1% Igepal®, 1 mM MgCl2, 0.5 mM EDTA, 0.1 mM EGTA, 1 mM DTT, phosphatase and protease inhibitors) and shaken vigorously. After incubation on ice and centrifugation at 14,000 g for 10 min at 4°C, supernatants were collected and protein concentration was determined with a Bradford-based assay (Bio-Rad Laboratories, Marnes-la-Coquette, France).
NF-κB activation was determined with the TransAM® ELISA kit (Active Motif Europe). In brief, 5 μg of nuclear extract was added to each well of a 96-well plate into which an oligonucleotide with a NF-κB consensus binding site had been immobilized. After 1 hour of incubation with smooth agitation, wells were washed three times with washing buffer (100 mM PBS, pH 7.5, 500 mM NaCl and 1% Tween 20) and then incubated with p65 antibody (dilution 1:1,000 in washing buffer) for 1 hour at 20°C. After three successive washings with buffer, the wells were finally incubated for 1 hour with diluted horseradish peroxidase-conjugated antibody (dilution 1:1,000 in washing buffer) before the addition of 100 μl of developing solution (3,3',5,5'-tetramethylbenzidine substrate solution diluted in 1% dimethylsulphoxide). After 5 min of incubation, the reaction was stopped by the addition of 100 μl of 0.5 M H2SO4 and the final A450 was read on a Multiskan® microplate reader.
Assays for PGE2 and 6-keto-PGF1α
Levels of PGE2 and 6-keto-PGF1α were determined in culture supernatants with Assay Design® ELISA kits (Oxford Biomedical Research, Ann Arbor, MI, USA) in accordance with manufacturer's instructions. Assays are based on the combined use of a monoclonal antibody against PGE2 or PGF1α and an alkaline phosphatase-conjugated polyclonal antibody. After the addition of p-nitrophenyl phosphate substrate, A405 was read at on a micro Multiskan® plate reader. The limits of detection were 10 pg/ml and 1.4 pg/ml for PGE2 and 6-keto-PGF1α, respectively, with negligible cross-reactivity with PGE1 and PGF2α, respectively (manufacturer's data). Positive controls were used in each experiment.
Western blot analysis
Cells, seeded in six-well plates and grown to 90% confluence, were washed twice with ice-cold PBS and scraped off the wells in 1 × Laemmli blue for PPARγ or in TBS containing 0.1% SDS for other proteins. Cells were disrupted by sonication (five pulses) and centrifuged at 800 g for 10 min, before determination of protein concentration with a Bradford-based assay. Protein samples (5 μg) were analysed by SDS-PAGE (10% acrylamide for COX-2 and PPARγ, 12% for β-actin, and 15% for mPGES-1), and electroblotted on a poly(vinylidene difluoride) membrane. After 1 hour in blocking buffer (TBS-Tween with 5% nonfat dried milk), membranes (Immobilon; Waters, Saint-Quentin en Yvelines, France) were blotted overnight at 4°C with antibodies against β-actin (dilution 1:500; Sigma), mPGES-1 (dilution 1:200; Cayman), COX-2 (dilution 1:1,000; Cayman) or PPARγ (a gift from Professor Michel Dauça, Université Henri Poincaré, Vandœuvre-lès-Nancy, France; dilution 1:1,000), diluted in TBS-Tween with 5% bovine serum albumin. After three washings with TBS-Tween, the blot was incubated for 1 hour at room temperature with anti-rabbit IgG conjugated with horseradish peroxidase (Cell Signaling, Beverly, MA, USA) at 1:2,000 dilution in TBS-Tween containing 5% nonfat dried milk. After four washings with TBS-Tween, protein bands were detected by chemiluminescence with the Phototope Detection system in accordance with the manufacturer's instructions (Cell Signaling).
Statistical analysis
Results are expressed as means ± SD for at least three assays. Comparisons were made by ANOVA, followed by the Fisher protected least-squares difference post-hoc test with Statview™ 5.0 software (SAS Institute Inc). A P value of less than 0.05 was considered significant.
Results
Kinetics of COX-2/mPGES-1 expression and prostaglandin production in IL-1β-stimulated rat chondrocytes
Under basal conditions, PGE2 and 6-keto-PGF1α production was almost undetectable (Fig. 1a), whereas COX-2 and mPGES-1 mRNAs were expressed at a very low level (Fig. 1b). In response to IL-1β, PGE2 levels increased earlier (6 hours) than 6-keto-PGF1α levels (12 hours), although both peaked at 24 hours (Fig. 1a). At the time of maximal production, PGE2 levels were increased 70-fold and 6-keto-PGF1α levels 11-fold. Under these experimental conditions, COX-2 and mPGES-1 expression was induced from 6 hours, with maximal induction at 12 hours and 24 hours, respectively, after challenge with IL-1β (Fig. 1b). At these times, the extent of gene variation was higher for mPGES-1 (68-fold) than for COX-2 (37-fold).

Time course of prostaglandins production, COX-2 and mPGES-1 mRNA expression, in IL-1β-stimulated chondrocytes. Rat cells were exposed to 10 ng/ml IL-1β for 6, 12, 24, 36 or 48 hours before total RNA extraction and collection of culture supernatant. (a) Prostaglandin levels (PGE2, 6-keto-PGF1α) assayed by ELISA in culture supernatant; (b) relative abundances of cyclo-oxygenase-2 (COX-2) and microsomal prostaglandin E synthase-1 (mPGES-1) mRNAs, analysed by real-time PCR and normalized to S29 mRNA. Prostaglandin levels and PCR COX-2/S29 or mPGES-1/S29 mRNA ratios presented in histograms are expressed as means ± SD for at least three independent experiments. Statistically significant differences (P < 0.05) from controls: * for PGE2 or COX-2; † for 6-keto-PGF1α or mPGES-1
Effect of PPARγ agonists on prostaglandin cascade in IL-1β-stimulated rat chondrocytes
As shown in Fig. 2a, IL-1β-induced PGE2 production was decreased by 92%, and 6-keto-PGF1α levels by 66%, by 10 μM 15d-PGJ2. The effect of 10 μM rosiglitazone on the stimulated levels of prostaglandins was less than the variation range of our biological system (-12% for PGE2 and +10% for 6-keto-PGF1α; Fig. 2a). Under IL-1-stimulated conditions, 10 μM 15d-PGJ2 decreased the expression of COX-2 and mPGES-1 by 40% and 92%, respectively, at the mRNA level (Fig. 2b) and by 52% and 73%, respectively, at the protein level (Fig. 2c). In contrast, 10 μM rosiglitazone increased COX-2 mRNAs by 37% and decreased mPGES-1 mRNAs by 10% (Fig. 2b), while leaving COX-2 protein unaffected and decreasing mPGES-1 protein by 36% (Fig. 2c). The inhibitory potency of 15d-PGJ2 on PGE2 levels was dose-related (-8% at 0.1 μM and -42% at 10 μM), whereas rosiglitazone was still ineffective at lower concentrations (-2% at 0.1 μM and -6% at 10 μM). As shown in Table 1, the proliferation of chondrocytes was increased by challenge with IL-1β but this effect was reduced neither by 15d-PGJ2 nor by rosiglitazone. Under IL-1-stimulated conditions, the PPARγ agonist troglitazone (10 μM) had a potency similar to that of rosiglitazone on mPGES-1 mRNAs (-12%), although its induction of COX-2 mRNAs was less (+25% versus +37%) and it was more inhibitory towards PGE2 levels (-25% versus -12%; data not shown). The basal levels of prostaglandins were unaffected by PPARγ agonists (Fig. 2a) despite a moderate inducing effect of 15d-PGJ2 on COX-2 mRNAs (Fig. 2b) and protein (Fig. 2c).

Effect of PPARγ agonists on IL-1β-induced prostaglandins levels, COX-2 and mPGES-1 mRNAs. After 4 hours of pretreatment with 10 μM 15-deoxy-Δ12,14prostaglandin J2 (15d-PGJ2) or rosiglitazone, chondrocytes were incubated with 10 ng/ml IL-1β for 12 or 24 hours. (a) PGE2 and 6-keto-PGF1α levels assayed by ELISA in culture supernatant; (b) relative abundances of cyclo-oxygenase-2 (COX-2) and microsomal prostaglandin E synthase-1 (mPGES-1) mRNAs, analysed by real-time PCR and normalized to S29 mRNA (c) COX-2 and mPGES-1 protein levels assessed by western blotting and normalized to β-actin level. Results are expressed as means ± SD for at least three independent experiments. Statistically significant differences (P < 0.05): *, comparison with non-stimulated controls; #, comparison with IL-1β-stimulated cells.
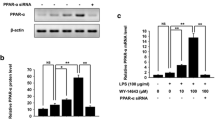
Effect of PPARγ blockade on inhibitory potency of 15d-PGJ2on stimulated prostaglandin cascade
When 10 μM 15d-PGJ2 was tested in combination with the PPARγ antagonist GW-9662 at 10 μM, its inhibitory effect on IL-1-induced PGE2 (-94% versus -95%) and 6-keto-PGF1α (-64% versus -58%) levels remained unchanged (Fig. 3a). Similarly, the strong decrease in mPGES-1 mRNA (-93% versus -87%; Fig. 3b) and protein (-70% versus -65%; Fig. 3c) levels was unaffected. In all experiments, the inducing effect of IL-1β on prostaglandin release and gene expression was not modified by GW-9662. Because of the low efficacy of chondrocyte transfection with a PPRE-luciferase construct as a gene reporter assay, the functionality of PPARγ ligands was controlled by measuring changes in adiponectin expression. As shown in Fig. 3d, the adiponectin mRNA level was increased by 10 μM 15d-PGJ2 or rosiglitazone and returned to the basal level in the presence of GW-9662.

Effect of PPARγ blockade on the inhibition of IL-1β-induced responses by 15d-PGJ2. Chondrocytes were pretreated for 4 hours with 10 μM 15-deoxy-Δ12,14prostaglandin J2 (15d-PGJ2) in the presence or absence of 10 μM GW9662 (a specific antagonist of peroxisome-proliferator-activated receptor γ (PPARγ)), then stimulated with 10 ng/ml IL-1β for 24 hours before analysis of prostaglandin production and mPGES-1 expression. (a) PGE2 and 6-keto-PGF1α levels assayed by ELISA in culture supernatant; (b) relative abundance of microsomal prostaglandin E synthase-1 (mPGES-1) mRNA analysed by real-time PCR and normalized to S29 mRNA; (c) mPGES-1 protein level assessed by western blotting and normalized to β-actin level; (d) modulation of adiponectin (a PPARγ target gene) mRNAs by PPARγ ligands, analysed by real-time PCR and normalized to S29 mRNA. Results are expressed as means ± SD for at least three independent experiments. Statistically significant differences (P < 0.05): *, comparison with non stimulated controls; #comparison with IL-1β-stimulated cells; †, comparison with PPARγ agonists alone or in combination with PPARγ antagonist.
Effect of PPARγ overexpression on inhibitory potency of 15d-PGJ2on stimulated prostaglandin cascade
Transfection of chondrocytes with a PPARγ expression vector did not change their response to IL-1β and provoked a limited increase in PGE2 level and mPGES-1 expression in resting cells (Fig. 4a, b). The inhibition of IL-1β-induced PGE2 release and mPGES-1 mRNA level by 10 μM 15d-PGJ2 was not impaired in cells overexpressing PPARγ (-88% versus -94% and -79% versus -82%, respectively; Fig. 4a, b). Control experiments showed that PPARγ protein was efficiently overexpressed (Fig. 4c), and that the level of adiponectin mRNA was enhanced by 15d-PGJ2 or rosiglitazone (Fig. 4d), in cells transfected with the PPARγ expression vector.

Effect of PPARγ overexpression on the inhibition of IL-1β-induced responses by 15d-PGJ2. Chondrocytes in six-well plates were transfected with pcDNA3.1 peroxisome-proliferator-activated receptor γ (PPARγ) construct (500 ng) for 36 hours. Thereafter, cells were pretreated for 4 hours with 10 μM 15-deoxy-Δ12,14prostaglandin J2 (15d-PGJ2), then stimulated with 10 ng/ml IL-1β for 24 hours before extraction of total RNA and collection of culture supernatant. (a) PGE2 levels assayed by ELISA in culture supernatant; (b) relative abundance of microsomal prostaglandin E synthase-1 (mPGES-1) mRNAs analysed by real-time PCR and normalized to S29 mRNA; (c) western blot control experiment of PPARγ and β-actin expression; (d) modulation of adiponectin (a PPARγ target gene) mRNAs by PPARγ agonists and pcDNA3.1 PPARγ construct, analysed by real-time PCR and normalized to S29 mRNA. Results are expressed as means ± SD for at least three independent experiments. Statistically significant differences (P < 0.05): *, comparison with non-stimulated controls; #, comparison with IL-1β-stimulated cells; †, comparison with PPARγ agonists alone or in combination with PPARγ plasmid.
Contribution of NF-κB pathway to regulation of stimulated prostaglandin cascade by IL-1β and 15d-PGJ2in rat chondrocytes
As shown in Fig. 5, transfection with a dominant-negative vector of NF-κB (IκBαΔN) almost completely eliminated the synthesis of PGE2 (Fig. 5a) and the expression of mPGES-1 (Fig. 5b) in IL-1β-stimulated chondrocytes. As with PPARγ, transient overexpression was associated with a negligible induction of PGE2 and mPGES-1 in resting cells (Fig. 5a, b). Gel-shift analysis (Fig. 5c) and TransAM® assay (Fig. 5d) confirmed that IL-1β induced NF-κB transactivation in rat chondrocytes and demonstrated that this activity was markedly decreased by 15d-PGJ2.

Contribution of NF-κB pathway to IL-1β-induced responses and 15d-PGJ2 inhibitory effects. In one set of experiments (a, b), chondrocytes cultured in six-well plates were transfected with 500 ng of IKBα dominant-negative (IκBαΔN) vector for 24 hours, then stimulated for 24 hours with 10 ng/ml IL-1β. (a) PGE2 levels in culture supernatant assayed by ELISA; (b) Relative abundance of microsomal prostaglandin E synthase-1 (mPGES-1) mRNAs analysed by real-time PCR and normalized to S29 mRNA. Results are expressed as means ± SD for at least three independent experiments. In another set of experiments (c, d), chondrocytes cultured in six-well plates were exposed to 10 ng/ml IL-1β for 15 min in the presence or absence of 10 μM 15-deoxy-Δ12,14prostaglandin J2 (15d-PGJ2) before extraction of nuclear proteins. Activation of NF-κB was determined by EMSA (c) and by ELISA with the TransAm® technology (d). Results in (d) are expressed as relative arbitrary units with IL-1β treatment set at 100, and are representative of three different experiments. Statistically significant differences (P < 0.05): *, comparison with non-stimulated controls; #, comparison with IL-1β-stimulated cells.
Discussion
Since the discovery of a preferential coupling between several inducible enzymes of the prostaglandin cascade [31], it has become necessary to re-evaluate which step is critical for the synthesis of mediators. COX and phospholipases A2 have long been considered the rate-limiting enzymes; this was confirmed indirectly by the successful launching of non-steroidal anti-inflammatory drugs for the treatment of inflammation, pain and fever. However, the discovery of inducible mPGES-1 opened new insights because it was expressed at a high level in joint tissues during experimental polyarthritis [32] as well as in periarticular soft tissues and brain during acute inflammation [33]. Moreover, PGE2 was shown to contribute to inflammation and hyperalgesia [34], and the pivotal role of mPGES-1 in its production was confirmed by the decrease in pain nociception and inflammatory reactions in mPGES-1-deficient mice [35]. Finally, in contrast with COX inhibition, blockade of mPGES-1 could theoretically favour the biotransformation of cyclic endoperoxide H2 into anti-inflammatory 15d-PGJ2 depending on the tissue expression of PGD synthase [36]. The pathophysiological role of mPGES-1 in inflammatory diseases is therefore worthy of study, and inhibitors of this enzyme might have potent therapeutical relevance [37].
In the present study we investigated first the respective time courses of prostaglandin production and induction of genes of the arachidonic acid cascade in chondrocytes activated with IL-1β, a pro-inflammatory cytokine with a central function in joint diseases [38]. We confirmed that normal rat chondrocytes were very sensitive to stimulation by IL-1β and produced large amounts of prostaglandins [39], with kinetics comparable to that of human osteoarthritic chondrocytes [19, 40] or the immortalized T/C-28a2 cell line [41]. As expected, resting and activated chondrocytes produced several types of prostaglandin, although the extent of variation was much higher for PGE2 than for 6-keto-PGF1α [39, 42]. Although IL-1β-induced PGE2 synthesis was associated with the induction of COX-2 expression in articular cells [19, 39], it has been shown that COX-2 and mPGES-1 are coordinately upregulated, but with different time courses [37, 39, 43], and that their subcellular localizations overlap in the perinuclear region [40, 43]. Our kinetics study confirmed an early induction of COX-2 and a delayed induction of mPGES-1 in IL-1β-stimulated chondrocytes [40], thereby mimicking the time course reported for inflamed rat tissues [33].
The increase in PGE2 level fitted well with the extent of mPGES-1 gene induction but not with that of COX-2, whereas changes in the 6-keto-PGF1α level were much smaller than the extent of COX-2 induction. Of course, each inducible enzyme of the arachidonic acid cascade is rate limiting in that it controls the bioavailability of substrate to downstream effectors [6, 7]. However, our results strongly support the contention that mPGES-1 expression is the most limiting step in PGE2 synthesis, consistent with previous experiments with MK-886 [40], a five-lipoxygenase activating protein (FLAP) inhibitor with in vitro inhibitory potency towards mPGES-1 [44].
Because the stimulated synthesis of 6-keto-PGF1α requires successive metabolization by COX-2 and prostacyclin synthase (PGIS), the lower than expected increase could reflect a limited induction of PGIS in rat chondrocytes. Thus, induction of PGIS by IL-1β was less than double that in rat non-articular cells [45] despite its selective upregulation by COX-2 induction in human endothelial cells [46]. A decrease in PGIS expression, contrasting with an increase in mPGES-1 expression, was also reported in inflamed tissues of rat with adjuvant polyarthritis [32]. Alternatively, other metabolic pathways might have been favoured such as the conversion of cyclic endoperoxides into other prostaglandins [42], depending on the substrate concentration dependences of the terminal synthases [6, 46]. Arachidonic acid could also have been transformed into hydroxylated non-prostaglandin metabolites, which can be synthesized in IL-1β-stimulated chondrocytes [25], depending on the balance between the COX and lipoxygenase pathways [47]. In all instances, IL-1β stimulated all inducible steps of the arachidonic acid cascade to produce PGE2 maximally in rat chondrocytes.
The study of the expression of COX-2 or mPGES-1 and the release of prostaglandins in activated chondrocytes showed that 15d-PGJ2 was strongly inhibitory, whereas the high-affinity PPARγ agonist rosiglitazone was marginally potent in the same concentration range. Although 15d-PGJ2 and rosiglitazone were able to induce adiponectin expression, thereby demonstrating their potency to activate PPARγ, these results, irrespective of the binding affinity of agonists to PPARγ [48], supported the idea that this isotype was not primarily involved. It is interesting to note that the inducing effect of rosiglitazone on COX-2 mRNA was not confirmed at the protein level and that it was slightly inhibitory on mPGES-1, resulting in an unchanged PGE2 level. When we tried to decrease the inhibitory potency of 15d-PGJ2 by antagonizing its binding to PPARγ with GW-9662, we failed to observe any changes in gene mRNAs and PGE2 levels. As a corollary, the efficient overexpression of PPARγ did not enhance the potency of 15d-PGJ2 in our experimental system. Finally, despite the existence of a PPRE consensus site in the promoter of human COX-2 [49] and evidence that 15d-PGJ2 stimulates COX-2 gene expression in rat chondrocytes as in human synovial fibroblasts [50], we failed to observe any change in the basal production of PGE2, as reported previously in human osteoarthritic chondrocytes [51].
Taken together, our data strongly support the contention that 15d-PGJ2 was acting independently of PPARγ. Very few data are available in the rat species, but a PPARγ-dependent inhibition of inducible arachidonic acid cascade was reported in cardiac myocytes stimulated with IL-1β [52]. Because the inhibitory potency of 15d-PGJ2 on the COX-2, mPGES-1 and PGE2 levels was closely similar in both studies, we suggest that this discrepancy might be supported by cell type specificities. Indeed, the decrease in the levels of prostacyclin metabolites was different between cardiac myocytes and chondrocytes (no inhibition versus -66%) for a comparable extent of COX-2 inhibition (-40 to -50%), whereas the synthetic PPARγ agonist troglitazone was much more inhibitory towards PGE2 levels in the former cell type. In human chondrocytes, the inhibitory potency of 15d-PGJ2 was similar to our results on PGE2 levels [51], although supported by a stronger inhibition of COX-2 and a PPARγ-dependent inhibition of mPGES-1 [53]. In this cell type, the dose-dependent effect of 15d-PGJ2 was also thought to be mainly supported by the activation of PPARγ for the control of other inflammatory mediators [54] and apoptosis [55]. The biological responses to PPAR agonists are well known to differ between species [56], but our data support the notion that the potency of PPARγ agonists on joint cells might be influenced by differences in both cell type and species. Consistently, 15d-PGJ2 and troglitazone were shown to inhibit PGE2 production and mPGES-1 expression in IL-1β-stimulated human synovial fibroblasts [57], whereas troglitazone was totally ineffective on LPS-induced COX-2 expression in rat cells [20]. Finally, one could underline that the contribution of PPARγ might also depend on 15d-PGJ2 concentration, because the inhibition of PGE2 production was reported to be PPARγ-dependent in the nanomolar range while becoming PPARγ-independent in the micromolar range [58]. Despite a variable contribution of the PPARγ isotype depending on the biological system used, the present study confirms that 15d-PGJ2 downregulates inducible steps of the arachidonic acid cascade in joint cells, thereby probably contributing to its anti-arthritic properties [10].
The inhibitory potency of 15d-PGJ2 was PPARγ-independent but dose-related, which does not favour non-specific activity. This led us to investigate whether 15d-PGJ2 could interact with the NF-κB pathway, which is known to be one of its major targets in many cell types [59, 60]. A previous study of the mouse mPGES-1 promoter indicated that it lacked binding sites for NF-κB, the cAMP-response element, and E-box, which have been implicated in COX-2 induction, implying that the mechanisms for inducible expression of COX-2 and mPGES-1 were distinct in this species [61]. In human synovial fibroblasts, transcriptional regulation of the mPGES-1 gene by IL-1β was shown to be closely dependent on the transcription factor early growth response factor-1 (Egr-1) [57], although activator protein-1 and specificity protein-1 binding sites were also found [62]. In human chondrocytes, IL-1β was demonstrated to use overlapping, but distinct, signalling pathways to induce COX-2 and mPGES-1, with a major role for ERK1/2 and p38β MAPK in controlling the latter [41]. However, in a non-articular human cell type, a substantial role for NF-κB was demonstrated recently in the coordinate induction of COX-2 and mPGES-1 by IL-1β [63]. As indicated previously, some of these signalling pathways can be inhibited in a PPARγ-dependent manner, possibly secondary to the squelching of transcription cofactors such as CBP/p300 by protein-protein interaction with PPARγ [64]. Consequently, such a mechanism is unlikely to explain the PPARγ-independent inhibitory potency of 15d-PGJ2 in our system.
Although the promoter of rat mPGES-1 has not so far been explored, our data with mutated IκBα are consistent with a major role of NF-κB in the control of its transcriptional activity. We showed further that 15d-PGJ2 inhibited IL-1β-induced NF-κB nuclear binding (with the use of EMSA) and transactivation (with a TransAM® assay). This inhibitory effect was consistent with the ability of 15d-PGJ2 to decrease IκB kinase (IKK) activity, by limiting the phosphorylation of its catalytic subunit IKKβ, and to prevent IκBα degradation by the proteasome [65]. Because of the high chemical reactivity of its cyclopentenone ring with substances containing nucleophilic groups, such as the cysteinyl thiol group of proteins [66], possible mechanisms may include covalent binding of 15d-PGJ2 to IKK [67] or alkylation of a conserved cysteine residue located in the p65 subunit DNA-binding domain of NF-κB [68]. A possible chemical interaction with NF-κB components is further sustained by the ability of 15d-PGJ2 to suppress the induction of COX-2 in PPARγ-deficient macrophages [14]. However, we did not investigate whether NF-κB binds directly to mPGES-1 rat promoter, and the delayed induction of mPGES-1 by IL-1β supports indirect regulation. NF-κB was consistently shown to regulate the early expression of Egr-1 [69], which has been implicated in the regulation of murine and human mPGES-1 [57, 61]. Alternatively, we cannot exclude the possibility that inhibition of COX-2 by 15d-PGJ2 might participate partly in its inhibitory potency towards mPGES-1, because PGE2 production associated with COX-2 is involved in the induction of mPGES-1 by IL-1β in rheumatoid synovial fibroblasts [43].
Conclusion
The data reported here demonstrate that IL-1β activates COX-2 and mPGES-1 sequentially in rat chondrocytes and that the production of large amounts of PGE2 depends mainly on the expression of mPGES-1. In our cell type, 15d-PGJ2 displayed a strong inhibitory effect on prostaglandin levels and gene expression, whereas rosiglitazone was poorly active in the same concentration range. Despite its efficient activation of PPARγ, the effect of 15d-PGJ2 occurred through a PPARγ-independent mechanism. The activation of the NF-κB pathway was critical for mediating the inducing effect of IL-1β on PGE2 levels and mPGES-1 expression in rat chondrocytes, and was abolished by 15d-PGJ2. On the basis of the pathophysiological role of PGE2 in rheumatic diseases, our data support the general meaning that 15d-PGJ2 could behave as an endogenous regulator of inflammation if it was synthesized in sufficient amounts within joint tissues.
Abbreviations
- COX-2:
-
cyclo-oxygenase-2
- DMEM:
-
Dulbecco's modified Eagle's medium
- DTT:
-
dithiothreitol
- EMSA:
-
electrophoretic mobility-shift assay
- FCS:
-
fetal calf serum
- IKK:
-
IκB kinase
- IL:
-
interleukin
- mPGES-1:
-
microsomal prostaglandin E synthase-1
- NF-κB:
-
nuclear factor-κB
- PBS:
-
phosphate-buffered saline
- PCR:
-
polymerase chain reaction
- PG:
-
prostaglandin
- PGI2:
-
prostacyclin
- PGIS:
-
prostacyclin synthase
- PPAR:
-
peroxisome-proliferator-activated receptor
- PPRE:
-
peroxisome-proliferator-response element.
References
Willoughby DA, Colville-Nash PR, Seed MP: Inflammation, prostaglandins, and loss of function. J Lipid Mediat. 1993, 6: 287-293.
Norrdin RW, Jee WS, High WB: The role of prostaglandins in bone in vivo. Prostaglandins Leukot Essent Fatty Acids. 1990, 41: 139-149. 10.1016/0952-3278(90)90081-U.
Matsumoto H, Naraba H, Murakami M, Kudo I, Yamaki K, Ueno A, Oh-ishi S: Concordant induction of prostaglandin E2 synthase with cyclooxygenase-2 leads to preferred production of prostaglandin E2 over thromboxane and prostaglandin D2 in lipopolysaccharide-stimulated rat peritoneal macrophages. Biochem Biophys Res Commun. 1997, 230: 110-114. 10.1006/bbrc.1996.5894.
Shinomiya S, Naraba H, Ueno A, Utsunomiya I, Maruyama T, Ohuchida S, Ushikubi F, Yuki K, Narumiya S, Sugimoto Y, et al: Regulation of TNFalpha and interleukin-10 production by prostaglandins I2 and E2: studies with prostaglandin receptor-deficient mice and prostaglandin E-receptor subtype-selective synthetic agonists. Biochem Pharmacol. 2001, 61: 1153-1160. 10.1016/S0006-2952(01)00586-X.
Harris SG, Padilla J, Koumas L, Ray D, Phipps RP: Prostaglandins as modulators of immunity. Trends Immunol. 2002, 23: 144-150. 10.1016/S1471-4906(01)02154-8.
Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LB, Lipsky PE: Cyclooxygenase in biology and disease. FASEB J. 1998, 12: 1063-1073.
Tanioka T, Nakatani Y, Semmyo N, Murakami M, Kudo I: Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. J Biol Chem. 2000, 275: 32775-32782. 10.1074/jbc.M003504200.
Gilroy DW, Colville-Nash PR, McMaster S, Sawatzky DA, Willoughby DA, Lawrence T: Inducible cyclooxygenase-derived 15-deoxyΔ12–14PGJ2 brings about acute inflammatory resolution in rat pleurisy by inducing neutrophil and macrophage apoptosis. FASEB J. 2003, 17: 2269-2271.
Cuzzocrea S, Wayman NS, Mazzon E, Dugo L, Di Paola R, Serraino I, Britti D, Chatterjee PK, Caputi AP, Thiemermann C: The cyclopentenone prostaglandin 15-deoxy-Δ12,14-prostaglandin J2 attenuates the development of acute and chronic inflammation. Mol Pharmacol. 2002, 61: 997-1007. 10.1124/mol.61.5.997.
Kawahito Y, Kondo M, Tsubouchi Y, Hashiramoto A, Bishop-Bailey D, Inoue K, Kohno M, Yamada R, Hla T, Sano H: 15-deoxy-Δ12,14-PGJ2 induces synoviocyte apoptosis and suppresses adjuvant-induced arthritis in rats. J Clin Invest. 2000, 106: 189-197.
Willoughby DA, Moore AR, Colville-Nash PR, Gilroy D: Resolution of inflammation. Int J Immunopharmacol. 2000, 22: 1131-1135. 10.1016/S0192-0561(00)00064-3.
Murakami M, Nakatani Y, Tanioka T, Kudo I: Prostaglandin E synthase. Prostaglandins Other Lipid Mediat. 2002, 68-69: 383-399. 10.1016/S0090-6980(02)00043-6.
Tanikawa N, Ohmiya Y, Ohkubo H, Hashimoto K, Kangawa K, Kojima M, Ito S, Watanabe K: Identification and characterization of a novel type of membrane-associated prostaglandin E synthase. Biochem Biophys Res Commun. 2002, 291: 884-889. 10.1006/bbrc.2002.6531.
Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM: PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. 2001, 7: 48-52. 10.1038/83336.
Chawla A, Schwarz EJ, Dimaculangan DD, Lazar MA: Peroxisome proliferator-activated receptor (PPAR) gamma: adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology. 1994, 135: 798-800. 10.1210/en.135.2.798.
Tontonoz P, Hu E, Spiegelman BM: Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994, 79: 1147-1156. 10.1016/0092-8674(94)90006-X.
Jiang C, Ting AT, Seed B: PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998, 391: 82-86. 10.1038/35154.
Colville-Nash PR, Qureshi SS, Willis D, Willoughby DA: Inhibition of inducible nitric oxide synthase by peroxisome proliferator-activated receptor agonists: correlation with induction of heme oxygenase 1. J Immunol. 1998, 161: 978-984.
Boyault S, Simonin MA, Bianchi A, Compe E, Liagre B, Mainard D, Becuwe P, Dauca M, Netter P, Terlain B, Bordji K: 15-Deoxy-Δ12,14-PGJ2, but not troglitazone, modulates IL-1β effects in human chondrocytes by inhibiting NF-κB and AP-1 activation pathways. FEBS Lett. 2001, 501: 24-30. 10.1016/S0014-5793(01)02614-X.
Simonin MA, Bordji K, Boyault S, Bianchi A, Gouze E, Becuwe P, Dauca M, Netter P, Terlain B: PPAR-gamma ligands modulate effects of LPS in stimulated rat synovial fibroblasts. Am J Physiol Cell Physiol. 2002, 282: C125-C133.
Cuzzocrea S, Mazzon E, Dugo L, Patel NS, Serraino I, Di Paola R, Genovese T, Britti D, De Maio M, Caputi AP, Thiemermann C: Reduction in the evolution of murine type II collagen-induced arthritis by treatment with rosiglitazone, a ligand of the peroxisome proliferator-activated receptor gamma. Arthritis Rheum. 2003, 48: 3544-3556. 10.1002/art.11351.
Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK: The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature. 1998, 391: 79-82. 10.1038/34178.
Spiegelman BM: PPARgamma in monocytes: less pain, any gain?. Cell. 1998, 93: 153-155. 10.1016/S0092-8674(00)81567-6.
Blanco FJ, Guitian R, Moreno J, de Toro FJ, Galdo F: Effect of antiinflammatory drugs on COX-1 and COX-2 activity in human articular chondrocytes. J Rheumatol. 1999, 26: 1366-1373.
Kerr JS, Stevens TM, Davis GL, McLaughlin JA, Harris RR: Effects of recombinant interleukin-1 beta on phospholipase A2 activity, phospholipase A2 mRNA levels, and eicosanoid formation in rabbit chondrocytes. Biochem Biophys Res Commun. 1989, 165: 1079-1084. 10.1016/0006-291X(89)92712-5.
Quraishi O, Mancini JA, Riendeau D: Inhibition of inducible prostaglandin E(2) synthase by 15-deoxy-Δ12,14-prostaglandin J2 and polyunsaturated fatty acids. Biochem Pharmacol. 2002, 63: 1183-1189. 10.1016/S0006-2952(02)00844-4.
Kuettner KE, Pauli BU, Gall G, Memoli VA, Schenk RK: Synthesis of cartilage matrix by mammalian chondrocytes in vitro. I. Isolation, culture characteristics, and morphology. J Cell Biol. 1982, 93: 743-750. 10.1083/jcb.93.3.743.
Hansen MB, Nielsen SE, Berg K: Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods. 1989, 119: 203-210. 10.1016/0022-1759(89)90397-9.
Yang B, Brown KK, Chen L, Carrick KM, Clifton LG, McNulty JA, Winegar DA, Strum JC, Stimpson SA, Pahel GL: Serum adiponectin as a biomarker for in vivo PPARgamma activation and PPARgamma agonist-induced efficacy on insulin sensitization/lipid lowering in rats. BMC Pharmacol. 2004, 4: 23-10.1186/1471-2210-4-23.
Dignam JD, Lebovitz RM, Roeder RG: Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983, 11: 1475-1489.
Kudo I, Murakami M: Diverse functional coupling of prostanoid biosynthetic enzymes in various cell types. Adv Exp Med Biol. 1999, 469: 29-35.
Claveau D, Sirinyan M, Guay J, Gordon R, Chan CC, Bureau Y, Riendeau D, Mancini JA: Microsomal prostaglandin E synthase-1 is a major terminal synthase that is selectively up-regulated during cyclooxygenase-2-dependent prostaglandin E2 production in the rat adjuvant-induced arthritis model. J Immunol. 2003, 170: 4738-4744.
Guay J, Bateman K, Gordon R, Mancini J, Riendeau D: Carrageenan-induced paw edema in rat elicits a predominant prostaglandin E2 (PGE2) response in the central nervous system associated with the induction of microsomal PGE2 synthase-1. J Biol Chem. 2004, 279: 24866-24872. 10.1074/jbc.M403106200.
Portanova JP, Zhang Y, Anderson GD, Hauser SD, Masferrer JL, Seibert K, Gregory SA, Isakson PC: Selective neutralization of prostaglandin E2 blocks inflammation, hyperalgesia, and interleukin 6 production in vivo. J Exp Med. 1996, 184: 883-891. 10.1084/jem.184.3.883.
Kamei D, Yamakawa K, Takegoshi Y, Mikami-Nakanishi M, Nakatani Y, Oh-Ishi S, Yasui H, Azuma Y, Hirasawa N, Ohuchi K, et al: Reduced pain hypersensitivity and inflammation in mice lacking microsomal prostaglandin E synthase-1. J Biol Chem. 2004, 279: 33684-33695. 10.1074/jbc.M400199200.
Murakami Y, Akahoshi T, Hayashi I, Endo H, Hashimoto A, Kono S, Kondo H, Kawai S, Inoue M, Kitasato H: Inhibition of monosodium urate monohydrate crystal-induced acute inflammation by retrovirally transfected prostaglandin D synthase. Arthritis Rheum. 2003, 48: 2931-2941. 10.1002/art.11271.
Stichtenoth DO, Thoren S, Bian H, Peters-Golden M, Jakobsson PJ, Crofford LJ: Microsomal prostaglandin E synthase is regulated by proinflammatory cytokines and glucocorticoids in primary rheumatoid synovial cells. J Immunol. 2001, 167: 469-474.
Dayer JM: The pivotal role of interleukin-1 in the clinical manifestations of rheumatoid arthritis. Rheumatology (Oxford). 2003, 42 (Suppl 2): ii3-ii10.
Nedelec E, Abid A, Cipolletta C, Presle N, Terlain B, Netter P, Jouzeau J: Stimulation of cyclooxygenase-2-activity by nitric oxide-derived species in rat chondrocyte: lack of contribution to loss of cartilage anabolism. Biochem Pharmacol. 2001, 61: 965-978. 10.1016/S0006-2952(01)00559-7.
Kojima F, Naraba H, Miyamoto S, Beppu M, Aoki H, Kawai S: Membrane-associated prostaglandin E synthase-1 is upregulated by proinflammatory cytokines in chondrocytes from patients with osteoarthritis. Arthritis Res Ther. 2004, 6: R355-R365. 10.1186/ar1195.
Masuko-Hongo K, Berenbaum F, Humbert L, Salvat C, Goldring MB, Thirion S: Up-regulation of microsomal prostaglandin E synthase 1 in osteoarthritic human cartilage: critical roles of the ERK-1/2 and p38 signaling pathways. Arthritis Rheum. 2004, 50: 2829-2838. 10.1002/art.20437.
Knott I, Dieu M, Burton M, Houbion A, Remacle J, Raes M: Induction of cyclooxygenase by interleukin 1: comparative study between human synovial cells and chondrocytes. J Rheumatol. 1994, 21: 462-466.
Kojima F, Naraba H, Sasaki Y, Beppu M, Aoki H, Kawai S: Prostaglandin E2 is an enhancer of interleukin-1beta-induced expression of membrane-associated prostaglandin E synthase in rheumatoid synovial fibroblasts. Arthritis Rheum. 2003, 48: 2819-2828. 10.1002/art.11261.
Mancini JA, Blood K, Guay J, Gordon R, Claveau D, Chan CC, Riendeau D: Cloning, expression, and up-regulation of inducible rat prostaglandin E synthase during lipopolysaccharide-induced pyresis and adjuvant-induced arthritis. J Biol Chem. 2001, 276: 4469-4475. 10.1074/jbc.M006865200.
Itoh A, Nishihira J, Makita H, Miyamoto K, Yamaguchi E, Nishimura M: Effects of IL-1beta, TNF-alpha, and macrophage migration inhibitory factor on prostacyclin synthesis in rat pulmonary artery smooth muscle cells. Respirology. 2003, 8: 467-472. 10.1046/j.1440-1843.2003.00491.x.
Caughey GE, Cleland LG, Penglis PS, Gamble JR, James MJ: Roles of cyclooxygenase (COX)-1 and COX-2 in prostanoid production by human endothelial cells: selective up-regulation of prostacyclin synthesis by COX-2. J Immunol. 2001, 167: 2831-2838.
Martel-Pelletier J, Mineau F, Fahmi H, Laufer S, Reboul P, Boileau C, Lavigne M, Pelletier JP: Regulation of the expression of 5-lipoxygenase-activating protein/5-lipoxygenase and the synthesis of leukotriene B4 in osteoarthritic chondrocytes: role of transforming growth factor beta and eicosanoids. Arthritis Rheum. 2004, 50: 3925-3933. 10.1002/art.20632.
Willson TM, Cobb JE, Cowan DJ, Wiethe RW, Correa ID, Prakash SR, Beck KD, Moore LB, Kliewer SA, Lehmann JM: The structure-activity relationship between peroxisome proliferator-activated receptor gamma agonism and the antihyperglycemic activity of thiazolidinediones. J Med Chem. 1996, 39: 665-668. 10.1021/jm950395a.
Meade EA, McIntyre TM, Zimmerman GA, Prescott SM: Peroxisome proliferators enhance cyclooxygenase-2 expression in epithelial cells. J Biol Chem. 1999, 274: 8328-8334. 10.1074/jbc.274.12.8328.
Kalajdzic T, Faour WH, He QW, Fahmi H, Martel-Pelletier J, Pelletier JP, Di Battista JA: Nimesulide, a preferential cyclooxygenase 2 inhibitor, suppresses peroxisome proliferator-activated receptor induction of cyclooxygenase 2 gene expression in human synovial fibroblasts: evidence for receptor antagonism. Arthritis Rheum. 2002, 46: 494-506. 10.1002/art.10055.
Fahmi H, Pelletier JP, Mineau F, Martel-Pelletier J: 15d-PGJ2 is acting as a 'dual agent' on the regulation of COX-2 expression in human osteoarthritic chondrocytes. Osteoarthritis Cartilage. 2002, 10: 845-848. 10.1053/joca.2002.0835.
Mendez M, LaPointe MC: PPARgamma inhibition of cyclooxygenase-2, PGE2 synthase, and inducible nitric oxide synthase in cardiac myocytes. Hypertension. 2003, 42: 844-850. 10.1161/01.HYP.0000085332.69777.D1.
Li X, Afif H, Cheng S, Martel-Pelletier J, Pelletier JP, Ranger P, Fahmi H: Expression and regulation of microsomal prostaglandin E synthase-1 in human osteoarthritic cartilage and chondrocytes. J Rheumatol. 2005, 32: 887-895.
Fahmi H, Di Battista JA, Pelletier JP, Mineau F, Ranger P, Martel-Pelletier J: Peroxisome proliferator-activated receptor gamma activators inhibit interleukin-1beta-induced nitric oxide and matrix metalloproteinase 13 production in human chondrocytes. Arthritis Rheum. 2001, 44: 595-607. 10.1002/1529-0131(200103)44:3<595::AID-ANR108>3.0.CO;2-8.
Shan ZZ, Masuko-Hongo K, Dai SM, Nakamura H, Kato T, Nishioka K: A potential role of 15-deoxy-Δ12,14-prostaglandin J2 for induction of human articular chondrocyte apoptosis in arthritis. J Biol Chem. 2004, 279: 37939-37950. 10.1074/jbc.M402424200.
Blanquart C, Barbier O, Fruchart JC, Staels B, Glineur C: Peroxisome proliferator-activated receptors: regulation of transcriptional activities and roles in inflammation. J Steroid Biochem Mol Biol. 2003, 85: 267-273. 10.1016/S0960-0760(03)00214-0.
Cheng S, Afif H, Martel-Pelletier J, Pelletier JP, Li X, Farrajota K, Lavigne M, Fahmi H: Activation of peroxisome proliferator-activated receptor gamma inhibits interleukin-1β-induced membrane-associated prostaglandin E2 synthase-1 expression in human synovial fibroblasts by interfering with Egr-1. J Biol Chem. 2004, 279: 22057-22065. 10.1074/jbc.M402828200.
Berry EB, Keelan JA, Helliwell RJ, Gilmour RS, Mitchell MD: Nanomolar and micromolar effects of 15-deoxy-delta 12,14-prostaglandin J2 on amnion-derived WISH epithelial cells: differential roles of peroxisome proliferator-activated receptors gamma and delta and nuclear factor kappa B. Mol Pharmacol. 2005, 68: 169-178.
Nosjean O, Boutin JA: Natural ligands of PPARγ: are prostaglandin J2 derivatives really playing the part?. Cell Signal. 2002, 14: 573-583. 10.1016/S0898-6568(01)00281-9.
Scher JU, Pillinger MH: 15d-PGJ2: the anti-inflammatory prostaglandin?. Clin Immunol. 2005, 114: 100-109. 10.1016/j.clim.2004.09.008.
Naraba H, Yokoyama C, Tago N, Murakami M, Kudo I, Fueki M, Oh-Ishi S, Tanabe T: Transcriptional regulation of the membrane-associated prostaglandin E2 synthase gene. Essential role of the transcription factor Egr-1. J Biol Chem. 2002, 277: 28601-28608. 10.1074/jbc.M203618200.
Ekstrom L, Lyrenas L, Jakobsson PJ, Morgenstern R, Kelner MJ: Basal expression of the human MAPEG members microsomal glutathione transferase 1 and prostaglandin E synthase genes is mediated by Sp1 and Sp3. Biochim Biophys Acta. 2003, 1627: 79-84.
Catley MC, Chivers JE, Cambridge LM, Holden N, Slater DM, Staples KJ, Bergmann MW, Loser P, Barnes PJ, Newton R: IL-1β-dependent activation of NF-κB mediates PGE2 release via the expression of cyclooxygenase-2 and microsomal prostaglandin E synthase. FEBS Lett. 2003, 547: 75-79. 10.1016/S0014-5793(03)00672-0.
Subbaramaiah K, Lin DT, Hart JC, Dannenberg AJ: Peroxisome proliferator-activated receptor gamma ligands suppress the transcriptional activation of cyclooxygenase-2. Evidence for involvement of activator protein-1 and CREB-binding protein/p300. J Biol Chem. 2001, 276: 12440-12448. 10.1074/jbc.M007237200.
Boyault S, Bianchi A, Moulin D, Morin S, Francois M, Netter P, Terlain B, Bordji K: 15-Deoxy-Δ12,14-prostaglandin J2 inhibits IL-1β-induced IKK enzymatic activity and IκBα degradation in rat chondrocytes through a PPARγ-independent pathway. FEBS Lett. 2004, 572: 33-40. 10.1016/j.febslet.2004.06.090.
Fukushima M: Biological activities and mechanisms of action of PGJ2 and related compounds: an update. Prostaglandins Leukot Essent Fatty Acids. 1992, 47: 1-12. 10.1016/0952-3278(92)90178-L.
Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG: Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IκB kinase. Nature. 2000, 403: 103-108. 10.1038/47520.
Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, Sengchanthalangsy LL, Ghosh G, Glass CK: 15-deoxy-Δ12,14-prostaglandin J2 inhibits multiple steps in the NF-κB signaling pathway. Proc Natl Acad Sci USA. 2000, 97: 4844-4849. 10.1073/pnas.97.9.4844.
Thyss R, Virolle V, Imbert V, Peyron JF, Aberdam D, Virolle T: NF-kappaB/Egr-1/Gadd45 are sequentially activated upon UVB irradiation to mediate epidermal cell death. EMBO J. 2005, 24: 128-137. 10.1038/sj.emboj.7600501.
Acknowledgements
This work was supported by grants from the Association de la Recherche contre la Polyarthite and the Communauté Urbaine du Grand Nancy.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
AB and DM performed the molecular studies and drafted the manuscript. SS and MK performed the immunoassays and the statistical analysis. MMG and PN supervised the study design and the manuscript. BT and JYJ conceived the study and participated in its design and final presentation. All authors read and approved the final manuscript.
Arnaud Bianchi, David Moulin contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Bianchi, A., Moulin, D., Sebillaud, S. et al. Contrasting effects of peroxisome-proliferator-activated receptor (PPAR)γ agonists on membrane-associated prostaglandin E2 synthase-1 in IL-1β-stimulated rat chondrocytes: evidence for PPARγ-independent inhibition by 15-deoxy-Δ12,14prostaglandin J2. Arthritis Res Ther 7, R1325 (2005). https://doi.org/10.1186/ar1830
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar1830