
Abstract
Inflammation and inflammatory responses are modulated by a bidirectional communication between the neuroendocrine and immune system. Many lines of research have established the numerous routes by which the immune system and the central nervous system (CNS) communicate. The CNS signals the immune system through hormonal pathways, including the hypothalamic–pituitary–adrenal axis and the hormones of the neuroendocrine stress response, and through neuronal pathways, including the autonomic nervous system. The hypothalamic–pituitary–gonadal axis and sex hormones also have an important immunoregulatory role. The immune system signals the CNS through immune mediators and cytokines that can cross the blood–brain barrier, or signal indirectly through the vagus nerve or second messengers. Neuroendocrine regulation of immune function is essential for survival during stress or infection and to modulate immune responses in inflammatory disease. This review discusses neuroimmune interactions and evidence for the role of such neural immune regulation of inflammation, rather than a discussion of the individual inflammatory mediators, in rheumatoid arthritis.
Similar content being viewed by others


Introduction
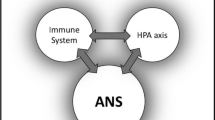
The inflammatory response is modulated in part by a bidirectional communication between the brain and the immune systems. This involves hormonal and neuronal mechanisms by which the brain regulates the function of the immune system and, in the reverse, cytokines, which allow the immune system to regulate the brain. In a healthy individual this bidirectional regulatory system forms a negative feedback loop, which keeps the immune system and central nervous system (CNS) in balance. Perturbations of these regulatory systems could potentially lead to either overactivation of immune responses and inflammatory disease, or oversuppression of the immune system and increased susceptibility to infectious disease. Many lines of research have recently established the numerous routes by which the immune system and the CNS communicate. This review will focus on these regulatory systems and their involvement in the pathogenesis of inflammatory diseases such as rheumatoid arthritis (RA). For other reviews on the involvement of these regulatory pathways in RA and other inflammatory diseases, see reviews by Eijsbouts and Murphy [1], Crofford [2], and Imrich [3].
There are two major pathways by which the CNS regulates the immune system: the first is the hormonal response, mainly through the hypothalamic–pituitary–adrenal (HPA) axis, as well as the hypothalamic–pituitary–gonadal (HPG), the hypothalamic–pituitary–thyroid (HPT) and the hypothalamic–growth-hormone axes; the second is the autonomic nervous system, through the release of norepinephrine (noradrenaline) and acetylcholine from sympathetic and parasympathetic nerves. In turn, the immune system can also regulate the CNS through cytokines.
Conversely, cytokines released in the periphery change brain function, whereas cytokines produced within the CNS act more like growth factors. Thus, cytokines produced at inflammatory sites signal the brain to produce sickness-related behavior including depression and other symptoms such as fever [4–7]. In addition, cytokines produced locally exert paracrine/autocrine effects on hormone secretion and cell proliferation [8, 9].
The interactions between the neuroendocrine and immune systems provide a finely tuned regulatory system required for health. Disturbances at any level can lead to changes in susceptibility to or severity of infectious, inflammatory or autoimmune diseases.
Regulation of the immune system by the CNS
Hormonal pathways
HPA axis
On stimulation, corticotropin-releasing hormone (CRH) is secreted from the paraventricular nucleus of the hypothalamus into the hypophyseal portal blood supply. CRH then stimulates the expression and release of adrenocorticotropin (ACTH) from the anterior pituitary gland. Arginine vasopressin (AVP) synergistically enhances CRH-stimulated ACTH release [10, 11] ACTH in turn induces the expression and release of glucocorticoids from the adrenal glands.
Glucocorticoids regulate a wide variety of immune-related genes and immune cell expression and function. For example, glucocorticoids modulate the expression of cytokines, adhesion molecules, chemoattractants and other inflammatory mediators and molecules and affect immune cell trafficking, migration, maturation, and differentiation [12, 13]. Glucocorticoids cause a Th1 (cellular immunity) to Th2 (humoral immunity) shift in the immune response, from a proinflammatory cytokine pattern with increased interleukin (IL)-1 and tumor necrosis factor (TNF)-α to an anti-inflammatory cytokine pattern with increased IL-10 and IL-4 [14, 15]. Pharmacological doses and preparations of glucocorticoids cause a general suppression of the immune system, whereas physiological doses and preparations of glucocorticoids are not completely immunosuppressive but can enhance and specifically regulate the immune response under certain circumstances. For example, physiological concentrations of natural glucocorticoids (i.e. corticosterone) stimulate delayed-type hypersensitivity reactions acutely, whereas pharmacological preparations (i.e. dexamethasone) are immunosuppressive [16].
Glucocorticoids exert these immunomodulatory effects through a cytosolic receptor, the glucocorticoid receptor (GR). This is a ligand-dependent transcription factor that, after binding of the ligand, dissociates from a protein complex, dimerizes, and translocates to the nucleus, where it binds to specific DNA sequences (glucocorticoid response elements) to regulate gene transcription [17]. GR can also interfere with other signaling pathways, such as nuclear factor (NF)-κB and activator protein-1 (AP-1), to repress gene transcription; it is through these mechanisms that most of the anti-inflammatory actions are mediated [18–21]. A splice variant of GR, GRβ, that is unable to bind ligand but is able to bind to DNA and cannot activate gene transcription [22] (although this is still under some dispute), has been suggested to be able to act as a dominant repressor of GR [23, 24]. Increased GRβ expression has been shown in several inflammatory diseases including asthma [25–28], inflammatory bowel disease/ulcerative colitis [29, 30], and RA [31].
HPG axis
In addition to the HPA axis, other central hormonal systems, such as the HPG axis and in particular estrogen, also modulate the immune system [32]. In general, physiological concentrations of estrogen enhance immune responses [33, 34] whereas physiological concentrations of androgens, such as testosterone and dehydroepiandrosterone (DHEA), are immunosuppressive [34]. Females of all species exhibit a greater risk of developing many autoimmune/inflammatory diseases, such as systemic lupus erythematosus, RA and multiple sclerosis, ranging from a 2-fold to a 10-fold higher risk compared with males [35, 36]. Animal models have provided evidence for the importance of in vivo modulation of the immune system by the estrogen receptors [37, 38]. Knockout mouse models indicate that both estrogen receptors α and β are important for thymus development and atrophy in a gender-specific manner [39].
In contrast, immune stress, such as occurs during inflammation, has an inhibitory effect on the HPG axis and thus gonadal function is reduced in conditions associated with severe inflammation such as sepsis and trauma. This effect is mediated either through a direct cytokine effect on hypothalamic neurons secreting luteinizing hormone releasing hormone [40, 41] or through other factors such as CRH [42, 43] and endogenous opioids [44]. Cytokines also affect gonadal sex steroid production by acting directly on the gonads [45].
Hypothalamic–growth-hormone axis
Growth hormone (GH) is a modulator of the immune system [46, 47]. The effects of GH are mediated primarily through insulin-like growth factor-1 (IGF-1). GH and IGF-1 have been shown to modulate the immune system by inducing the survival and proliferation of lymphoid cells [48], leading some to suggest that GH functions as a cytokine [49]. Thus, immune cells including T and B lymphocytes [50] and mononuclear cells [51] express IGF-1 receptor. After binding to these receptors, GH activates the phosphoinositide 3-kinase/Akt and NF-κB signal transduction pathways, leading to the expression of genes involved in the cell cycle. The NF-κB pathway is also important in immunity, and therefore some of the GH effects on the immune system might be mediated through this signal transduction pathway [49]. However, the role of GH in regulation of the immune system is somewhat controversial. Studies in GH knockout animals have shown that this hormone is only minimally required for immune function [52], leading to an alternative hypothesis in which the primary role of GH is proposed to be protection from the immunosuppressive effects of glucocorticoids during stress [53].
GH might also modulate immune function indirectly by interacting with other hormonal systems. Thus, short-term increases in glucocorticoids increase GH production [54], whereas long-term high doses result in a decrease in the hypothalamic–GH axis and even growth impairment [55]. Conversely, prolonged HPA axis activation and resultant excessive glucocorticoid production, as occurs during chronic stress, also inhibits the hypothalamic–GH axis [56–58]. Consistent with this is the observation that children with chronic inflammatory disease exhibit growth retardation. During the early phase of inflammatory reactions, the concentration of GH is increased. In spite of an initial rise in GH secretion, GH action is reduced because of GH and IGF-1 resistance induced by inflammation. IL-1α initially stimulates GH [59], but subsequently inhibits its secretion [60].
HPT axis
As with the interaction between the HPA axis and the immune system, there is a bidirectional interaction between the HPT axis and immune system [61]. The HPT axis has an immunomodulatory effect on most aspects of the immune system. Thyrotropin-releasing hormone (TRH), thyroid-stimulating hormone (TSH), and the thyroid hormones triiodothyronine (T3) and thyroxine (T4) all have stimulatory effects on immune cells [62–64]. As for GH, the role of thyroid hormones in the regulation of immunity is somewhat controversial, and for the same reasons the alternative hypothesis of protection from the immunosuppressive effects of glucocorticoids has also been suggested for thyroid hormones [53]. Inflammation inhibits TSH secretion because of the inhibitory effect of cytokines on TRH [62]. IL-1 has been shown to suppress TSH secretion [59], whereas IL-2 has been shown to stimulate the pituitary–thyroid axis [65]. IL-6 and its receptor have been shown to be involved in developing euthyroid sick syndrome in patients with acute myocardial infarction [66].
In addition to direct effects of thyroid hormones on immune response, there is also interaction between the HPA and HPT axes. Hyperthyroid and hypothyroid states in rats have been shown to alter responses of the HPA axis, with hypothyroidism resulting in a reduced HPA axis response and hyperthyroidism resulting in an increased HPA axis response [67]. In agreement with this, administration of thyroxine, inducing a hyperthyroid state, has been shown to activate the HPA axis and be protective against an inflammatory challenge in rats [68], and hypothyroidism has been shown to cause a reduction in CRH gene expression [69]. Chronic HPA axis activation also represses TSH production and inhibits the conversion of inactive T4 to the active T3 [70].
Neural pathways
Sympathetic nervous system
The sympathetic nervous system regulates the immune system at regional, local, and systemic levels. Immune organs including thymus, spleen, and lymph nodes are innervated by sympathetic nerves [71–73]. Immune cells also express neurotransmitter receptors, such as adrenergic receptors on lymphocytes, that allow them to respond to neurotransmitters released from these nerves.
Catecholamines inhibit production of proinflammatory cytokines, such as IL-12, TNF-α, and interferon-γ, and stimulate the production of anti-inflammatory cytokines, such as IL-10 and transforming growth factor-β [15]. Through this mechanism, systemic catecholamines can cause a selective suppression of Th1 responses and enhance Th2 responses [15, 74]. However, in certain local responses and under certain conditions, catecholamines can enhance regional immune responses by inducing the production of IL-1, TNF-α, and IL-8 [75]. Interruption of sympathetic innervation of immune organs has been shown to modulate the outcome of, and susceptibility to, inflammatory and infectious disease. Denervation of lymph node noradrenergic fibers is associated with exacerbation of inflammation [76, 77], whereas systemic sympathectomy or denervation of joints is associated with decreased severity of inflammation [77]. However, mice lacking β2-adrenergic receptor from early development (β2AR-/- mice) maintain their immune homeostasis [78]. Therefore, dual activation of the sympathetic nervous system and HPA axis is required for full modulation of host defenses to infection [16, 79].
Opioids
Opioids suppress many aspects of immune responses, including antimicrobial resistance, antibody production, and delayed-type hypersensitivity. This occurs in part through the desensitization of chemokine receptors on neutrophils, monocytes, and lymphocytes [80, 81]. Morphine decreases mitogen responsiveness and natural killer cell activity [82–86]. In addition to these direct effects, morphine could also affect immune responses indirectly through adrenergic effects, because it increases concentrations of catecholamines in the plasma [87].
Parasympathetic nervous system
Activation of the parasympathetic nervous system results in the activation of cholinergic nerve fibers of the efferent vagus nerve and the release of acetylcholine at the synapses. Together with the inflammation-activated sensory nerve fibers of the vagus nerve (discussed below) this forms the so-called 'inflammatory reflex'. This is a rapid mechanism by which inflammatory signals reach the brain; the brain responds with a rapid anti-inflammatory action through cholinergic nerve fibers [88].
Acetylcholine attenuates the release of proinflammatory cytokines (TNF, IL-1β, IL-6, and IL-18) but not the anti-inflammatory cytokine IL-10, in lipopolysaccharide-stimulated human macrophage cultures through the post-transcriptional suppression of protein synthesis. This effect seems, at least in part, to be independent of the HPA axis, because direct electrical stimulation of the peripheral vagus nerve does not stimulate the HPA axis but decreases hepatic lipopolysaccharide-stimulated TNF synthesis and the development of shock during lethal endotoxemia [89].
Peripheral nervous system
The peripheral nervous system regulates immunity locally, at sites of inflammation, through neuropeptides such as substance P, peripherally released CRH, and vasoactive intestinal polypeptide. These molecules are released from nerve endings or synapses, or they may be synthesized and released by immune cells and have immunomodulatory and generally proinflammatory effects [90–92].
Neuropeptides
The HPA axis is also subject to regulation by both neurotransmitters and neuropeptides from within the CNS. CRH is positively regulated by serotonergic [93–95], cholinergic [96, 97], and catecholaminergic [98] systems. Other neuropeptides, such as γ-aminobutyric acid/benzodiazepines (GABA/BZD) have been shown to inhibit the serotonin-induced secretion of CRH [99].
Regulation of the CNS by the immune system
Cytokines
Cytokines are important factors connecting and modulating the immune and neuroendrocrine systems. Cytokines and their receptors are expressed in the neuroendocrine system and exert their effects both centrally and peripherally [100–102].
Systemic cytokines can affect the brain through several mechanisms, including active transport across the blood–brain barrier [103], through leaky areas in the blood–brain barrier in the circumventricular organs [104] or through the activation of neural pathways such as the vagal nerve [105]. The blood–brain barrier is absent or imperfect in several small areas of the brain, the so-called circumventricular organs, which are located at various sites within the walls of the cerebral ventricles. These include the median eminence, the organum vasculosum of the laminae terminalis (OVLT), the subfornical organ, the choroid plexus, the neural lobe of the pituitary, and the area postrema. In addition, in the presence of inflammation, the permeability of the blood–brain barrier might be generally altered [106–108]. Moreover, circulating IL-1 can interact with IL-1 receptors on endothelial cells of the vasculature and thereby stimulate signaling molecules such as nitric oxide or prostaglandins, which can locally influence neurons [109].
Cytokines signal the brain not only to activate the HPA axis but also to facilitate pain and induce a series of mood and behavioral responses generally termed sickness behavior [110, 111]. Cytokines, such as IL-1, IL-6, and TNF-α, are also produced in the brain [112–114]. Thus, these brain-derived cytokines can stimulate the HPA axis. For example, IL-1 stimulates the expression of the gene encoding CRH and thereby the release of the hormone from the hypothalamus [115], the release of AVP from the hypothalamus [116], and the release of ACTH from the anterior pituitary [117]. IL-2 stimulates AVP secretion from the hypothalamus [118]. IL-6 [119] and TNF-α [120] also stimulate ACTH secretion. In chronic inflammation there seems to be a shift from CRH-driven to AVP-driven HPA axis response [121].
However, in contrast to these effects of peripheral cytokines on neuroendocrine responses in the CNS, cytokines produced within the brain by resident glia or invading immune cells act more like growth factors protecting from or enhancing neuronal cell death. Cytokines might therefore have a pathological consequence, because cytokine-mediated neuronal cell death is thought to be important in several neurodegenerative diseases such as neuroAIDS, Alzheimer's disease, multiple sclerosis, stroke, and nerve trauma [100–102]. In contrast, activated immune cells and cytokines might also protect neuronal survival after trauma and contribute to neural repair [122].
Vagus nerve
The vagus nerve is involved in signaling of the CNS to the immune system. The vagus innervates most visceral structures such as the lung and the gastrointestinal tract, where there may be frequent contact with pathogens. Immune stimuli activate vagal sensory neurons, possibly after binding to receptors in cells in paraganglial structures [123–126]. Administration of endotoxins and IL-1 has been shown to induce Fos expression in the vagal sensory ganglia, and vagotomy abolishes this early activation gene response [124–126]. Vagal afferents terminate in the dorsal vagal complex of the caudal medulla, which consists of the area postrema, the nucleus of the solitary tract, and the dorsal motor nucleus of the vagus. These nuclei integrate sensory signals and control visceral reflexes, and also relay visceral sensory information to the central autonomic network [127]. Subdiaphragmatic vagotomy inhibits activation of the paraventricular nucleus and subsequent secretion of ACTH in response to lipopolysccharides and IL-1 [128, 129].
Correlation between blunted HPA axis and disease
A blunted HPA axis has been associated with increased susceptibility to autoimmune/inflammatory disease in a variety of animal models and human studies. In general, at the baseline the HPA axis parameters do not differ in individuals susceptible and resistant to inflammatory disease. However, differences become apparent with stimulation of the axis.
Animal models
A blunted HPA axis has been associated with susceptibility to autoimmune/inflammatory diseases in several animal models. These include the Obese strain (OS) chickens, a model for thyroiditis [130]; MRL mice, which develop lupus [131]; and Lewis (LEW/N) rats. A region on rat chromosome 10 that links to the innate carrageenan inflammation [132] is syntenic with a region on human chromosome 17 that is known to link to susceptibility to a variety of autoimmune diseases [133] and is also syntenic with one of the 20 different regions on 15 different chromosomes shown to link to inflammatory arthritis in other linkage studies [134–136]. Several candidate genes within the rat chromosome 10 linkage region are known to have a role in hypothalamic CRH regulation as well as inflammation, including the CRH R1 receptor, angiotensin-converting enzyme, and STAT3 and STAT5a/5b [132]. However, these candidate genes either show no mutation in the coding region and no differences in regulation between susceptible and resistant strains, or show a mutation in the coding region that does not seem to have a role in expression of the inflammatory trait [137]. As in most complex illnesses and traits, the genotypic contribution to variance in the trait is small: about 35%, which is consistent with such multigenic and polygenic conditions.
Inbred rat strains provide a genetically uniform system that can be systemically manipulated to test the role of neuro-endocrine regulation of various aspects of immunity. Lewis (LEW/N) rats are highly susceptible to the development of a wide range of autoimmune diseases in response to a variety of proinflammatory/antigenic stimuli. Fischer (F344/N) rats are relatively resistant to development of these illnesses after exposure to the same dose of antigens or proinflammatory stimuli. These two strains also show related differences in HPA axis responsiveness. The inflammatory-susceptible LEW/N rats exhibit a blunted HPA axis response, compared with inflammatory-resistant F344/N rats with an exaggerated HPA axis response [138–140]. Differences in the expression of hypothalamic CRH [141], pro-opiomelanocortin, corticosterone-binding globulin [142] and glucocorticoid expression and activation [143, 144] have been shown in these two rat strains.
Disruptions of the HPA axis in inflammatory resistant animals, through genetic, surgical, or pharmacological interventions, have been shown to be associated with enhanced susceptibility to, or increased severity of, inflammatory disease [139, 145–148]. Reconstitution of the HPA axis in these inflammatory-susceptible animals, either pharmacologically with glucocorticoids or surgically by intracerebral fetal hypothalamic tissue transplantation, has been shown to attenuate inflammatory disease [139, 149].
Animal models of arthritis
Several animal models exist for RA in rodents. Lewis rats develop arthritis in response to streptococcal cell walls [138, 139], heterologous (but not homologous) type II collagen in incomplete Freund's adjuvant (IFA) [150], and various adjuvant oils – including mycobacteria (MTB-AIA) [109], pristine [151], and avridine, but not IFA alone [152]. Inbred dark Agouti (DA) rats develop arthritis in response to heterologous and homologous type II collagen in IFA [153–156], cartilage oligomeric matrix protein [109], MTB-AIA [152], pristine, avridine [157], and ovalbumin-induced arthritis. DBA mice develop arthritis in response to type II collagen in complete Freund's adjuvant [158, 159]. For specific reviews on animal models for RA, refer to reviews by Morand and Leech [160] and Joe and Wilder [161].
A premorbid blunting of normal diurnal corticosterone levels in both Lewis and DA rats has been shown in animals susceptible to experimentally induced arthritis [162]. In adjuvant-induced arthritis, chronic activation of the HPA axis is seen 7–21 days after adjuvant injection, together with loss of circadian rhythm [163]. This chronic activation of the HPA axis was shown to be due to increased corticosterone secretion due to an increase in the pulse frequency of secretion in adjuvant-induced arthritis [164]. During this chronic activation of the HPA axis, rats with adjuvant-induced arthritis are incapable of mounting an HPA axis response to acute stress (such as noise) but are still able to respond to an acute immunological stress [165]. Adrenalectomy or glucocorticoid receptor blockade exacerbates the disease state and results in death or disease expression in surviving animals [139, 166, 167]. It has been suggested that mortality from such shock-like responses is due to the increased cytokine production that occurs in adrenalectomized animals exposed to proinflammatory stimuli [166, 168].
In addition to the role of HPA axis dysregulation, a dual role for the sympathetic nervous system in animal models of RA has been suggested. Activation of β-adrenoceptors or A2 receptors by high concentrations of norepinephrine or adenosine results in increased intracellular concentrations of cAMP and anti-inflammatory responses, whereas activation of α2-adrenoceptors and A1 receptors by low concentrations of norepinephrine or adenosine results in proinflammatory events, such as the release of substance P [169]. Consistent with this is the observation that β-adrenergic agonists attenuate RA in animal models [170, 171]. Rolipram, an inhibitor of the PDE-IV phophodiesterase, an enzyme that degrades cAMP, has been shown reduce inflammation in several rodent models [170, 172–174]. The effects of rolipram have also been suggested to be mediated by catecholamines [175] or by the stimulation of the adrenal and HPA axis [176, 177]. There is also a loss of sympathetic nerve fibers during adjuvant-induced arthritis [178]. The peripheral natural anti-inflammatory agent, vasoactive intestinal peptide, has been shown to reduce the severity of arthritis symptoms in the mouse model of collagen-induced arthritis [179, 180].
In addition to the sympathetic nervous system, the parasympathetic nervous system is also important in immune regulation. A role of the cholinergic parasympathetic nervous system in an animal model of RA was suggested because direct stimulation of the vagus nerve was shown to inhibit the inflammatory response [181]. Impairment of the cholinergic regulation also exacerbates an inflammatory response to adjuvant in the knees of rats [182].
Summary of animal model studies and therapeutic correlates
Thus, animal models for arthritis have shown a role for the HPA axis, sympathetic, parasympathetic, and peripheral nervous systems. They have shown the necessity of endogenous glucocorticoids in regulating the immune response after exposure to antigenic or proinflammatory stimuli, and severity of inflammatory/autoimmune disease or mortality after removal of these endogenous glucocorticoids by adrenalectomy or GR blockade. Animal models have enabled genetic linkage studies, which have demonstrated the multigenic, polygenic nature of such inflammatory diseases with genes on more than 20 different chromosomes being linked to inflammatory arthritis. Finally, animal models have shown defects in the sympathetic and parasympathetic nervous system in arthritis. These findings have led to the development and testing of novel therapies (see the penultimate section, 'New therapies').
Human studies
In humans, ovine CRH, hypoglycemia, or psychological stresses have been used to stimulate the HPA axis. In such studies, blunted HPA axis responses have been shown in a variety of autoimmune/inflammatory or allergic diseases such as allergic asthma and atopic dermatitis [183–186], fibromyalgia [187–190], chronic fatigue syndrome [188, 189, 191, 192], Sjögren's syndrome [2, 193], systemic lupus erythematosus [2, 194], multiple sclerosis [195, 196], and RA [1, 197–202]. Conversely, chronic stimulation of the stress hormone response, such as experienced by caregivers of Alzheimer's patients, students taking examinations, couples during marital conflict, and Army Rangers undergoing extreme exercise, results in chronically elevated glucocorticoids, causing a shift from Th1 to Th2 immune response, and is associated with an enhanced susceptibility to viral infection, prolonged wound healing, or decreased antibody production in response to vaccination [203–206].
Rheumatoid arthritis
RA is more common in women than in men, with onset usually occurring between menarche and menopause [207, 208]. However, the incidence of RA becomes much less gender specific in elderly men and women [207]. In women, RA activity is reduced during pregnancy but returns postpartum, suggesting a role for the hormones that are fluctuating at this time (cortisol, progesterone, and estrogen) in the regulation of RA activity [33, 209–212].
Glucocorticoids have been used for therapy for RA since the 1950s [213, 214], when the Nobel Prize was awarded for the discovery of this effect. They are effective because of their anti-inflammatory actions in the suppression of many inflammatory immune molecules and cells. In patients with RA, administration of glucocorticoids decreases the release of TNF-α into the bloodstream [215]; however, there are many debilitating side effects including weight gain, bone loss, and mood changes.
The HPA axis in RA
Human clinical studies are much more difficult to perform than animal models. However, some evidence exists supporting the involvement of the HPA axis in RA. Alterations in the diurnal rhythm of cortisol secretion have been documented in patients with RA [216, 217]. An association between the cortisol diurnal cycle and diurnal variations in RA activity has been made, although it still remains to be determined whether this is cause or effect [218]. One of the most pertinent observations for the regulation of RA by endogenous cortisol comes from a study in which RA was exacerbated by inhibition of adrenal glucocorticoid synthesis by the 11β-hydroxylase inhibitor metyrapone [219].
Several studies have looked for abnormalities in the HPA axis of patients with RA. In general, these point to an inappropriately low cortisol response. Subtle changes in cortisol responses have been reported in response to insulin-induced hypoglycemia [201]. However, another study, also using insulin-induced hypoglycemia, described a blunted HPA axis in patients with RA [220]. In one study, lower cortisol responses to surgical stress were shown in patients with RA compared with healthy controls and an inflammatory control group, whereas normal responses of ACTH and cortisol to ovine CRH were seen in the same patients [198]; however, these results are complicated by the steroid therapy that these patients were taking. Other studies have shown increased peripheral ACTH levels in patients with RA without increases in cortisol [221–223], whereas other studies have shown a normal HPA axis in patients with RA [200]. Some studies have suggested that, given the inflammatory state of RA, a normal cortisol response is in fact indicative of an under-responsive HPA axis [224, 225]. It has become generally accepted that lower than normal cortisol responses to stimulation are characteristic of RA [169, 197, 201, 216, 221, 223, 225–227]. Most recently Straub and colleagues have shown that the most sensitive indicator of blunted HPA axis responsiveness in early, untreated PA is an inappropriately low ratio of cortisol to IL-6 in these subjects [228].
Such defects in the stress response system are in agreement with patients' descriptions of RA 'flare up' during stress [229], which are likely to be caused by imbalances of the neuroendocrine and immune systems induced by psychosocial stressors [230]. It is worth noting that psychosocial stress is important in RA disease activity [231–233]. However, this will not be reviewed here and readers are referred to reviews by Walker and colleagues [234] and Herrmann and colleagues [235].
Glucocorticoid receptors in RA
Quantification of the numbers of GRs by ligand binding studies has produced contrasting results. In one study, normal or even slightly elevated numbers of GRs in peripheral blood mononuclear cells (PBMCs) were seen in untreated patients with RA [236], whereas other studies have shown a decrease in the number of GR molecules in the lymphocytes of patients with RA in comparison with controls [237]. Others have also shown a downregulation of GR during early RA [238, 239]. Recently, Neeck and colleagues, evaluating the expression of GR by immunoblot analysis, showed a higher expression of GR in untreated patients with RA in comparison with controls but a decreased GR expression in glucocorticoid-treated patients with RA in comparison with controls [202]. This has been confirmed by others [240]. A polymorphism in the 5' untranslated region of exon 9 of the GR gene, which is associated with enhanced stability of the dominant-negative spice variant, GRβ, has been shown in patients with RA [31]. Enhanced expression of GRβ has also been shown in the PBMCs of steroid-resistant patients with RA [241]. A polymorphism in the CRH gene has also been described as a susceptibility marker for RA in an indigenous South African population [242–244].
Other hormone measures in RA
Patients with RA also show abnormalities in other endocrine hormones. Like other inflammatory diseases, they have been shown to have low serum androgen levels but unchanged serum estrogen levels [245–252]. Growth retardation is a phenomenon seen in juvenile RA [253], and an impairment of the GH axis has been shown in patients with active and remitted RA [220, 225]. An increased expression of IGF-1-binding protein, resulting in a decreased concentration of free IGF-1, was also observed in patients with RA [254–256]. However, another study has attributed this difference in IGF-binding proteins to physical activity rather than inflammation [257].
An association between thyroid and rheumatoid disorders, such as RA and autoimmune thyroiditis, has been known for many years [258] although little is known about the thyroid involvement in RA. One study has shown that patients with RA have increased free T4 levels, and consequently lower free T3, than normal controls [259], although other studies were unable to confirm low levels in T3 patients with RA [260]. However, a higher incidence of thyroid dysfunction has been shown in women with RA [261, 262].
Sympathetic nervous system in RA
The extent to which the sympathetic nervous system is involved in human RA is unclear. In one study, a decreased number of β-adrenoceptors in the PBMCs and synovial lymphocytes of patients with RA was described, suggesting a shift to a proinflammatory state [263, 264]. Regional blockade of the sympathetic nervous system in patients with RA has been described to attenuate some of features of RA [265]. Others were unable to confirm this result but found defects in other aspects of this signaling pathway [266]. However, as in animal models, β-adrenergic agonists have been shown to attenuate RA in humans [267].
For the sympathetic nervous system to be able to modulate inflammation in RA it is necessary for the synovial tissue to be innervated by sympathetic nerve fibers. In patients with long-term RA there is a significant decrease in sympathetic nerve fibers but an increase in substance P-producing sensory nerve fibers [268, 269], suggesting a decrease in the anti-inflammatory effects of the sympathetic nervous system and an increase in the proinflammatory effects of the peripheral nervous system.
Peripheral neuropeptides in RA
Consistent with these changes in peripheral and autonomic innervation in RA are findings of altered peripheral neuropeptides in RA. proinflammatory CRH is locally secreted in the synovium of patients with RA and at a lower level than in osteoarthritis [199, 270]. Human T lymphocytes have been shown to synthesize and secrete CRH [271]. Inflammation in chronic RA has also been shown to be attenuated with the μ-opioid-specific agonist morphine [272]. In animal models, infusion of substance P into the knee exacerbated RA [273].
Summary of hormonal findings in RA
Studies of patients with RA are difficult to interpret and some might be tainted by a prior use of glucocorticoids used generally in the treatment of RA. However, these studies have generally shown a defect in cortisol secretion after HPA axis stimulation, decreased androgen levels, a blunted GH response, and dysregulation of the thyroid response. In addition there is evidence of an impaired response of the sympathetic nervous system and enhanced levels of the peripheral proinflammatory neuropeptides CRH and substance P. In some cases, a decrease in the number of GRs has been shown in RA, or reduced glucocorticoid sensitivity has been observed due to GRβ overexpression, which is consistent with relative glucocorticoid resistance in some patients. Furthermore, a polymorphism of the GRβ associated with the enhanced stability of that receptor has also been shown in RA [31]. It still remains to be fully determined whether these alterations in neuroendocrine pathways and receptors are involved in the pathogenesis of RA or whether they are a result of the inflammatory status of the disease.
New therapies
On the basis of the principles described above, new therapeutic modalities for inflammatory diseases are being investigated. For example, recent studies have indicated a potential therapeutic role for CRH type 1-specific receptor antagonist (antalarmin) in an animal model of adjuvant-induced arthritis [274], β-adrenergic agonists in both animal models of RA and in a human study [170, 171, 267], the μ-opioid-specific agonist morphine in chronic RA [272]), and the phophodiesterase inhibitor rolipram in several rodent models for RA [170, 172–174]. Androgen replacement, DHEA therapy, could be potentially therapeutic in RA, particularly in men [275], and has proved beneficial for inflammatory diseases [276].
Conclusion
The CNS and immune system communicate through multiple neuroanatomical and hormonal routes and molecular mechanisms. The interactions between the neuroendocrine and immune systems provide a finely tuned regulatory system required for health. Disturbances at any level can lead to changes in susceptibility to, and severity of, autoimmune/inflammatory disease. A thorough understanding of the mechanisms by which the CNS and immune systems communicate at all levels will provide many new insights into the bidirectional regulation of these systems and the disruptions in these communications that lead to disease, and ultimately will inform new avenues of therapy for autoimmune/inflammatory disease. Animal models of arthritis have shown changes in both the HPA axis and the sympathetic nervous system during inflammation. More importantly, these models have demonstrated the importance of endogenous glucocorticoids in the regulation of immunity and the prevention of lethality from an uncontrolled immune response. Furthermore, in both animals and humans, RA is associated with dysregulation of the HPA, HPT, HPG, and GH axes. There is also evidence of an impaired regulation of immunity by the sympathetic nervous system and of defects in glucocorticoid signaling. These principles are now being used to test novel therapies for RA based on addressing and correcting the dysregulation of these neural and neuroendocrine pathways.
Abbreviations
- ACTH:
-
adrenocorticotropin
- AVP:
-
arginine vasopressin
- CNS:
-
central nervous system
- CRH:
-
corticotropin-releasing hormone
- DHEA:
-
dehydroepiandrosterone
- GH:
-
growth hormone
- GR:
-
glucocorticoid receptor
- HPA:
-
hypothalamic–pituitary–adrenal
- HPG:
-
hypothalamic–pituitary–gonadal
- HPT:
-
hypothalamic–pituitary–thyroid
- IFA:
-
incomplete Freund's adjuvant
- IGF:
-
insulin-like growth factor
- IL:
-
interleukin
- NF-κB:
-
nuclear factor-κB
- PBMCs:
-
peripheral blood mononuclear cells
- RA:
-
rheumatoid arthritis
- T3:
-
triiodothyronine
- T4:
-
thyroxine
- Th:
-
T helper cells
- TNF:
-
tumor necrosis factor
- TRH:
-
thyrotropin-releasing hormone
- TSH:
-
thyroid-stimulating hormone.
References
Eijsbouts AM, Murphy EP: The role of the hypothalamic–pituitary–adrenal axis in rheumatoid arthritis. Baillieres Best Pract Res Clin Rheumatol. 1999, 13: 599-613. 10.1053/berh.1999.0048.
Crofford LJ: The hypothalamic–pituitary–adrenal axis in the pathogenesis of rheumatic diseases. Endocrinol Metab Clin North Am. 2002, 31: 1-13. 10.1016/S0889-8529(01)00004-4.
Imrich R: The role of neuroendocrine system in the pathogenesis of rheumatic diseases (minireview). Endocr Regul. 2002, 36: 95-106.
Morag M, Yirmiya R, Lerer B, Morag A: Influence of socioeconomic status on behavioral, emotional and cognitive effects of rubella vaccination: a prospective, double blind study. Psychoneuroendocrinology. 1998, 23: 337-351. 10.1016/S0306-4530(98)00012-2.
Reichenberg A, Kraus T, Haack M, Schuld A, Pollmacher T, Yirmiya R: Endotoxin-induced changes in food consumption in healthy volunteers are associated with TNF-alpha and IL-6 secretion. Psychoneuroendocrinology. 2002, 27: 945-956. 10.1016/S0306-4530(01)00101-9.
Watkins LR, Maier SF, Goehler LE: Cytokine-to-brain communication: a review and analysis of alternative mechanisms. Life Sci. 1995, 57: 1011-1026. 10.1016/0024-3205(95)02047-M.
Dantzer R, Bluthe RM, Laye S, Bret-Dibat JL, Parnet P, Kelley KW: Cytokines and sickness behavior. Ann N Y Acad Sci. 1998, 840: 586-590. 10.1111/j.1749-6632.1998.tb09597.x.
Ritchlin C, Haas-Smith SA: Expression of interleukin 10 mRNA and protein by synovial fibroblastoid cells. J Rheumatol. 2001, 28: 698-705.
Kurowska M, Rudnicka W, Kontny E, Janicka I, Chorazy M, Kowalczewski J, Ziolkowska M, Ferrari-Lacraz S, Strom TB, Maslinski W: Fibroblast-like synoviocytes from rheumatoid arthritis patients express functional IL-15 receptor complex: endogenous IL-15 in autocrine fashion enhances cell proliferation and expression of Bcl-xL and Bcl-2. J Immunol. 2002, 169: 1760-1767.
Lamberts SW, Verleun T, Oosterom R, de Jong F, Hackeng WH: Corticotropin-releasing factor (ovine) and vasopressin exert a synergistic effect on adrenocorticotropin release in man. J Clin Endocrinol Metab. 1984, 58: 298-303. 10.1210/jcem-58-2-298.
Antoni FA: Vasopressinergic control of pituitary adrenocorticotropin secretion comes of age. Front Neuroendocrinol. 1993, 14: 76-122. 10.1006/frne.1993.1004.
Barnes PJ: Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci (Lond). 1998, 94: 557-572.
Adcock IM, Ito K: Molecular mechanisms of corticosteroid actions. Monaldi Arch Chest Dis. 2000, 55: 256-266.
DeRijk R, Michelson D, Karp B, Petrides J, Galliven E, Deuster P, Paciotti G, Gold PW, Sternberg EM: Exercise and circadian rhythm-induced variations in plasma cortisol differentially regulate interleukin-1 beta (IL-1 beta), IL-6, and tumor necrosis factor-alpha (TNF alpha) production in humans: high sensitivity of TNF alpha and resistance of IL-6. J Clin Endocrinol Metab. 1997, 82: 2182-2191. 10.1210/jc.82.7.2182.
Elenkov IJ, Chrousos GP: Stress hormones, Th1/Th2 patterns, pro/anti-inflammatory cytokines and susceptibility to disease. Trends Endocrinol Metab. 1999, 10: 359-368. 10.1016/S1043-2760(99)00188-5.
Dhabhar FS, McEwen BS: Enhancing versus suppressive effects of stress hormones on skin immune function. Proc Natl Acad Sci U S A. 1999, 96: 1059-1064. 10.1073/pnas.96.3.1059.
Aranda A, Pascual A: Nuclear hormone receptors and gene expression. Physiol. Rev. 2001, 81: 1269-1304.
Karin M, Chang L: AP-1-glucocorticoid receptor crosstalk taken to a higher level. J Endocrinol. 2001, 169: 447-451. 10.1677/joe.0.1690447.
McKay LI, Cidlowski JA: Molecular control of immune/inflammatory responses: interactions between nuclear factor-kappa B and steroid receptor-signaling pathways. Endocr Rev. 1999, 20: 435-459. 10.1210/er.20.4.435.
Herrlich P: Cross-talk between glucocorticoid receptor and AP-1. Oncogene. 2001, 20: 2465-2475. 10.1038/sj.onc.1204388.
Adcock IM: Molecular mechanisms of glucocorticosteroid actions. Pulm Pharmacol Ther. 2000, 13: 115-126. 10.1006/pupt.2000.0243.
Encío IJ, Detera-Wadleigh SD: The genomic structure of the human glucocorticoid receptor. J Biol Chem. 1991, 266: 7182-7188.
Vottero A, Chrousos GP: Glucocorticoid receptor β: view I. Trends Endocrinol Metab. 1999, 10: 333-338. 10.1016/S1043-2760(99)00179-4.
Carlstedt-Duke J: Glucocorticoid receptor β: view II. Trends Endocrinol Metab. 1999, 10: 339-342. 10.1016/S1043-2760(99)00178-2.
Sousa AR, Lane SJ, Cidlowski JA, Staynov DZ, Lee TH: Glucocorticoid resistance in asthma is associated with elevated in vivo expression of the glucocorticoid receptor beta-isoform. J Allergy Clin Immunol. 2000, 105: 943-950. 10.1067/mai.2000.106486.
Strickland I, Kisich K, Hauk PJ, Vottero A, Chrousos GP, Klemm DJ, Leung DYM: High constitutive glucocorticoid receptor β in human neutrophils enables them to reduce their spontaneous rate of cell death in response to corticosteroids. J Exp Med. 2001, 193: 585-593. 10.1084/jem.193.5.585.
Leung DYM, Hamid Q, Vottero A, Szefler SJ, Surs W, Minshall E, Chrousos GP, Klemm DJ: Association of glucocorticoid insensitivity with increased expression of glucocorticoid receptor β. J Exp Med. 1997, 186: 1567-1574. 10.1084/jem.186.9.1567.
Hamid QA, Wenzel SE, Hauk PJ, Tsicopoulos A, Wallaert B, Lafitte J-J, Chrousos GP, Szefler SJ, Leung DYM: Increased glucocorticoid receptor β in airway cells of glucocorticoid-insensitive asthma. Am J Respir Crit Care Med. 1999, 159: 1600-1604.
Honda M, Orii F, Ayabe T, Imai S, Ashida T, Obara T, Kohgo Y: Expression of glucocorticoid receptor β in lymphocytes of patients with glucocorticoid-resistant ulcerative colitis. Gastroenterology. 2000, 118: 859-866. 10.1016/S0016-5085(00)70172-7.
Orii F, Ashida T, Nomura M, Maemoto A, Fujiki T, Ayabe T, Imai S, Saitoh Y, Kohgo Y: Quantitative analysis for human glucocorticoid receptor alpha/beta mRNA in IBD. Biochem Biophys Res Commun. 2002, 296: 1286-1294. 10.1016/S0006-291X(02)02030-2.
DeRijk RH, Schaaf MJ, Turner G, Datson NA, Vreugdenhil E, Cidetalowski J, de Kloet ER, Emery P, Sternberg EM, Detera-Wadleigh SD: A human glucocorticoid receptor gene variant that increases the stability of the glucocorticoid receptor beta-isoform mRNA is associated with rheumatoid arthritis. J Rheumatol. 2001, 28: 2383-2388.
Olsen NJ, Kovacs WJ: Hormones, pregnancy, and rheumatoid arthritis. J Gend Specif Med. 2002, 5: 28-37.
Cutolo M: The roles of steroid hormones in arthritis. Br J Rheumatol. 1998, 37: 597-599. 10.1093/rheumatology/37.6.597.
Cutolo M, Wilder RL: Different roles for androgens and estrogens in the susceptibility to autoimmune rheumatic diseases. Rheum Dis Clin North Am. 2000, 26: 825-839. 10.1016/S0889-857X(05)70171-X.
Olsen NJ, Kovacs WJ: Gonadal steroids and immunity. Endocr Rev. 1996, 17: 369-384. 10.1210/er.17.4.369.
Ahmed SA, Hissong BD, Verthelyi D, Donner K, Becker K, Karpuzoglu-Sahin E: Gender and risk of autoimmune diseases: possible role of estrogenic compounds. Environ Health Perspect. 1999, 107 (Suppl 5): 681-686. 10.2307/3434327.
Kincade PW, Medina KL, Payne KJ, Rossi MI, Tudor KS, Yamashita Y, Kouro T: Early B-lymphocyte precursors and their regulation by sex steroids. Immunol Rev. 2000, 175: 128-137. 10.1111/j.1600-065X.2000.imr017502.x.
Medina KL, Strasser A, Kincade PW: Estrogen influences the differentiation, proliferation, and survival of early B-lineage precursors. Blood. 2000, 95: 2059-2067.
Erlandsson MC, Ohlsson C, Gustafsson JA, Carlsten H: Role of oestrogen receptors alpha and beta in immune organ development and in oestrogen-mediated effects on thymus. Immunology. 2001, 103: 17-25. 10.1046/j.1365-2567.2001.01212.x.
Rettori V, Gimeno MF, Karara A, Gonzalez MC, McCann SM: Interleukin 1 alpha inhibits prostaglandin E2 release to suppress pulsatile release of luteinizing hormone but not follicle-stimulating hormone. Proc Natl Acad Sci U S A. 1991, 88: 2763-2767. 10.1073/pnas.88.7.2763.
Rivest S, Rivier C: Interleukin-1 beta inhibits the endogenous expression of the early gene c-fos located within the nucleus of LH-RH neurons and interferes with hypothalamic LH-RH release during proestrus in the rat. Brain Res. 1993, 613: 132-142. 10.1016/0006-8993(93)90463-W.
Rivier C, Rivier J, Vale W: Stress-induced inhibition of reproductive functions: role of endogenous corticotropin-releasing factor. Science. 1986, 231: 607-609. 10.1126/science.3003907.
Petraglia F, Sutton S, Vale W, Plotsky P: Corticotropin-releasing factor decreases plasma luteinizing hormone levels in female rats by inhibiting gonadotropin-releasing hormone release into hypophysial–portal circulation. Endocrinology. 1987, 120: 1083-1088. 10.1210/endo-120-3-1083.
Bonavera JJ, Kalra SP, Kalra PS: Mode of action of interleukin-1 in suppression of pituitary LH release in castrated male rats. Brain Res. 1993, 612: 1-8. 10.1016/0006-8993(93)91637-8.
Rivier C, Vale W: In the rat, interleukin-1 alpha acts at the level of the brain and the gonads to interfere with gonadotropin and sex steroid secretion. Endocrinology. 1989, 124: 2105-2109. 10.1210/endo-124-5-2105.
Johnson RW, Arkins S, Dantzer R, Kelley KW: Hormones, lymphohemopoietic cytokines and the neuroimmune axis. Comp Biochem Physiol A Physiol. 1997, 116: 183-201. 10.1016/S0300-9629(96)00277-0.
Clark R: The somatogenic hormones and insulin-like growth factor-1:stimulators of lymphopoiesis and immune function. Endocr Rev. 1997, 18: 157-179. 10.1210/er.18.2.157.
van Buul-Offers SC, Kooijman R: The role of growth hormone and insulin-like growth factors in the immune system. Cell Mol Life Sci. 1998, 54: 1083-1094. 10.1007/s000180050237.
Jeay S, Sonenshein GE, Postel-Vinay MC, Kelly PA, Baixeras E: Growth hormone can act as a cytokine controlling survival and proliferation of immune cells: new insights into signaling pathways. Mol Cell Endocrinol. 2002, 188: 1-7. 10.1016/S0303-7207(02)00014-X.
Stuart CA, Meehan RT, Neale LS, Cintron NM, Furlanetto RW: Insulin-like growth factor-I binds selectively to human peripheral blood monocytes and B-lymphocytes. J Clin Endocrinol Metab. 1991, 72: 1117-1122. 10.1210/jcem-72-5-1117.
Kooijman R, Willems M, De Haas CJ, Rijkers GT, Schuurmans AL, Van Buul-Offers SC, Heijnen CJ, Zegers BJ: Expression of type I insulin-like growth factor receptors on human peripheral blood mononuclear cells. Endocrinology. 1992, 131: 2244-2250. 10.1210/en.131.5.2244.
Dorshkind K, Horseman ND: The roles of prolactin, growth hormone, insulin-like growth factor-I, and thyroid hormones in lymphocyte development and function: insights from genetic models of hormone and hormone receptor deficiency. Endocr Rev. 2000, 21: 292-312. 10.1210/er.21.3.292.
Dorshkind K, Horseman ND: Anterior pituitary hormones, stress, and immune system homeostasis. BioEssays. 2001, 23: 288-294. 10.1002/1521-1878(200103)23:3<288::AID-BIES1039>3.0.CO;2-P.
Veldhuis JD, Lizarralde G, Iranmanesh A: Divergent effects of short term glucocorticoid excess on the gonadotropic and somatotropic axes in normal men. J Clin Endocrinol Metab. 1992, 74: 96-102. 10.1210/jc.74.1.96.
Hochberg Z: Mechanisms of steroid impairment of growth. Horm Res. 2002, 58 (Suppl 1): 33-38. 10.1159/000064764.
Armario A, Marti O, Gavalda A, Giralt M, Jolin T: Effects of chronic immobilization stress on GH and TSH secretion in the rat: response to hypothalamic regulatory factors. Psychoneuroendocrinology. 1993, 18: 405-413. 10.1016/0306-4530(93)90015-D.
Marti O, Gavalda A, Jolin T, Armario A: Effect of regularity of exposure to chronic immobilization stress on the circadian pattern of pituitary adrenal hormones, growth hormone, and thyroid stimulating hormone in the adult male rat. Psychoneu-roendocrinology. 1993, 18: 67-77. 10.1016/0306-4530(93)90056-Q.
Wu H, Wang J, Cacioppo JT, Glaser R, Kiecolt-Glaser JK, Malarkey WB: Chronic stress associated with spousal caregiving of patients with Alzheimer's dementia is associated with downregulation of B-lymphocyte GH mRNA. J Gerontol A Biol Sci Med Sci. 1999, 54: M212-M215. 10.1093/gerona/54.4.M212.
Rettori V, Jurcovicova J, McCann SM: Central action of interleukin-1 in altering the release of TSH, growth hormone, and prolactin in the male rat. J Neurosci Res. 1987, 18: 179-183. 10.1002/jnr.490180125.
Rettori V, Belova N, Yu WH, Gimeno M, McCann SM: Role of nitric oxide in control of growth hormone release in the rat. Neuroimmunomodulation. 1994, 1: 195-200. 10.1159/000097160.
Klecha AJ, Genaro AM, Lysionek AE, Caro RA, Coluccia AG, Cremaschi GA: Experimental evidence pointing to the bidirectional interaction between the immune system and the thyroid axis. Int J Immunopharmacol. 2000, 22: 491-500. 10.1016/S0192-0561(00)00012-6.
Pawlikowski M, Stepien H, Komorowski J: Hypothalamic–pituitary–thyroid axis and the immune system. Neuroimmunomodulation. 1994, 1: 149-152. 10.1159/000097154.
Kruger TE: Immunomodulation of peripheral lymphocytes by hormones of the hypothalamus–pituitary–thyroid axis. Adv Neuroimmunol. 1996, 6: 387-395. 10.1016/S0960-5428(97)00033-2.
Wang HC, Klein JR: Immune function of thyroid stimulating hormone and receptor. Crit Rev Immunol. 2001, 21: 323-337.
Witzke O, Winterhagen T, Saller B, Roggenbuck U, Lehr I, Philipp T, Mann K, Reinhardt W: Transient stimulatory effects on pituitary–thyroid axis in patients treated with interleukin-2. Thyroid. 2001, 11: 665-670. 10.1089/105072501750362736.
Kimura T, Kanda T, Kotajima N, Kuwabara A, Fukumura Y, Kobayashi I: Involvement of circulating interleukin-6 and its receptor in the development of euthyroid sick syndrome in patients with acute myocardial infarction. Eur J Endocrinol. 2000, 143: 179-184. 10.1530/eje.0.1430179.
Kamilaris TC, DeBold CR, Johnson EO, Mamalaki E, Listwak SJ, Calogero AE, Kalogeras KT, Gold PW, Orth DN: Effects of short and long duration hypothyroidism and hyperthyroidism on the plasma adrenocorticotropin and corticosterone responses to ovine corticotropin-releasing hormone in rats. Endocrinology. 1991, 128: 2567-2576. 10.1210/endo-128-5-2567.
Rittenhouse PA, Redei E: Thyroxine administration prevents streptococcal cell wall-induced inflammatory responses. Endocrinology. 1997, 138: 1434-1439. 10.1210/en.138.4.1434.
Shi ZX, Levy A, Lightman SL: Thyroid hormone-mediated regulation of corticotropin-releasing hormone messenger ribonucleic acid in the rat. Endocrinology. 1994, 134: 1577-1580. 10.1210/en.134.3.1577.
Benker G, Raida M, Olbricht T, Wagner R, Reinhardt W, Reinwein D: TSH secretion in Cushing's syndrome: relation to glucocorticoid excess, diabetes, goitre, and the 'sick euthyroid syndrome'. Clin Endocrinol (Oxf). 1990, 33: 777-786. 10.1111/j.1365-2265.1990.tb03915.x.
Ackerman KD, Felten SY, Dijkstra CD, Livnat S, Felten DL: Parallel development of noradrenergic innervation and cellular compartmentation in the rat spleen. Exp Neurol. 1989, 103: 239-255. 10.1016/0014-4886(89)90048-4.
Felten DL, Ackerman KD, Wiegand SJ, Felten SY: Noradrenergic sympathetic innervation of the spleen: I. Nerve fibers associate with lymphocytes and macrophages in specific compartments of the splenic white pulp. J Neurosci Res. 1987, 18: 28-36. 10.1002/jnr.490180107.
Felten SY, Felten DL, Bellinger DL, Carlson SL, Ackerman KD, Madden KS, Olschowka JA, Livnat S: Noradrenergic sympathetic innervation of lymphoid organs. Prog Allergy. 1988, 43: 14-36.
Madden KS, Sanders VM, Felten DL: Catecholamine influences and sympathetic neural modulation of immune responsiveness. Annu Rev Pharmacol Toxicol. 1995, 35: 417-448. 10.1146/annurev.pa.35.040195.002221.
ThyagaRajan S, Madden KS, Stevens SY, Felten DL: Effects of L-deprenyl treatment on noradrenergic innervation and immune reactivity in lymphoid organs of young F344 rats. J Neuroimmunol. 1999, 96: 57-65. 10.1016/S0165-5728(99)00017-X.
Lorton D, Bellinger D, Duclos M, Felten SY, Felten DL: Application of 6-hydroxydopamine into the fatpads surrounding the draining lymph nodes exacerbates adjuvant-induced arthritis. J Neuroimmunol. 1996, 64: 103-113. 10.1016/0165-5728(95)00150-6.
Lorton D, Lubahn C, Klein N, Schaller J, Bellinger DL: Dual role for noradrenergic innervation of lymphoid tissue and arthritic joints in adjuvant-induced arthritis. Brain Behav Immun. 1999, 13: 315-334. 10.1006/brbi.1999.0564.
Sanders VM, Kasprowicz DJ, Swanson-Mungerson MA, Podojil JR, Kohm AP: Adaptive immunity in mice lacking the β2-adrenergic receptor. Brain Behav Immun. 2003, 17: 55-67. 10.1016/S0889-1591(02)00056-9.
Hermann G, Beck FM, Tovar CA, Malarkey WB, Allen C, Sheridan JF: Stress-induced changes attributable to the sympathetic nervous system during experimental influenza viral infection in DBA/2 inbred mouse strain. J Neuroimmunol. 1994, 53: 173-180. 10.1016/0165-5728(94)90027-2.
Grimm MC, Ben-Baruch A, Taub DD, Howard OM, Resau JH, Wang JM, Ali H, Richardson R, Snyderman R, Oppenheim JJ: Opiates transdeactivate chemokine receptors: delta and mu opiate receptor-mediated heterologous desensitization. J Exp Med. 1998, 188: 317-325. 10.1084/jem.188.2.317.
Rogers TJ, Steele A, Howard OM, Oppenheim JJ: Bidirectional heterologous desensitization of opioid and chemokine receptors. Ann N Y Acad Sci. 2000, 917: 19-28. 10.1111/j.1749-6632.2000.tb05369.x.
Gomez-Flores R, Suo JL, Weber RJ: Suppression of splenic macrophage functions following acute morphine action in the rat mesencephalon periaqueductal gray. Brain Behav Immun. 1999, 13: 212-224. 10.1006/brbi.1999.0563.
Gomez-Flores R, Weber RJ: Inhibition of interleukin-2 production and downregulation of IL-2 and transferrin receptors on rat splenic lymphocytes following PAG morphine administration: a role in natural killer and T cell suppression. J Interferon Cytokine Res. 1999, 19: 625-630. 10.1089/107999099313767.
Mellon RD, Bayer BM: Role of central opioid receptor subtypes in morphine-induced alterations in peripheral lymphocyte activity. Brain Res. 1998, 789: 56-67. 10.1016/S0006-8993(97)01529-1.
Mellon RD, Bayer BM: The effects of morphine, nicotine and epibatidine on lymphocyte activity and hypothalamic–pituitary–adrenal axis responses. J Pharmacol Exp Ther. 1999, 288: 635-642.
Houghtling RA, Mellon RD, Tan RJ, Bayer BM: Acute effects of morphine on blood lymphocyte proliferation and plasma IL-6 levels. Ann N Y Acad Sci. 2000, 917: 771-777. 10.1111/j.1749-6632.2000.tb05442.x.
Gomez-Flores R, Weber RJ: Differential effects of buprenorphine and morphine on immune and neuroendocrine functions following acute administration in the rat mesencephalon periaqueductal gray. Immunopharmacology. 2000, 48: 145-156. 10.1016/S0162-3109(00)00198-3.
Tracey KJ: The inflammatory reflex. Nature. 2002, 420: 853-859. 10.1038/nature01321.
Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ: Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000, 405: 458-462. 10.1038/35013070.
Payan DG, Goetzl EJ: Dual roles of substance P: modulator of immune and neuroendocrine functions. Ann N Y Acad Sci. 1987, 512: 465-475. 10.1111/j.1749-6632.1987.tb24981.x.
Crofford LJ, Sano H, Karalis K, Webster EA, Friedman TC, Chrousos GP, Wilder RL: Local expression of corticotropin-releasing hormone in inflammatory arthritis. Ann N Y Acad Sci. 1995, 771: 459-471. 10.1111/j.1749-6632.1995.tb44702.x.
Dorsam G, Voice J, Kong Y, Goetzl EJ: Vasoactive intestinal peptide mediation of development and functions of T lymphocytes. Ann N Y Acad Sci. 2000, 921: 79-91. 10.1111/j.1749-6632.2000.tb06953.x.
Bagdy G, Calogero AE, Murphy DL, Szemeredi K: Serotonin agonists cause parallel activation of the sympathoadrenomedullary system and the hypothalamo-pituitary-adrenocortical axis in conscious rats. Endocrinology. 1989, 125: 2664-2669. 10.1210/endo-125-5-2664.
Calogero AE, Bernardini R, Margioris AN, Bagdy G, Gallucci WT, Munson PJ, Tamarkin L, Tomai TP, Brady L, Gold PW, Chrousos GP: Effects of serotonergic agonists and antagonists on corticotropin-releasing hormone secretion by explanted rat hypothalami. Peptides. 1989, 10: 189-200. 10.1016/0196-9781(89)90096-X.
Calogero AE, Bagdy G, Szemeredi K, Tartaglia ME, Gold PW, Chrousos GP: Mechanisms of serotonin receptor agonist-induced activation of the hypothalamic–pituitary–adrenal axis in the rat. Endocrinology. 1990, 126: 1888-1894. 10.1210/endo-126-4-1888.
Calogero AE, Gallucci WT, Bernardini R, Saoutis C, Gold PW, Chrousos GP: Effect of cholinergic agonists and antagonists on rat hypothalamic corticotropin-releasing hormone secretion in vitro. Neuroendocrinology. 1988, 47: 303-308. 10.1159/000124929.
Calogero AE, Kamilaris TC, Gomez MT, Johnson EO, Tartaglia ME, Gold PW, Chrousos GP: The muscarinic cholinergic agonist arecoline stimulates the rat hypothalamic–pituitary–adrenal axis through a centrally-mediated corticotropin-releasing hormone-dependent mechanism. Endocrinology. 1989, 125: 2445-2453. 10.1210/endo-125-5-2445.
Calogero AE, Gallucci WT, Chrousos GP, Gold PW: Catecholamine effects upon rat hypothalamic corticotropin-releasing hormone secretion in vitro. J Clin Invest. 1988, 82: 839-846. 10.1172/JCI113687.
Calogero AE, Gallucci WT, Chrousos GP, Gold PW: Interaction between GABAergic neurotransmission and rat hypothalamic corticotropin-releasing hormone secretion in vitro. Brain Res. 1988, 463: 28-36. 10.1016/0006-8993(88)90523-9.
Hopkins SJ, Rothwell NJ: Cytokines and the nervous system. I: Expression and recognition. Trends Neurosci. 1995, 18: 83-88. 10.1016/0166-2236(95)93881-W.
Rothwell NJ, Hopkins SJ: Cytokines and the nervous system II: Actions and mechanisms of action. Trends Neurosci. 1995, 18: 130-136. 10.1016/0166-2236(95)93890-A.
Benveniste EN: Cytokine actions in the central nervous system. Cytokine Growth Factor Rev. 1998, 9: 259-275. 10.1016/S1359-6101(98)00015-X.
Banks WA, Ortiz L, Plotkin SR, Kastin AJ: Human interleukin (IL) 1 alpha, murine IL-1 alpha and murine IL-1 beta are transported from blood to brain in the mouse by a shared saturable mechanism. J Pharmacol Exp Ther. 1991, 259: 988-996.
Blatteis CM: Role of the OVLT in the febrile response to circulating pyrogens. Prog Brain Res. 1992, 91: 409-412. full_text.
Fleshner M, Goehler LE, Hermann J, Relton JK, Maier SF, Watkins LR: Interleukin-1 beta induced corticosterone elevation and hypothalamic NE depletion is vagally mediated. Brain Res Bull. 1995, 37: 605-610. 10.1016/0361-9230(95)00051-F.
Quagliarello VJ, Wispelwey B, Long WJ, Scheld WM: Recombinant human interleukin-1 induces meningitis and blood-brain barrier injury in the rat. Characterization and comparison with tumor necrosis factor. J Clin Invest. 1991, 87: 1360-1366. 10.1172/JCI115140.
Claudio L, Martiney JA, Brosnan CF: Ultrastructural studies of the blood-retina barrier after exposure to interleukin-1 beta or tumor necrosis factor-alpha. Lab Invest. 1994, 70: 850-861.
Lustig S, Danenberg HD, Kafri Y, Kobiler D, Ben-Nathan D: Viral neuroinvasion and encephalitis induced by lipopolysaccharide and its mediators. J Exp Med. 1992, 176: 707-712. 10.1084/jem.176.3.707.
Tilders FJ, DeRijk RH, Van Dam AM, Vincent VA, Schotanus K, Persoons JH: Activation of the hypothalamus-pituitary-adrenal axis by bacterial endotoxins: routes and intermediate signals. Psychoneuroendocrinology. 1994, 19: 209-232. 10.1016/0306-4530(94)90010-8.
Watkins LR, Maier SF: The pain of being sick: implications of immune-to-brain communication for understanding pain. Annu Rev Psychol. 2000, 51: 29-57. 10.1146/annurev.psych.51.1.29.
Dantzer R: Cytokine-induced sickness behavior: mechanisms and implications. Ann N Y Acad Sci. 2001, 933: 222-234. 10.1111/j.1749-6632.2001.tb05827.x.
Hetier E, Ayala J, Denefle P, Bousseau A, Rouget P, Mallat M, Prochiantz A: Brain macrophages synthesize interleukin-1 and interleukin-1 mRNAs in vitro. J Neurosci Res. 1988, 21: 391-397. 10.1002/jnr.490210230.
Sebire G, Emilie D, Wallon C, Hery C, Devergne O, Delfraissy JF, Galanaud P, Tardieu M: In vitro production of IL-6, IL-1 beta, and tumor necrosis factor-alpha by human embryonic microglial and neural cells. J Immunol. 1993, 150: 1517-1523.
Breder CD, Dinarello CA, Saper CB: Interleukin-1 immunoreactive innervation of the human hypothalamus. Science. 1988, 240: 321-324. 10.1126/science.3258444.
Suda T, Tozawa F, Ushiyama T, Sumitomo T, Yamada M, Demura H: Interleukin-1 stimulates corticotropin-releasing factor gene expression in rat hypothalamus. Endocrinology. 1990, 126: 1223-1228. 10.1210/endo-126-2-1223.
Chover-Gonzalez AJ, Lightman SL, Harbuz MS: An investigation of the effects of interleukin-1 beta on plasma arginine vasopressin in the rat: role of adrenal steroids. J Endocrinol. 1994, 142: 361-366. 10.1677/joe.0.1420361.
Kehrer P, Turnill D, Dayer JM, Muller AF, Gaillard RC: Human recombinant interleukin-1 beta and -alpha, but not recombinant tumor necrosis factor alpha stimulate ACTH release from rat anterior pituitary cells in vitro in a prostaglandin E2 and cAMP independent manner. Neuroendocrinology. 1988, 48: 160-166. 10.1159/000125004.
Hillhouse EW: Interleukin-2 stimulates the secretion of argi-nine vasopressin but not corticotropin-releasing hormone from rat hypothalamic cells in vitro. Brain Res. 1994, 650: 323-325. 10.1016/0006-8993(94)91799-X.
Lyson K, McCann SM: The effect of interleukin-6 on pituitary hormone release in vivo and in vitro. Neuroendocrinology. 1991, 54: 262-266. 10.1159/000125884.
Sharp BM, Matta SG, Peterson PK, Newton R, Chao C, McAllen K: Tumor necrosis factor-alpha is a potent ACTH secretagogue: comparison to interleukin-1 beta. Endocrinology. 1989, 124: 3131-3133. 10.1210/endo-124-6-3131.
Lightman SL, Windle RJ, Ma XM, Harbuz MS, Shanks NM, Julian MD, Wood SA, Kershaw YM, Ingram CD: Hypothalamic–pituitary–adrenal function. Arch Physiol Biochem. 2002, 110: 90-93. 10.1076/apab.110.1.90.899.
Schwartz M, Hauben E: T cell-based therapeutic vaccination for spinal cord injury. Prog Brain Res. 2002, 137: 401-406. 10.1016/S0079-6123(02)37031-6.
Goehler LE, Relton JK, Dripps D, Kiechle R, Tartaglia N, Maier SF, Watkins LR: Vagal paraganglia bind biotinylated interleukin-1 receptor antagonist: a possible mechanism for immune-to-brain communication. Brain Res Bull. 1997, 43: 357-364. 10.1016/S0361-9230(97)00020-8.
Goehler LE, Gaykema RP, Hammack SE, Maier SF, Watkins LR: Interleukin-1 induces c-Fos immunoreactivity in primary afferent neurons of the vagus nerve. Brain Res. 1998, 804: 306-310. 10.1016/S0006-8993(98)00685-4.
Gaykema RP, Goehler LE, Tilders FJ, Bol JG, McGorry M, Fleshner M, Maier SF, Watkins LR: Bacterial endotoxin induces fos immunoreactivity in primary afferent neurons of the vagus nerve. Neuroimmunomodulation. 1998, 5: 234-240. 10.1159/000026343.
Watkins LR, Goehler LE, Relton JK, Tartaglia N, Silbert L, Martin D, Maier SF: Blockade of interleukin-1 induced hyperthermia by subdiaphragmatic vagotomy: evidence for vagal mediation of immune–brain communication. Neurosci Lett. 1995, 183: 27-31. 10.1016/0304-3940(94)11105-R.
Saper CB: Central nervous system. In The Rat Nervous System. Edited by: Paxinos G. 1995, San Diego: Academic Press, 107-135.
Gaykema RP, Dijkstra I, Tilders FJ: Subdiaphragmatic vagotomy suppresses endotoxin-induced activation of hypothalamic corticotropin-releasing hormone neurons and ACTH secretion. Endocrinology. 1995, 136: 4717-4720. 10.1210/en.136.10.4717.
Kapcala LP, He JR, Gao Y, Pieper JO, DeTolla LJ: Subdiaphragmatic vagotomy inhibits intra-abdominal interleukin-1 beta stimulation of adrenocorticotropin secretion. Brain Res. 1996, 728: 247-254. 10.1016/0006-8993(96)00511-2.
Dietrich HM, Oliveira-dos-Santos AJ, Wick G: Development of spontaneous autoimmune thyroiditis in Obese strain (OS) chickens. Vet Immunol Immunopathol. 1997, 57: 141-146. 10.1016/S0165-2427(96)05762-5.
Lechner O, Hu Y, Jafarian-Tehrani M, Dietrich H, Schwarz S, Herold M, Haour F, Wick G: Disturbed immunoendocrine communication via the hypothalamo-pituitary-adrenal axis in murine lupus. Brain Behav Immun. 1996, 10: 337-350. 10.1006/brbi.1996.0030.
Listwak S, Barrientos RM, Koike G, Ghosh S, Gomez M, Misiewicz B, Sternberg EM: Identification of a novel inflammation-protective locus in the Fischer rat. Mamm Genome. 1999, 10: 362-365. 10.1007/s003359901002.
Becker KG, Simon RM, Bailey-Wilson JE, Freidlin B, Biddison WE, McFarland HF, Trent JM: Clustering of non-major histo-compatibility complex susceptibility candidate loci in human autoimmune diseases. Proc Natl Acad Sci U S A. 1998, 95: 9979-9984. 10.1073/pnas.95.17.9979.
Furuya T, Salstrom JL, McCall-Vining S, Cannon GW, Joe B, Remmers EF, Griffiths MM, Wilder RL: Genetic dissection of a rat model for rheumatoid arthritis: significant gender influences on autosomal modifier loci. Hum Mol Genet. 2000, 9: 2241-2250.
Lorentzen JC, Glaser A, Jacobsson L, Galli J, Fakhrai-rad H, Klareskog L, Luthman H: Identification of rat susceptibility loci for adjuvant-oil-induced arthritis. Proc Natl Acad Sci U S A. 1998, 95: 6383-6387. 10.1073/pnas.95.11.6383.
Remmers EF, Longman RE, Du Y, O'Hare A, Cannon GW, Grif-fiths MM, Wilder RL: A genome scan localizes five non-MHC loci controlling collagen-induced arthritis in rats. Nat Genet. 1996, 14: 82-85. 10.1038/ng0996-82.
Jafarian-Tehrani M, Listwak S, Barrientos RM, Michaud A, Corvol P, Sternberg EM: Exclusion of angiotensin I-converting enzyme as a candidate gene involved in exudative inflammatory resistance in F344/N rats. Mol Med. 2000, 6: 319-331.
Wilder RL, Calandra GB, Garvin AJ, Wright KD, Hansen CT: Strain and sex variation in the susceptibility to streptococcal cell wall-induced polyarthritis in the rat. Arthritis Rheum. 1982, 25: 1064-1072. 10.1002/art.1780250906.
Sternberg EM, Hill JM, Chrousos GP, Kamilaris T, Listwak SJ, Gold PW, Wilder RL: Inflammatory mediator-induced hypothalamic–pituitary–adrenal axis activation is defective in streptococcal cell wall arthritis-susceptible Lewis rats. Proc Natl Acad Sci U S A. 1989, 86: 2374-2378. 10.1073/pnas.86.7.2374.
Moncek F, Kvetnansky R, Jezova D: Differential responses to stress stimuli of Lewis and Fischer rats at the pituitary and adrenocortical level. Endocr Regul. 2001, 35: 35-41.
Sternberg EM, Young WS, Bernardini R, Calogero AE, Chrousos GP, Gold PW, Wilder RL: A central nervous system defect in biosynthesis of corticotropin-releasing hormone is associated with susceptibility to streptococcal cell wall-induced arthritis in Lewis rats. Proc Natl Acad Sci U S A. 1989, 86: 4771-4775. 10.1073/pnas.86.12.4771.
Dhabhar FS, McEwen BS, Spencer RL: Stress response, adrenal steroid receptor levels and corticosteroid-binding globulin levels – a comparison between Sprague–Dawley, Fischer 344 and Lewis rats. Brain Res. 1993, 616: 89-98. 10.1016/0006-8993(93)90196-T.
Smith CC, Omeljaniuk RJ, Whitfield HJ, Aksentijevich S, Fellows MQ, Zelazowska E, Gold PW, Sternberg EM: Differential mineralocorticoid (type 1) and glucocorticoid (type 2) receptor expression in Lewis and Fischer rats. Neuroimmunomodulation. 1994, 1: 66-73. 10.1159/000097092.
Dhabhar FS, Miller AH, McEwen BS, Spencer RL: Differential activation of adrenal steroid receptors in neural and immune tissues of Sprague Dawley, Fischer 344, and Lewis rats. J Neuroimmunol. 1995, 56: 77-90. 10.1016/0165-5728(94)00135-B.
MacPhee IA, Antoni FA, Mason DW: Spontaneous recovery of rats from experimental allergic encephalomyelitis is dependent on regulation of the immune system by endogenous adrenal corticosteroids. J Exp Med. 1989, 169: 431-445. 10.1084/jem.169.2.431.
Edwards CK, Yunger LM, Lorence RM, Dantzer R, Kelley KW: The pituitary gland is required for protection against lethal effects of Salmonella typhimurium. Proc Natl Acad Sci U S A. 1991, 88: 2274-2277. 10.1073/pnas.88.6.2274.
Ruzek MC, Pearce BD, Miller AH, Biron CA: Endogenous glucocorticoids protect against cytokine-mediated lethality during viral infection. J Immunol. 1999, 162: 3527-3533.
Gomez SA, Fernandez GC, Vanzulli S, Dran G, Rubel C, Berki T, Isturiz MA, Palermo MS: Endogenous glucocorticoids attenuate Shiga toxin-2-induced toxicity in a mouse model of haemolytic uraemic syndrome. Clin Exp Immunol. 2003, 131: 217-224. 10.1046/j.1365-2249.2003.02057.x.
Misiewicz B, Poltorak M, Raybourne RB, Gomez M, Listwak S, Sternberg EM: Intracerebroventricular transplantation of embryonic neuronal tissue from inflammatory resistant into inflammatory susceptible rats suppresses specific components of inflammation. Exp Neurol. 1997, 146: 305-314. 10.1006/exnr.1997.6446.
Trentham DE, Dynesius-Trentham RA: Attenuation of an adjuvant arthritis by type II collagen. J Immunol. 1983, 130: 2689-2692.
Vingsbo C, Sahlstrand P, Brun JG, Jonsson R, Saxne T, Holmdahl R: Pristane-induced arthritis in rats: a new model for rheumatoid arthritis with a chronic disease course influenced by both major histocompatibility complex and non-major histocompatibility complex genes. Am J Pathol. 1996, 149: 1675-1683.
Kleinau S, Erlandsson H, Holmdahl R, Klareskog L: Adjuvant oils induce arthritis in the DA rat. I. Characterization of the disease and evidence for an immunological involvement. J Autoimmun. 1991, 4: 871-880. 10.1016/0896-8411(91)90050-M.
Wooley PH, Chapedelaine JM: Immunogenetics of collagen-induced arthritis. Crit Rev Immunol. 1987, 8: 1-22.
Wooley PH: Animal models of rheumatoid arthritis. Curr Opin Rheumatol. 1991, 3: 407-420. 10.1097/00002281-199106000-00013.
Trentham DE, Townes AS, Kang AH: Autoimmunity to type II collagen an experimental model of arthritis. J Exp Med. 1977, 146: 857-868. 10.1084/jem.146.3.857.
Griffiths MM: Immunogenetics of collagen-induced arthritis in rats. Int Rev Immunol. 1988, 4: 1-15. 10.3109/08830188809044766.
Vingsbo C, Jonsson R, Holmdahl R: Avridine-induced arthritis in rats; a T cell-dependent chronic disease influenced both by MHC genes and by non-MHC genes. Clin Exp Immunol. 1995, 99: 359-363. 10.1111/j.1365-2249.1995.tb05558.x.
Doncarli A, Stasiuk LM, Fournier C, Abehsira-Amar O: Conversion in vivo from an early dominant Th0/Th1 response to a Th2 phenotype during the development of collagen-induced arthritis. Eur J Immunol. 1997, 27: 1451-1458. 10.1002/eji.1830270623.
Mauri C, Williams RO, Walmsley M, Feldmann M: Relationship between Th1/Th2 cytokine patterns and the arthritogenic response in collagen-induced arthritis. Eur J Immunol. 1996, 26: 1511-1518. 10.1002/eji.1830260716.
Morand EF, Leech M: Hypothalamic–pituitary–adrenal axis regulation of inflammation in rheumatoid arthritis. Immunol Cell Biol. 2001, 79: 395-399. 10.1046/j.1440-1711.2001.01028.x.
Joe B, Wilder RL: Animal models of rheumatoid arthritis. Mol Med Today. 1999, 5: 367-369. 10.1016/S1357-4310(99)01528-2.
Wilder RL, Griffiths MM, Cannon GW, Caspi R, Remmers EF: Susceptibility to autoimmune disease and drug addiction in inbred rats. Are there mechanistic factors in common related to abnormalities in hypothalamic–pituitary–adrenal axis and stress response function?. Ann N Y Acad Sci. 2000, 917: 784-796. 10.1111/j.1749-6632.2000.tb05444.x.
Sarlis NJ, Chowdrey HS, Stephanou A, Lightman SL: Chronic activation of the hypothalamo-pituitary-adrenal axis and loss of circadian rhythm during adjuvant-induced arthritis in the rat. Endocrinology. 1992, 130: 1775-1779. 10.1210/en.130.4.1775.
Windle RJ, Wood SA, Kershaw YM, Lightman SL, Ingram CD, Harbuz MS: Increased corticosterone pulse frequency during adjuvant-induced arthritis and its relationship to alterations in stress responsiveness. J Neuroendocrinol. 2001, 13: 905-911. 10.1046/j.1365-2826.2001.00715.x.
Harbuz MS, Windle RJ, Jessop DS, Renshaw D, Ingram CD, Lightman SL: Differential effects of psychological and immunological challenge on the hypothalamo-pituitary-adrenal axis function in adjuvant-induced arthritis. Ann N Y Acad Sci. 1999, 876: 43-52. 10.1111/j.1749-6632.1999.tb07621.x.
Perretti M, Mugridge KG, Becherucci C, Parente L: Evidence that interleukin-1 and lipoxygenase metabolites mediate the lethal effect of complete Freund's adjuvant in adrenalectomized rats. Lymphokine Cytokine Res. 1991, 10: 239-243.
Harbuz MS, Rees RG, Lightman SL: HPA axis responses to acute stress and adrenalectomy during adjuvant-induced arthritis in the rat. Am J Physiol. 1993, 264: R179-R185.
Sarlis NJ, Stephanou A, Knight RA, Lightman SL, Chowdrey HS: Effects of glucocorticoids and chronic inflammatory stress upon anterior pituitary interleukin-6 mRNA expression in the rat. Br J Rheumatol. 1993, 32: 653-657. 10.1093/rheumatology/32.8.653.
Straub RH, Cutolo M: Involvement of the hypothalamic–pituitary–adrenal/gonadal axis and the peripheral nervous system in rheumatoid arthritis: viewpoint based on a systemic pathogenetic role. Arthritis Rheum. 2001, 44: 493-507. 10.1002/1529-0131(200103)44:3<493::AID-ANR95>3.3.CO;2-L.
Laemont KD, Schaefer CJ, Juneau PL, Schrier DJ: Effects of the phosphodiesterase inhibitor rolipram on streptococcal cell wall-induced arthritis in rats. Int J Immunopharmacol. 1999, 21: 711-725. 10.1016/S0192-0561(99)00046-6.
Malfait AM, Malik AS, Marinova-Mutafchieva L, Butler DM, Maini RN, Feldmann M: The beta2-adrenergic agonist salbutamol is a potent suppressor of established collagen-induced arthritis: mechanisms of action. J Immunol. 1999, 162: 6278-6283.
Nyman U, Mussener A, Larsson E, Lorentzen J, Klareskog L: Amelioration of collagen II-induced arthritis in rats by the type IV phosphodiesterase inhibitor Rolipram. Clin Exp Immunol. 1997, 108: 415-419. 10.1046/j.1365-2249.1997.3931291.x.
Ross SE, Williams RO, Mason LJ, Mauri C, Marinova-Mutafchieva L, Malfait AM, Maini RN, Feldmann M: Suppression of TNF-alpha expression, inhibition of Th1 activity, and amelioration of collagen-induced arthritis by rolipram. J Immunol. 1997, 159: 6253-6259.
Sekut L, Yarnall D, Stimpson SA, Noel LS, Bateman-Fite R, Clark RL, Brackeen MF, Menius JA, Connolly KM: Anti-inflammatory activity of phosphodiesterase (PDE)-IV inhibitors in acute and chronic models of inflammation. Clin Exp Immunol. 1995, 100: 126-132. 10.1111/j.1365-2249.1995.tb03613.x.
Nielson CP, Vestal RE, Sturm RJ, Heaslip R: Effects of selective phosphodiesterase inhibitors on the polymorphonuclear leukocyte respiratory burst. J Allergy Clin Immunol. 1990, 86: 801-808. 10.1016/S0091-6749(05)80186-1.
Pettipher ER, Labasi JM, Salter ED, Stam EJ, Cheng JB, Griffiths RJ: Regulation of tumour necrosis factor production by adrenal hormones in vivo: insights into the antiinflammatory activity of rolipram. Br J Pharmacol. 1996, 117: 1530-1534.
Kumari M, Cover PO, Poyser RH, Buckingham JC: Stimulation of the hypothalamo-pituitary-adrenal axis in the rat by three selective type-4 phosphodiesterase inhibitors: in vitro and in vivo studies. Br J Pharmacol. 1997, 121: 459-468. 10.1038/sj.bjp.0701158.
Imai S, Tokunaga Y, Konttinen YT, Maeda T, Hukuda S, Santavirta S: Ultrastructure of the synovial sensory peptidergic fibers is distinctively altered in different phases of adjuvant induced arthritis in rats: ultramorphological characterization combined with morphometric and immunohistochemical study for substance P, calcitonin gene related peptide, and protein gene product 9.5. J Rheumatol. 1997, 24: 2177-2187.
Delgado M, Abad C, Martinez C, Leceta J, Gomariz RP: Vasoactive intestinal peptide prevents experimental arthritis by downregulating both autoimmune and inflammatory components of the disease. Nat Med. 2001, 7: 563-568. 10.1038/87887.
Delgado M, Abad C, Martinez C, Juarranz MG, Arranz A, Gomariz RP, Leceta J: Vasoactive intestinal peptide in the immune system: potential therapeutic role in inflammatory and autoimmune diseases. J Mol Med. 2002, 80: 16-24. 10.1007/s00109-001-0291-5.
Borovikova LV, Ivanova S, Nardi D, Zhang M, Yang H, Ombrellino M, Tracey KJ: Role of vagus nerve signaling in CNI-1493-mediated suppression of acute inflammation. Auton Neurosci. 2000, 85: 141-147. 10.1016/S1566-0702(00)00233-2.
McDougall JJ, Elenko RD, Bray RC: Cholinergic vasoregulation in normal and adjuvant monoarthritic rat knee joints. J Auton Nerv Syst. 1998, 72: 55-60. 10.1016/S0165-1838(98)00087-3.
Buske-Kirschbaum A, Jobst S, Psych D, Wustmans A, Kirschbaum C, Rauh W, Hellhammer D: Attenuated free cortisol response to psychosocial stress in children with atopic dermatitis. Psychosom Med. 1997, 59: 419-426.
Straub RH, Herfarth H, Falk W, Andus T, Scholmerich J: Uncoupling of the sympathetic nervous system and the hypothalamic–pituitary–adrenal axis in inflammatory bowel disease?. J Neuroimmunol. 2002, 126: 116-125. 10.1016/S0165-5728(02)00047-4.
Niess JH, Monnikes H, Dignass AU, Klapp BF, Arck PC: Review on the influence of stress on immune mediators, neuropeptides and hormones with relevance for inflammatory bowel disease. Digestion. 2002, 65: 131-140. 10.1159/000064933.
Buske-Kirschbaum A, Geiben A, Hollig H, Morschhauser E, Hell-hammer D: Altered responsiveness of the hypothalamus–pituitary–adrenal axis and the sympathetic adrenomedullary system to stress in patients with atopic dermatitis. J Clin Endocrinol Metab. 2002, 87: 4245-4251. 10.1210/jc.2001-010872.
Crofford LJ, Pillemer SR, Kalogeras KT, Cash JM, Michelson D, Kling MA, Sternberg EM, Gold PW, Chrousos GP, Wilder RL: Hypothalamic–pituitary–adrenal axis perturbations in patients with fibromyalgia. Arthritis Rheum. 1994, 37: 1583-1592. 10.1002/art.1780371105.
Demitrack MA, Crofford LJ: Evidence for and pathophysiologic implications of hypothalamic–pituitary–adrenal axis dysregulation in fibromyalgia and chronic fatigue syndrome. Ann N Y Acad Sci. 1998, 840: 684-697. 10.1111/j.1749-6632.1998.tb09607.x.
Neeck G, Crofford LJ: Neuroendocrine perturbations in fibromyalgia and chronic fatigue syndrome. Rheum Dis Clin North Am. 2000, 26: 989-1002. 10.1016/S0889-857X(05)70180-0.
Crofford LJ, Clauw DJ: Fibromyalgia: where are we a decade after the American College of Rheumatology classification criteria were developed?. Arthritis Rheum. 2002, 46: 1136-1138. 10.1002/art.10217.
Demitrack MA, Dale JK, Straus SE, Laue L, Listwak SJ, Kruesi MJ, Chrousos GP, Gold PW: Evidence for impaired activation of the hypothalamic–pituitary–adrenal axis in patients with chronic fatigue syndrome. J Clin Endocrinol Metab. 1991, 73: 1224-1234. 10.1210/jcem-73-6-1224.
Gaab J, Huster D, Peisen R, Engert V, Heitz V, Schad T, Schurmeyer TH, Ehlert U: Hypothalamic–pituitary–adrenal axis reactivity in chronic fatigue syndrome and health under psychological, physiological, and pharmacological stimulation. Psychosom Med. 2002, 64: 951-962. 10.1097/01.PSY.0000038937.67401.61.
Johnson EO, Vlachoyiannopoulos PG, Skopouli FN, Tzioufas AG, Moutsopoulos HM: Hypofunction of the stress axis in Sjogren's syndrome. J Rheumatol. 1998, 25: 1508-1514.
Gutierrez MA, Garcia ME, Rodriguez JA, Rivero S, Jacobelli S: Hypothalamic–pituitary–adrenal axis function and prolactin secretion in systemic lupus erythematosus. Lupus. 1998, 7: 404-408. 10.1191/096120398678920343.
Michelson D, Stone L, Galliven E, Magiakou MA, Chrousos GP, Sternberg EM, Gold PW: Multiple sclerosis is associated with alterations in hypothalamic–pituitary–adrenal axis function. J Clin Endocrinol Metab. 1994, 79: 848-853. 10.1210/jc.79.3.848.
Wei T, Lightman SL: The neuroendocrine axis in patients with multiple sclerosis. Brain. 1997, 120: 1067-1076. 10.1093/brain/120.6.1067.
Cash JM, Crofford LJ, Gallucci WT, Sternberg EM, Gold PW, Chrousos GP, Wilder RL: Pituitary–adrenal axis responsiveness to ovine corticotropin releasing hormone in patients with rheumatoid arthritis treated with low dose prednisone. J Rheumatol. 1992, 19: 1692-1696.
Chikanza IC, Petrou P, Kingsley G, Chrousos G, Panayi GS: Defective hypothalamic response to immune and inflammatory stimuli in patients with rheumatoid arthritis. Arthritis Rheum. 1992, 35: 1281-1288.
Crofford LJ, Sano H, Karalis K, Friedman TC, Epps HR, Remmers EF, Mathern P, Chrousos GP, Wilder RL: Corticotropin-releasing hormone in synovial fluids and tissues of patients with rheumatoid arthritis and osteoarthritis. J Immunol. 1993, 151: 1587-1596.
Crofford LJ, Kalogeras KT, Mastorakos G, Magiakou MA, Wells J, Kanik KS, Gold PW, Chrousos GP, Wilder RL: Circadian relationships between interleukin (IL)-6 and hypothalamic–pituitary–adrenal axis hormones: failure of IL-6 to cause sustained hypercortisolism in patients with early untreated rheumatoid arthritis. J Clin Endocrinol Metab. 1997, 82: 1279-1283. 10.1210/jc.82.4.1279.
Gutierrez MA, Garcia ME, Rodriguez JA, Mardonez G, Jacobelli S, Rivero S: Hypothalamic–pituitary–adrenal axis function in patients with active rheumatoid arthritis: a controlled study using insulin hypoglycemia stress test and prolactin stimulation. J Rheumatol. 1999, 26: 277-281.
Neeck G, Kluter A, Dotzlaw H, Eggert M: Involvement of the glucocorticoid receptor in the pathogenesis of rheumatoid arthritis. Ann N Y Acad Sci. 2002, 966: 491-495. 10.1111/j.1749-6632.2002.tb04252.x.
Glaser R, Kiecolt-Glaser JK: Stress-associated immune modulation: relevance to viral infections and chronic fatigue syndrome. Am J Med. 1998, 105: 35S-42S. 10.1016/S0002-9343(98)00160-0.
Rozlog LA, Kiecolt-Glaser JK, Marucha PT, Sheridan JF, Glaser R: Stress and immunity: implications for viral disease and wound healing. J Periodontol. 1999, 70: 786-792. 10.1902/jop.1999.70.7.786.
Vedhara K, Cox NK, Wilcock GK, Perks P, Hunt M, Anderson S, Lightman SL, Shanks NM: Chronic stress in elderly carers of dementia patients and antibody response to influenza vaccination. Lancet. 1999, 353: 627-631. 10.1016/S0140-6736(98)06098-X.
Friedl KE, Moore RJ, Hoyt RW, Marchitelli LJ, Martinez-Lopez LE, Askew EW: Endocrine markers of semistarvation in healthy lean men in a multistressor environment. J Appl Physiol. 2000, 88: 1820-1830.
Wilder RL: Adrenal and gonadal steroid hormone deficiency in the pathogenesis of rheumatoid arthritis. J Rheumatol Suppl. 1996, 44: 10-12.
Lahita RG: Sex steroids and the rheumatic diseases. Arthritis Rheum. 1985, 28: 121-126. 10.1002/art.1780280202.
Wilder RL, Elenkov IJ: Hormonal regulation of tumor necrosis factor-alpha, interleukin-12 and interleukin-10 production by activated macrophages. A disease-modifying mechanism in rheumatoid arthritis and systemic lupus erythematosus?. Ann N Y Acad Sci. 1999, 876: 14-31. 10.1111/j.1749-6632.1999.tb07619.x.
Wilder RL: Hormones, pregnancy, and autoimmune diseases. Ann N Y Acad Sci. 1998, 840: 45-50. 10.1111/j.1749-6632.1998.tb09547.x.
Barrett JH, Brennan P, Fiddler M, Silman AJ: Does rheumatoid arthritis remit during pregnancy and relapse postpartum? Results from a nationwide study in the United Kingdom performed prospectively from late pregnancy. Arthritis Rheum. 1999, 42: 1219-1227. 10.1002/1529-0131(199906)42:6<1219::AID-ANR19>3.0.CO;2-G.
Pope RM, Yoshinoya S, Rutstein J, Persellin RH: Effect of pregnancy on immune complexes and rheumatoid factors in patients with rheumatoid arthritis. Am J Med. 1983, 74: 973-979. 10.1016/0002-9343(83)90794-5.
Hench PS, Kendall EC, Slocumb CH, Polley HF: Effects of cortisone acetate and primary ACTH on rheumatoid arthritis, rheumatic fever and certain other conditions. Arch Intern Med. 1950, 85: 545-666.
Neeck G: Fifty years of experience with cortisone therapy in the study and treatment of rheumatoid arthritis. Ann N Y Acad Sci. 2002, 966: 28-38. 10.1111/j.1749-6632.2002.tb04199.x.
Steer JH, Kroeger KM, Abraham LJ, Joyce DA: Glucocorticoids suppress tumor necrosis factor-alpha expression by human monocytic THP-1 cells by suppressing transactivation through adjacent NF-kappa B and c-Jun-activating transcription factor-2 binding sites in the promoter. J Biol Chem. 2000, 275: 18432-18440. 10.1074/jbc.M906304199.
Neeck G, Federlin K, Graef V, Rusch D, Schmidt KL: Adrenal secretion of cortisol in patients with rheumatoid arthritis. J Rheumatol. 1990, 17: 24-29.
Dekkers JC, Geenen R, Godaert GL, van Doornen LJ, Bijlsma JW: Diurnal rhythm of salivary cortisol levels in patients with recent-onset rheumatoid arthritis. Arthritis Rheum. 2000, 43: 465-467. 10.1002/1529-0131(200002)43:2<465::AID-ANR31>3.0.CO;2-Q.
Harkness JA, Richter MB, Panayi GS, Van de Pette K, Unger A, Pownall R, Geddawi M: Circadian variation in disease activity in rheumatoid arthritis. Br Med J (Clin Res Ed). 1982, 284: 551-554. 10.1136/bmj.284.6315.551.
Saldanha C, Tougas G, Grace E: Evidence for anti-inflammatory effect of normal circulating plasma cortisol. Clin Exp Rheumatol. 1986, 4: 365-366.
Demir H, Kelestimur F, Tunc M, Kirnap M, Ozugul Y: Hypothalamo-pituitary-adrenal axis and growth hormone axis in patients with rheumatoid arthritis. Scand J Rheumatol. 1999, 28: 41-46. 10.1080/03009749950155779.
Kanik KS, Chrousos GP, Schumacher HR, Crane ML, Yarboro CH, Wilder RL: Adrenocorticotropin, glucocorticoid, and androgen secretion in patients with new onset synovitis/ rheumatoid arthritis: relations with indices of inflammation. J Clin Endocrinol Metab. 2000, 85: 1461-1466. 10.1210/jc.85.4.1461.
Hall J, Morand EF, Medbak S, Zaman M, Perry L, Goulding NJ, Maddison PJ, O'Hare JP: Abnormal hypothalamic–pituitary–adrenal axis function in rheumatoid arthritis. Effects of nonsteroidal antiinflammatory drugs and water immersion. Arthritis Rheum. 1994, 37: 1132-1137. 10.1002/art.1780370804.
Gudbjornsson B, Skogseid B, Oberg K, Wide L, Hallgren R: Intact adrenocorticotropic hormone secretion but impaired cortisol response in patients with active rheumatoid arthritis. Effect of glucocorticoids. J Rheumatol. 1996, 23: 596-602.
Cutolo M, Foppiani L, Prete C, Ballarino P, Sulli A, Villaggio B, Seriolo B, Giusti M, Accardo S: Hypothalamic–pituitary–adrenocortical axis function in premenopausal women with rheumatoid arthritis not treated with glucocorticoids. J Rheumatol. 1999, 26: 282-288.
Templ E, Koeller M, Riedl M, Wagner O, Graninger W, Luger A: Anterior pituitary function in patients with newly diagnosed rheumatoid arthritis. Br J Rheumatol. 1996, 35: 350-356. 10.1093/rheumatology/35.4.350.
Dekkers JC, Geenen R, Godaert GL, Glaudemans KA, Lafeber FP, van Doornen LJ, Bijlsma JW: Experimentally challenged reactivity of the hypothalamic pituitary adrenal axis in patients with recently diagnosed rheumatoid arthritis. J Rheumatol. 2001, 28: 1496-1504.
van den Brink HR, Blankenstein MA, Koppeschaar HP, Bijlsma JW: Influence of disease activity on steroid hormone levels in peripheral blood of patients with rheumatoid arthritis. Clin Exp Rheumatol. 1993, 11: 649-652.
Straub RH, Paimela L, Peltomaa R, Scholmerich J, Leirisalo-Repo M: Inadequately low serum levels of steroid hormones in relation to interleukin-6 and tumor necrosis factor in untreated patients with early rheumatoid arthritis and reactive arthritis. Arthritis Rheum. 2002, 46: 654-662. 10.1002/art.10177.
Affleck G, Pfeiffer C, Tennen H, Fifield J: Attributional processes in rheumatoid arthritis patients. Arthritis Rheum. 1987, 30: 927-931. 10.1002/art.1780300813.
Zautra AJ, Hoffman JM, Matt KS, Yocum D, Potter PT, Castro WL, Roth S: An examination of individual differences in the relationship between interpersonal stress and disease activity among women with rheumatoid arthritis. Arthritis Care Res. 1998, 11: 271-279. 10.1002/art.1790110408.
Affleck G, Tennen H, Urrows S, Higgins P: Neuroticism and the pain–mood relation in rheumatoid arthritis: insights from a prospective daily study. J Consult Clin Psychol. 1992, 60: 119-126. 10.1037//0022-006X.60.1.119.
Dwyer KA: Psychosocial factors and health status in women with rheumatoid arthritis: predictive models. Am J Prev Med. 1997, 13: 66-72.
Harrington L, Affleck G, Urrows S, Tennen H, Higgins P, Zautra A, Hoffman S: Temporal covariation of soluble interleukin-2 receptor levels, daily stress, and disease activity in rheumatoid arthritis. Arthritis Rheum. 1993, 36: 199-203.
Walker JG, Littlejohn GO, McMurray NE, Cutolo M: Stress system response and rheumatoid arthritis: a multilevel approach. Rheumatology (Oxford). 1999, 38: 1050-1057. 10.1093/rheumatology/38.11.1050.
Herrmann M, Scholmerich J, Straub RH: Stress and rheumatic diseases. Rheum Dis Clin North Am. 2000, 26: 737-763. 10.1016/S0889-857X(05)70167-8.
Sanden S, Tripmacher R, Weltrich R, Rohde W, Hiepe F, Burmester GR, Buttgereit F: Glucocorticoid dose dependent downregulation of glucocorticoid receptors in patients with rheumatic diseases. J Rheumatol. 2000, 27: 1265-1270.
Schlaghecke R, Kornely E, Wollenhaupt J, Specker C: Glucocorticoid receptors in rheumatoid arthritis. Arthritis Rheum. 1992, 35: 740-744. 10.1002/art.1780350704.
Huisman AM, Van Everdingen AA, Wenting MJ, Siewertsz Van Reesema DR, Lafeber FP, Jacobs JW, Bijlsma JW: Glucocorticoid receptor downregulation in early diagnosed rheumatoid arthritis. Ann N Y Acad Sci. 2002, 966: 64-67. 10.1111/j.1749-6632.2002.tb04202.x.
van Everdingen AA, Huisman AM, Wenting MJ, van Reesema S, Jacobs JW, Bijlsma JW: Down regulation of glucocorticoid receptors in early-diagnosed rheumatoid arthritis. Clin Exp Rheumatol. 2002, 20: 463-468.
Eggert M, Kluter A, Rusch D, Schmidt KL, Dotzlaw H, Schulz M, Pabst W, Boke J, Renkawitz R: Expression analysis of the glucocorticoid receptor and the nuclear factor-κB subunit p50 in lymphocytes from patients with rheumatoid arthritis. J Rheumatol. 2002, 29: 2500-2506.
Chikanza IC: Mechanisms of corticosteroid resistance in rheumatoid arthritis: a putative role for the corticosteroid receptor beta isoform. Ann N Y Acad Sci. 2002, 966: 39-48. 10.1111/j.1749-6632.2002.tb04200.x.
Baerwald CG, Panayi GS, Lanchbury JS: A new XmnI polymorphism in the regulatory region of the corticotropin releasing hormone gene. Hum Genet. 1996, 97: 697-698. 10.1007/s004390050121.
Baerwald CG, Panayi GS, Lanchbury JS: Corticotropin releasing hormone promoter region polymorphisms in rheumatoid arthritis. J Rheumatol. 1997, 24: 215-216.
Baerwald CG, Mok CC, Fife MS, Tikly M, Lau CS, Wordsworth BP, Ollier B, Panayi GS, Lanchbury JS: Distribution of corticotropin-releasing hormone promoter polymorphism in different ethnic groups: evidence for natural selection in human populations. Immunogenetics. 1999, 49: 894-899. 10.1007/s002510050570.
Cutolo M, Foppiani L, Minuto F: Hypothalamic–pituitary–adrenal axis impairment in the pathogenesis of rheumatoid arthritis and polymyalgia rheumatica. J Endocrinol Invest. 2002, 25: 19-23.
Cutolo M, Balleari E, Giusti M, Monachesi M, Accardo S: Sex hormone status of male patients with rheumatoid arthritis: evidence of low serum concentrations of testosterone at baseline and after human chorionic gonadotropin stimulation. Arthritis Rheum. 1988, 31: 1314-1317. 10.1002/art.1780311015.
Chikanza IC: Neuroendocrine immune features of pediatric inflammatory rheumatic diseases. Ann N Y Acad Sci. 1999, 876: 71-80. 10.1111/j.1749-6632.1999.tb07624.x. discussion 80–82.
Castagnetta L, Cutolo M, Granata OM, Di Falco M, Bellavia V, Carruba G: Endocrine end-points in rheumatoid arthritis. Ann N Y Acad Sci. 1999, 876: 180-192. 10.1111/j.1749-6632.1999.tb07637.x.
Masi AT, Josipovic DB, Jefferson WE: Low adrenal androgenic-anabolic steroids in women with rheumatoid arthritis (RA): gas-liquid chromatographic studies of RA patients and matched normal control women indicating decreased 11-deoxy-17-ketosteroid excretion. Semin Arthritis Rheum. 1984, 14: 1-23. 10.1016/0049-0172(84)90005-2.
Sambrook PN, Eisman JA, Champion GD, Pocock NA: Sex hormone status and osteoporosis in postmenopausal women with rheumatoid arthritis. Arthritis Rheum. 1988, 31: 973-978. 10.1002/art.1780310805.
Spector TD, Perry LA, Tubb G, Silman AJ, Huskisson EC: Low free testosterone levels in rheumatoid arthritis. Ann Rheum Dis. 1988, 47: 65-68. 10.1136/ard.47.1.65.
Seriolo B, Cutolo M, Garnero A, Accardo S: Relationships between serum 17 beta-oestradiol and anticardiolipin antibody concentrations in female patients with rheumatoid arthritis. Rheumatology (Oxford). 1999, 38: 1159-1161. 10.1093/rheumatology/38.11.1159.
Woo PM: Growth retardation and osteoporosis in juvenile chronic arthritis. Clin Exp Rheumatol. 1994, 12 (Suppl 10): S87-S90.
Matsumoto T, Tsurumoto T: Inappropriate serum levels of IGF-I and IGFBP-3 in patients with rheumatoid arthritis. Rheumatology (Oxford). 2002, 41: 352-353. 10.1093/rheumatology/41.3.352.
Neidel J: Changes in systemic levels of insulin-like growth factors and their binding proteins in patients with rheumatoid arthritis. Clin Exp Rheumatol. 2001, 19: 81-84.
Tavera C, Abribat T, Reboul P, Dore S, Brazeau P, Pelletier JP, Martel-Pelletier J: IGF and IGF-binding protein system in the synovial fluid of osteoarthritic and rheumatoid arthritic patients. Osteoarthritis Cartilage. 1996, 4: 263-274. 10.1016/S1063-4584(05)80104-9.
Lemmey A, Maddison P, Breslin A, Cassar P, Hasso N, McCann R, Whellams E, Holly J: Association between insulin-like growth factor status and physical activity levels in rheumatoid arthritis. J Rheumatol. 2001, 28: 29-34.
Grennan DM, Sanders PA, Thomson W, Dyer PA: Rheumatoid arthritis: inheritance and association with other autoimmune diseases. Dis Markers. 1986, 4: 157-162.
Bianchi G, Marchesini G, Zoli M, Falasconi MC, Iervese T, Vecchi F, Magalotti D, Ferri S: Thyroid involvement in chronic inflammatory rheumatological disorders. Clin Rheumatol. 1993, 12: 479-484. 10.1007/BF02231775.
Wellby ML, Kennedy JA, Pile K, True BS, Barreau P: Serum interleukin-6 and thyroid hormones in rheumatoid arthritis. Metabolism. 2001, 50: 463-467. 10.1053/meta.2001.21034.
Andonopoulos AP, Siambi V, Makri M, Christofidou M, Markou C, Vagenakis AG: Thyroid function and immune profile in rheumatoid arthritis. A controlled study. Clin Rheumatol. 1996, 15: 599-603. 10.1007/BF02238551.
Shiroky JB, Cohen M, Ballachey ML, Neville C: Thyroid dysfunction in rheumatoid arthritis: a controlled prospective survey. Ann Rheum Dis. 1993, 52: 454-456. 10.1136/ard.52.6.454.
Baerwald C, Graefe C, von Wichert P, Krause A: Decreased density of beta-adrenergic receptors on peripheral blood mononuclear cells in patients with rheumatoid arthritis. J Rheumatol. 1992, 19: 204-210.
Baerwald CG, Laufenberg M, Specht T, von Wichert P, Burmester GR, Krause A: Impaired sympathetic influence on the immune response in patients with rheumatoid arthritis due to lymphocyte subset-specific modulation of beta 2-adrenergic receptors. Br J Rheumatol. 1997, 36: 1262-1269. 10.1093/rheumatology/36.12.1262.
Levine JD, Fye K, Heller P, Basbaum AI, Whiting-O'Keefe Q: Clinical response to regional intravenous guanethidine in patients with rheumatoid arthritis. J Rheumatol. 1986, 13: 1040-1043.
Lombardi MS, Kavelaars A, Schedlowski M, Bijlsma JW, Okihara KL, Van de Pol M, Ochsmann S, Pawlak C, Schmidt RE, Heijnen CJ: Decreased expression and activity of G-protein-coupled receptor kinases in peripheral blood mononuclear cells of patients with rheumatoid arthritis. FASEB J. 1999, 13: 715-725.
Trang LE, Lovgren O, Roch-Norlund AE, Horn RS, Walaas O: Cyclic nucleotides in joint fluid in rheumatoid arthritis and in Reiter's syndrome. Scand J Rheumatol. 1979, 8: 91-96. 10.3109/03009747909105343.
Miller LE, Justen HP, Scholmerich J, Straub RH: The loss of sympathetic nerve fibers in the synovial tissue of patients with rheumatoid arthritis is accompanied by increased norepinephrine release from synovial macrophages. FASEB J. 2000, 14: 2097-2107. 10.1096/fj.99-1082com.
Pereira da Silva JA, Carmo-Fonseca M: Peptide containing nerves in human synovium: immunohistochemical evidence for decreased innervation in rheumatoid arthritis. J Rheumatol. 1990, 17: 1592-1599.
Nishioka T, Kurokawa H, Takao T, Kumon Y, Nishiya K, Hashimoto K: Differential changes of corticotropin releasing hormone (CRH) concentrations in plasma and synovial fluids of patients with rheumatoid arthritis (RA). Endocr J. 1996, 43: 241-247. 10.1507/endocrj.43.241.
Ekman R, Servenius B, Castro MG, Lowry PJ, Cederlund AS, Bergman O, Sjogren HO: Biosynthesis of corticotropin-releasing hormone in human T-lymphocytes. J Neuroimmunol. 1993, 44: 7-13. 10.1016/0165-5728(93)90262-W.
Stein A, Yassouridis A, Szopko C, Helmke K, Stein C: Intraarticular morphine versus dexamethasone in chronic arthritis. Pain. 1999, 83: 525-532. 10.1016/S0304-3959(99)00156-6.
Levine JD, Clark R, Devor M, Helms C, Moskowitz MA, Basbaum AI: Intraneuronal substance P contributes to the severity of experimental arthritis. Science. 1984, 226: 547-549. 10.1126/science.6208609.
Webster EL, Barrientos RM, Contoreggi C, Isaac MG, Ligier S, Gabry KE, Chrousos GP, McCarthy EF, Rice KC, Gold PW, Sternberg EM: Corticotropin releasing hormone (CRH) antagonist attenuates adjuvant induced arthritis: role of CRH in peripheral inflammation. J Rheumatol. 2002, 29: 1252-1261.
Cutolo M, Balleari E, Giusti M, Intra E, Accardo S: Androgen replacement therapy in male patients with rheumatoid arthritis. Arthritis Rheum. 1991, 34: 1-5. 10.1002/art.1780340102.
van Vollenhoven RF, Engleman EG, McGuire JL: Dehydroepiandrosterone in systemic lupus erythematosus. Results of a double-blind, placebo-controlled, randomized clinical trial. Arthritis Rheum. 1995, 38: 1826-1831. 10.1002/art.1780381216.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None declared.
Rights and permissions
About this article
Cite this article
Eskandari, F., Webster, J.I. & Sternberg, E.M. Neural immune pathways and their connection to inflammatory diseases. Arthritis Res Ther 5, 251 (2003). https://doi.org/10.1186/ar1002
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar1002