
Abstract
The pathology of Alzheimer's disease (AD) is characterized by amyloid plaques (aggregates of amyloid-β (Aβ)) and neurofibrillary tangles (aggregates of tau) and is accompanied by mitochondrial dysfunction, but the mechanisms underlying this dysfunction are poorly understood. In this review, we discuss the critical role of mitochondria and the close inter-relationship of this organelle with the two main pathological features in the pathogenic process underlying AD. Moreover, we summarize evidence from AD post-mortem brain as well as cellular and animal AD models showing that Aβ and tau protein trigger mitochondrial dysfunction through a number of pathways, such as impairment of oxidative phosphorylation, elevation of reactive oxygen species production, alteration of mitochondrial dynamics, and interaction with mitochondrial proteins. A vicious cycle as well as several vicious circles within the cycle, each accelerating the other, can be drawn, emphasizing the synergistic deterioration of mitochondria by tau and Aβ.
Similar content being viewed by others

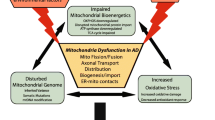

Introduction
With the increasing average lifespan of humans, Alzheimer's disease (AD) is the most common neurodegenerative disorder among elderly individuals. It accounts for up to 80% of all dementia cases and ranks as the fourth leading cause of death amongst those above 65 years of age [1]. Although the hallmark lesions of the disease were already described by Alois Alzheimer in 1906 - amyloid-β (Aβ)-containing plaques and microtubule-associated protein tau-containing neurofibrillary tangles (NFTs) - the underlying molecular mechanisms that cause the formation of these end-stage lesions are not known [2]. Moreover, as only a small fraction of AD is caused by autosomal dominant mutations, this comes down to a question of what is causing the prevalent sporadic cases in the first place. A growing body of evidence supports mitochondrial dysfunction as a prominent and early, chronic oxidative stress-associated event that contributes to synaptic abnormalities and, ultimately, selective neuronal degeneration in AD [3–9]. Is oxidative stress accelerating the NFT and Aβ pathologies, are these lesions causing oxidative stress themselves, or are there other mechanisms involved? Within the past few years, several cell culture models as well as single, double and, more recently, triple transgenic mouse models have been developed that reproduce diverse aspects of AD. These models help in understanding the pathogenic mechanisms that lead to mitochondrial failure in AD, and in particular the interplay of AD-related cellular modifications within this process [10].
Mitochondria: paradoxical organelles
Mitochondria play a pivotal role in cell survival and death by regulating both energy metabolism and apoptotic pathways (Figure 1); they contribute to many cellular functions, including intracellular calcium homeostasis, the alteration of the cellular reduction-oxidation potential, cell cycle regulation and synaptic plasticity [11]. They are the 'powerhouses of cells', providing energy via ATP generation, which is accomplished through oxidative phosphorylation from nutritional sources [12]. Neurons have particularly high numbers of mitochondria, and they are especially enriched in synapses. Due to their limited glycolytic capacity, neurons are highly dependent on mitochondrial function for energy production [13]. However, when mitochondria fulfil their physiological function, it is as if Pandora's box has been opened, as this vital organelle contains potentially harmful proteins and biochemical reaction centres: mitochondria are the major producers of reactive oxygen species (ROS) and at the same time targets of ROS toxicity. These include mitochondrial DNA, lipids of the mitochondrial membrane, and mitochondrial proteins. Dysregulation of mitochondrial function because of these insults leads to synaptic stress, disruption of synaptic transmission, apoptosis and, ultimately, neurodegeneration [14, 15]. Thus, it is important to understand the mechanisms of mitochondrial stress related to the pathogenesis of AD and to exploit this insight for developing therapeutic strategies for AD.

Amyloid-β-related mitochondrial impairment. Mitochondria were found to be the target for amyloid-β (Aβ), which interacts with several proteins, leading to mitochondrial dysfunction. Indeed, Aβ was found in the outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM) as well as in the matrix. The interaction of Aβ with the OMM affects the transport of nuclear-encoded mitochondrial proteins, such as subunits of the electron transport chain complex IV, into the organelle via the translocase of the outer membrane (TOM) import machinery. Moreover, Aβ disturbs the activity of several enzymes, such as pyruvate dehydrogenase (PDH) and α-ketoglutarate dehydrogenase (αKGDH), decreasing NADH reduction, and the electron transport chain enzyme complex IV, reducing the amount of hydrogen that is translocated from the matrix to the intermembrane space (IMS), thus impairing the mitochondrial membrane potential (MMP). Taken together, these events cause abnormal mitochondrial electron activities, leading to decreased complex V activity and so to a drop in ATP levels, in addition to increasing reactive oxygen species (ROS) generation. Moreover, ROS induce peroxidation of several mitochondrial macromolecules, such as mitochondrial DNA (mtDNA) and mitochondrial lipids, contributing to mitochondrial impairment in the mitochondrial matrix. The complex of Aβ bound to binding alcohol dehydrogenase (ABAD) impairs the binding of NAD+ to ABAD, changes mitochondrial membrane permeability and reduces activities of respiratory enzymes, inducing further ROS production and leading to mitochondrial failure. Aβ binding also activates Fis1 (fission protein) and promotes increased mitochondrial fragmentation; this increased mitochondrial fragmentation produces defective mitochondria that ultimately damage neurons. Furthermore, Aβ binding to cyclophilin D (CypD) enhances the protein translocation to the inner membrane, favouring the opening of the mitochondrial permeability transition pore, formed by the adenine nucleotide translocator (ANT) and voltage-dependent anion channels (VDACs). Cyt C, cytochrome C; DLP1, dynamin-like protein 1; PDH, pyruvate dehydrogenase; ProAp, proapoptotic factors; SOD, superoxide dismutase; TCA, tricarboxylic acid; TIM, translocase of the inner membrane.
Evidence of mitochondrial dysfunction in post-mortem AD brain and peripheral cells
Mitochondrial dysfunction has been proposed as an underlying mechanism in the early stages of AD since energy deficiency is a fundamental characteristic of the AD brain [16, 17] as well as of peripheral cells derived from AD patients [18]. The similar findings in peripheral tissue highlight the potential of using surrogate tissue in the diagnosis of AD (Figure 2).

Mitochondrial impairment in the brain and peripheral cells of Alzheimer's disease patients. AD, Alzheimer's disease; ETC, electron transport chain; ROS, reactive oxygen species; TCA, tricarboxylic acid.
Brain imaging studies have demonstrated defects in glucose use in living AD patients, an abnormality that may occur well before the onset of clinical symptoms [17]. Furthermore, several lines of evidence suggest that mitochondria intervene in the mechanism by which Aβ, which is derived from amyloid precursor protein (APP), triggers synaptic failure and neurodegeneration. Crucial to this finding was the notion that both Aβ and the full length protein, APP, accumulate within mitochondria in the brains of AD patients [6, 19–21].
Furthermore, morphometric analyses revealed that mitochondria display structural abnormalities, as illustrated by the accumulation of mitochondrial DNA and proteins in the cytoplasm and vacuoles; this accelerates mitochondrial degradation and thereby leads to a significant decrease in mitochondrial numbers in AD [22]. The mitochondrial accumulation of APP and Aβ in brain tissues causes both altered activities in key mitochondrial enzymes, including cytochrome c oxidase (COX, complex IV), pyruvate dehydrogenase (PDH) and α-ketoglutarate dehydrogenase complex (αKGDH), and import of nuclear-encoded proteins [3, 18, 21, 23]. The activity of αKGDH, but not of PDH, has been found to be reduced in cultured skin fibroblasts from sporadic AD patients and in a subset of patients with presenilin-1 mutations. Sorbi and colleagues [24] observed a reduction in PHD, ATP-citrate lyase and acetoacetyl-CoA thiolase in post-mortem brain tissues from AD-affected subjects and correlated the decrease of these enzymes to the decreased production of acetyl-coenzyme A and cholinergic defects that are observed in AD.
The impairment of mitochondrial oxidative phosphorylation in the AD brain was found to be proportional to clinical disability [3]. The most consistent defect in mitochondrial electron transport enzymes in AD is a deficiency in COX, a finding reported in AD post-mortem brain as well as in platelets and fibroblasts from AD patients [25, 26]. Dysfunction of COX increases ROS production, reduces energy stores, and disturbs energy metabolism [27]. Altered levels of antioxidant enzymes, such as catalase, copper/zinc superoxide dismutase (Cu/Zn-SOD) and manganese superoxide dismutase (Mn-SOD) have been found in AD brain, supporting increased oxidative stress as a pathomechanism in AD [28].
Laser-capture microdissection has been applied to investigate distinct brain areas as some undergo neurodegeneration whereas others do not: the analysis of the expression of 80 metabolically relevant nuclear-encoded electron transport chain genes from non-NFT-bearing AD neurons revealed that 60 to 70% of these were significantly lower in metabolically affected areas such as the posterior cingulate cortex, the middle temporal gyrus, and the hippocampal CA1 region [29]. Post-mortem tissue also provides strong evidence for increased levels of cellular oxidative stress in vulnerable regions of AD brains compared to aged controls [30]. Increased protein oxidation, protein nitration, and lipid peroxidation were detected in brain areas with overt NFTs and amyloid plaques [31].
More recently, abnormal mitochondrial dynamics has been identified in fibroblasts from sporadic and familial AD cases [32], a distortion probably mediated by altered expression of dynamin-like protein 1 (DLP1), a regulator of mitochondrial fission and distribution, due to elevated oxidative and/or Aβ-induced stress. Moreover, deregulated mitochondrial dynamics might be due to enhanced nitrosative stress generated by Aβ, such as S-nitrosylation of DLP1 in AD brain [33]. This modification can disturb the balance between fission and fusion of mitochondria in favour of mitochondrial fission followed by mitochondrial depletion from axons and dendrites and, subsequently, synaptic loss.
Altogether, these data indicate that mitochondrial dysfunction is a highly relevant event in AD patho-physiology. Post-mortem studies in AD brain allow the identification of long-term synergistic in vivo effects of tau and Aβ in human because, in the majority of AD cases, both pathologies persist in parallel for many years.
Mitochondrial dysfunction in cellular and transgenic mouse AD models
Understanding the molecular pathways by which the various pathological alterations including Aβ and tau compromise neuronal integrity and lead to clinical symptoms has been a long-standing goal of AD research. The successful development of cell and mouse models that mimic diverse aspects of the disease process have facilitated this effort and assisted in understanding of the interplay of Aβ and tau on bioenergetics processes in vivo [10] (Figure 3).

Mitochondrial impairment in Alzheimer's disease cell culture and transgenic mice models. The successful development of Alzheimer's disease (AD) cell culture models as well as single, double and triple transgenic mice models that mimic diverse aspects of the disease have been helpful in understanding the pathogenic mechanisms on bioenergetic processes in vivo, particularly the interplay of AD-related amyloid-β (Aβ) and tau proteins, leading to the mitochondrial failures underlying AD. APP, amyloid precursor protein; LTP, long-term potentiation; MMP, mitochondrial membrane potential; NO, nitric oxide; PDH, pyruvate dehydrogenase; ROS, reactive oxygen species.
Mitochondria and Aβ
Aβ may exert its toxicity via a plethora of pathways and thereby induce synaptic and neuronal degeneration [2].
Using neuronal PC12 cells, we found that expression of one known APP gene mutation, the 'Swedish APP (APPSw)' double mutation (KM670/671NL), leads to an enhanced vulnerability of these cells to oxidative stress and mitochondrial dysfunction, mediated by different caspases and the stress-activated protein kinase pathway [34–36] (Figure 3). Of note, we are able to study the effects of Aβ in a dose-dependent manner as cells transfected with APPSw had fivefold higher Aβ levels than wild-type APP (APPwt) transfected cells, similar to the increase found in human carriers with the APPSw mutation compared to sporadic AD cases. The data suggest that the mitochondrial dysfunction induced by Aβ is probably mediated via enhanced nitric oxide production that reduces complex IV activity. This reduction leads to a depletion of intracellular ATP levels, finally initiating cell death [37]. When intracellular Aβ production is prevented by a γ-secretase inhibitor, this restores nitric oxide and ATP levels, indicating a direct involvement of Aβ in these mechanisms.
Mitochondria were found to be a target for APP toxicity as both the full-length protein and Aβ accumulate in the mitochondrial import channels, and both lead to mitochondrial dysfunction. Recently, APP has been demonstrated as a substrate of the mitochondrial γ-secretase in cultured SH-SY5Y human neuroblastoma cells [38]. Pavlov and colleagues [38] proposed that APP processing in mitochondria may lead to local Aβ production, thereby contributing to mitochondrial dysfunction. Recently, our group investigated specific effects of Aβ on mitochondrial function under physiological conditions by analysing mitochondrial respiratory function and energy homeostasis in both control and APPwt-expressing SH-SY5Y cells [8]. One possibility as to how chronic exposure to soluble Aβ may result in an impairment of energy homeostasis is through decreased respiratory capacity of the mitochondrial respiratory chain, mainly of complex IV, which, in turn, may accelerate neuronal demise.
Eventually, APP, through Aβ production, causes an imbalance of mitochondrial fission/fusion that results in mitochondrial fragmentation and abnormal distribution, which contributes to mitochondrial and neuronal dysfunction [39].
An interesting observation was made when the toxic effects of Aβ were compared with human amylin (HA), a protein aggregating in the pancreas of type 2 diabetic patients [40]. Both agents share amyloidogenic properties and are toxic to primary neuronal cultures, whereas the non-amyloidogenic rat amylin is not [41]. In a next step, HA and Aβ toxicity to SH-SY5Y neuroblastoma cells was assessed by iTRAQ quantitative proteomics [42]. This revealed the surprising observation that identical proteins are deregulated by HA and Aβ, but not rat amylin, and a quarter of these were mitochondrial, supporting the notion that mitochondrial dysfunction is a common target in these two amyloidoses. Functional validation revealed that Aβ and HA both exert a shared toxicity, at least in part, via deregulation of identical mitochondrial proteins and impairment of complex IV activity [42].
AD is believed to be a disease of the synapses, characterized by massive synaptic and, eventually, neuronal loss, and these features have been detected in AD transgenic mouse models (Figure 3). A decrease of the mitochondrially localized chaperone hsp70, which stabilizes proteins during their import into mitochondria, has been observed in synaptosomal fractions derived from the APPSw transgenic strain Tg2576, pointing to a mitochondrial stress response [43]. Significant differences were revealed in the protein subunit composition of respiratory chain complexes I and III. Moreover, endogenous Aβ was found to be associated with brain mitochondria in these mice. Cu2+ ions have been shown to render Aβ more toxic, possibly in its dimeric conformation, as a potent inhibitor of COX, thereby contributing to neurodegeneration in AD [44]. In APPSw transgenic mice, reduced glucose metabolism was observed after [14C]-2-deoxyglucose infusion as well as impaired Cu/Zn SOD activity, both of which contribute to oxidative damage. In another transgenic mouse model, APPS/L, which combines the Swedish (K670M/N671L) with the London (V717I) mutation in the human APP gene, an early energy dysfunction was found as shown by a decreased mitochondrial membrane potential as well as decreased ATP levels at 3 months of age, when Aβ levels are elevated but plaques are not yet present [5, 14, 37]. A stronger reduction in mitochondrial membrane potential and in ATP levels was found in double transgenic APP/PS1 (APPS/L/PS1 M141L) mice, which generate higher levels of Aβ compared to their single transgenic littermates (APPS/L), and exhibit Aβ plaques already at an age of 3 months [4]. Together, this implies that an Aβ-dependent mitochondrial dysfunction starts at a very young age and accelerates substantially with increasing age, as does the Aβ plaque load [4]. Consistent with this observation, APP/PS2 (APPSw/PS2 N141I) double transgenic mice display age-related cognitive deficits associated with discrete brain Aβ deposition and inflammation [45]. Furthermore, an Aβ insult in APPS/L mice caused early deficits in synaptic mitochondria, as shown by increased mitochondrial permeability transition, a decline in both respiratory function and activity of COX, and increased mitochondrial oxidative stress [46].
Of note, age-dependent impairment of oxygen consumption, such as a decrease of state 3 and of uncoupled respiration, was observed in several APP transgenic mouse models compared to aged-matched controls [5, 14, 43, 47]. This indicates that mitochondrial deregulation is a common feature in Aβ-generating mice and independent of the mouse model used.
There is broad experimental proof that Aβ is indeed present in mitochondria. Aβ binds specifically to the mitochondrial Aβ-binding alcohol dehydrogenase (ABAD) [6], a mitochondrial matrix protein that is up-regulated in the temporal lobe of AD patients as well as in APP transgenic mice [48]. The Aβ-ABAD interaction causes elevated ROS production, cell death, as well as spatial learning and memory deficits in 5-month-old APP/ABAD double transgenic mice [49]. The investigation of the crystal structure of ABAD-Aβ demonstrated that the formation of the complex prevents the binding of NAD+ to ABAD, thereby changing the mitochondrial membrane permeability and reducing the activities of respiratory enzymes, which then may lead to mitochondrial failure [6]. Aβ in mitochondria further interacts with cyclophilin D (CypD), an integral part of the mitochondrial permeability transition pore that potentiates free radical production, causes synaptic failure, and promotes opening of the pore leading to apoptosis [50]. It has been demonstrated previously that CypD is capable of forming complexes with Aβ within mitochondria of cortical neurons from APP mutant mice, increasing the translocation of CypD from the matrix to the inner membrane. Furthermore, in APP transgenic mice, the abrogation of CypD was capable of attenuating Aβ-mediated abnormal mitochondrial dysfunction, such as calcium-induced mitochondrial swelling, and it lowered mitochondrial calcium uptake capacity and impaired mitochondrial respiratory function. Aβ impaired calcium storage in mitochondria, altering neuronal function, as it is exported to the cytosol, together with other apoptotic factors (ProAp), such as cytochrome c (Figure 1). Finally, Anandatheerthavarada and Devi [51] showed that APP contains a mitochondrial targeting sequence and that an accumulation of APP in mitochondrial membranes leads to mitochondrial dysfunction in Tg2576 neurons. Taken together, these findings are in line with the recently proposed hypothesis of an intracellular Aβ toxicity cascade, which suggests that the toxic Aβ species that cause molecular and biochemical abnormalities are in fact intracellular oligomeric aggregates rather than the extracellular, insoluble plaques [6, 20].
Mitochondria and tau protein
How does tau interfere with mitochondrial function? In its hyperphosphorylated form, tau, which forms the second hallmark lesion in AD, the NFTs, has been shown to block mitochondrial transport, which results in energy deprivation and oxidative stress at the synapse and, hence, neurodegeneration [52–55]. In transgenic pR5 mice that overexpress human P301L mutant tau [56], the biochemical consequences of tau pathology have been intensively investigated using proteomics followed by functional validation [57, 58]. A mass-spectrometric analysis of the brain proteins from these mice revealed mainly a deregulation of mitochondrial respiratory chain complex components (including complex V), antioxidant enzymes, and synaptic proteins (Figure 4). The functional analysis demonstrated a mitochondrial dysfunction in the mice, together with reduced NADH-ubiquinone oxidoreductase (complex I) activity and, with age, impaired mitochondrial respiration and ATP synthesis. Mitochondrial dysfunction was associated with higher levels of ROS in aged transgenic mice. Increased tau pathology as in aged homozygous pR5 mice revealed modified lipid peroxidation levels and the upregulation of antioxidant enzymes in response to oxidative stress [57]. Thus, this evidence demonstrated for the first time that not only Aβ but also tau pathology can lead to metabolic impairment and oxidative stress as in AD.

Differential expression of ascertained subunits of the electron transport chain. A quantitative mass-tag labelling proteomic technique, iTRAQ, and mass-spectrometric analysis of the brain proteins from single, double and triple transgenic mouse models revealed massive deregulation of 24 components mainly related to the mitochondrial respiratory chain complex (including complex V), antioxidant enzymes, and synaptic proteins. Functional analysis validated the proteomic approach by confirming the strongest defects of the respiratory capacity mainly at complexes I, IV and V in transgenic mice, at both the protein and activity level. Significantly changed expression of nuclear-encoded (Ndufs2, Ndufs8, Ndufb10, IV, Va, Vb, VIIa and D) and mitochondrial-encoded (COX II) complex subunits are highlighted in red. Complex I subunits: Ndufs2, NADH dehydrogenase (ubiquinone) Fe-S protein 2; Ndufs8, NADH dehydrogenase (ubiquinone) Fe-S protein 8; Ndufb10, NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 10. Complex IV subunits: II, Cytochrome c oxidase subunit 2; IV, Cytochorme c oxidase subunit IV isoform 1; Va, Cytochrome c oxidase polypeptide Va; Vb, Cytochrome c oxidase polypeptide Va; VIIa, Cytochrome c oxidase polypeptide VIIa-liver/heart. Complex V subunits: D, ATP synthase D chain. IMM, inner mitochondrial membrane; IMS, intermembrane space.
Consistent with observations of a cytosolic accumulation of the α-chain of ATP synthase observed at early stages of neurofibrillary degeneration in AD, one mechanism proposed is that tau accumulation could have direct consequences on mitochondrial activity through the cytosolic accumulation of the α-chain of ATP synthase. Reciprocally, hyperphosphorylation of tau may be directly attributable to mitochondrial oxidative stress in a mouse model lacking the mitochondrial detoxifying enzyme superoxide dismutase 2 (Sod2-/-), consistent with a synergistic interaction of APP and mitochondrial oxidative stress in contributing to an AD-like neocortical pathology [59]. Furthermore, chronic respiratory chain dysfunction through inhibition of complex I led, besides a concentration-dependent decrease in ATP levels, to a redistribution of tau from the axon to the cell body, the retrograde transport of mitochondria and, finally, cell death [60]. Together, these findings support the notion that tau pathology involves a mitochondrial and oxidative stress disorder possibly distinct from that caused by Aβ.
Pathogenic convergence of Aβ and tau on mitochondria
Although Aβ and tau pathologies are both known hall-marks in AD, the underlying mechanisms of the interplay between plaques and NFTs (or Aβ and tau) have remained unresolved. However, a close relationship between mitochondrial impairment and Aβ on the one hand and tau on the other has been established. How do both features in AD relate to each other and might the two molecules synergistically affect mitochondrial integrity? Several studies suggest that Aβ aggregates and hyperphosphorylated tau may block mitochondrial carriage to the synapse, leading to energy defects and neurodegeneration [61]. Moreover, transport of APP into axons and dendrites may be inhibited by enhanced tau levels, causing impaired axonal transport, which suggests a link between tau and APP [52, 55]. Remarkably, Aβ injections amplify a pre-existing tau pathology in several transgenic mouse models [56, 62, 63], whereas lack of tau abrogates Aβ toxicity [54, 64]. Our findings indicate that mitochondria in tau transgenic pR5 mice display enhanced vulnerability towards Aβ insult in vitro [57, 65], suggesting a synergistic action of tau and Aβ pathologies on this organelle (Figure 3). The Aβ insult caused a greater reduction of mitochondrial membrane potential in cerebral cells from pR5 mice [57]. Furthermore, incubation of isolated mitochondria from pR5 mice with either oligomeric or fibrillar Aβ preparations resulted in impairment of the mitochondrial membrane potential and of respiration. Interestingly, aging particularly increased the sensitivity of mitochondria to oligomeric Aβ insult compared to that of fibrillar Aβ. This suggests that while both oligomeric and fibrillar Aβ are toxic, they exert different degrees of toxicity [65]. In a related study, Crouch and colleagues [66] demonstrated that increasing the bioavailability of intracellular copper can restore cognitive function by inhibiting the accumulation of neurotoxic Aβ trimers and phosphorylated tau in APPSw/PS1 transgenic mice. In yet another study, it was shown that exposure to Aβ induces tau hyperphosphorylation by promoting glycogen synthase kinase (GSK)3β activity. This demonstrates an intimate relationship between Aβ accumulation and abnormal tau phosphorylation in causing the cognitive deficits that characterize AD, and highlights GSK3β activity as an important intermediate [67]. In contrast, overexpression of the longest form of human wild-type tau (tau441) in mouse neuroblastoma (N2A) showed an anti-apoptotic protective function of tau phosphorylation, which likely inhibited competitively phosphorylation of β-catenin by GSK-3β, facilitating the function of β-catenin. Thus, overexpression of tau seems to attenuate Aβ-mediated cell death via suppression of the mitochondria-caspase-3 pathway [68, 69]. Taken together, these studies illustrate a complex interplay between the two key proteins in AD.
In a step further, triple transgenic mouse models have recently been established that combine Aβ and tau pathologies, and the contributions of both AD-related proteins to the mitochondrial respiratory machinery and energy homeostasis have been investigated in vivo (Figure 3). Indeed, our group demonstrated a mitochondrial dysfunction in a novel triple transgenic mouse model (pR5/APPSw/PS2 N141I) - tripleAD mice - using proteomics followed by functional validation [9]. Notably, deregulation of complex I was found to be tau-dependent, while deregulation of complex IV was Aβ-dependent, at both the protein and activity level (Figure 5). Whereas down-regulation of several subunits of complex I was observed in tripleAD mice compared to both pR5 and APPSw/PS2 mice, deregulation of several subunits of complex V was seen in tripleAD mice compared to pR5 mice but not compared to APPSw/PS2 mice (Figure 4). The convergent effects of Aβ and tau led already at the age of 8 months to depolarized mitochondrial membrane potential in tripleAD mice. Additionally, we found that age-related oxidative stress at 12 months of age may exaggerate the disturbances in the respiratory system and synthesis of ATP and, in turn, take part in the vicious cycle that finally leads to cell death.

Impairment of the electron transport chain in Alzheimer's disease. Complexes I (NADH:ubiquinone oxidoreductase) and II (succinate dehydrogenase, belongs to the tricarboxylic acid (TCA) cycle) receive electrons from NADH and FADH2, respectively. Electrons are then driven from complexes by the mobile carrier molecules coenzyme Q/ubiquinone (UQ) and cytochrome c (Cyt c) to the final acceptor, molecular oxygen (O2). Electron flow is coupled to proton movement across the inner mitochondrial membrane (IMM) in complexes I, III and IV. The resulting proton gradient is harvested by complex V to generate ATP. In Alzheimer's disease, abnormal mitochondrial electron activities have been observed, predominantly in complexes I and IV, leading to impaired mitochondrial membrane potential, decreased production of ATP (complex V) and enhanced reactive oxygen species (ROS) levels. Interestingly, deregulation of complex I is mainly tau-dependent, while deregulation of complex IV is amyloid-β (Aβ)-dependent, at both the protein and activity level. IMS, intermembrane space.
Together, our studies highlight the key role of mitochondria in AD pathogenesis, and the close interrelationship of this organelle and the two main pathological hallmarks of this disease. Our data complement those obtained in another triple transgenic mouse model, 3xTg-AD (P301Ltau/APPSw/PS1 M146L) [70]. Mitochondrial dysfunction was evidenced by age-related decreased activity of regulatory enzymes of the oxidative phosphorylation system, and of PDH and COX in 3xTg-AD mice aged from 3 to 12 months [71]. These mice also exhibited increased oxidative stress and lipid peroxidation. Most of the effects on mitochondria were seen at the age of 9 months, whereas mitochondrial respiration was significantly decreased at 12 months of age. Importantly, mitochondrial bioenergetics deficits were found to precede the development of AD pathology in the 3xTg-AD mice. More recently, Hyun and colleagues [72] demonstrated that the plasma membrane redox system (PMRS) is impaired in the 3xTg-AD mice and that the activities of PMRS enzymes may protect neurons against Aβ toxicity, suggesting enhancement of PMRS function as a novel approach for protecting neurons against oxidative damage in AD and related disorders. Collectively, these recent data consolidate the idea that a synergistic effect of tau and Aβ augments the pathological deterioration of mitochondria at an early stage of AD.
Conclusion
We discuss here the critical role of mitochondria and the close interrelationship of this organelle with the two main pathological features in the pathogenic process underlying AD.
Mitochondrial impairment has been recognized as a prominent and early event in AD in terms of metabolic energy production associated with respiratory chain dysfunction and the regulation of levels of second messengers such as ROS or Ca2+. This is partially due to either accumulated damage to mitochondrial DNA or direct harmful effects of oxidative stress and/or Aβ on mitochondrial components. Also, mitochondrial trafficking as well as mitochondrial fusion/fission are known to be altered in AD, compromising normal neurophysiology and a healthy cellular mitochondrial pool, which eventually leads to apoptosis. Specifically, the mitochondrial impairment integrates the close interplay of the two common hallmarks of AD, plaques and NFTs, or Aβ and tau, which act independently as well as synergistically on this vital organelle. With regards to the involvement of AD-related proteins in pathogenesis, a vicious cycle as well as several vicious circles within the cycle, each accelerating the other, can be drawn, emphasizing the potency of the synergistic destruction in mitochondria.
Finally, the key role of mitochondrial dysfunction in the early pathogenic pathways by which Aβ leads to neuronal dysfunction in AD is particularly challenging with respect to establishing therapeutic treatments. Besides the modulation and/or removal of both Aβ and tau pathology, strategies involving efforts to protect cells at the mitochondrial level by stabilizing or restoring mitochondrial function or by interfering with energy metabolism appear to be promising; these efforts also emphasize the importance of mitochondria in the pathogenesis of AD. Cellular models for AD as well as transgenic mice, and particularly triple transgenic models that combine both pathologies, are valuable tools in monitoring therapeutic interventions at the mitochondrial level. Eventually, this may lead to therapies that prevent the progression of Aβ deposits and tau hyper-phosphorylation at an early stage of disease.
Abbreviations
- Aβ:
-
amyloid-β
- ABAD:
-
Aβ-binding alcohol dehydrogenase
- AD:
-
Alzheimer's disease
- APP:
-
amyloid precursor protein
- APPSw :
-
Swedish amyloid precursor protein
- APPwt :
-
wild-type amyloid precursor protein
- αKGDH:
-
α-ketoglutarate dehydrogenase complex
- COX:
-
cytochrome c oxidase
- CypD:
-
cyclophilin D
- DLP1:
-
dynamin-like protein 1
- GSK:
-
glycogen synthase kinase
- HA:
-
human amylin
- NFT:
-
neurofibrillary tangle
- PDH:
-
pyruvate dehydrogenase
- PMRS:
-
plasma membrane redox system
- ROS:
-
reactive oxygen species
- SOD:
-
superoxide dismutase.
References
Nussbaum RL, Ellis CE: Alzheimer's disease and Parkinson's disease. N Engl J Med. 2003, 348: 1356-1364. 10.1056/NEJM2003ra020003.
Gotz J, Ittner LM: Animal models of Alzheimer's disease and frontotemporal dementia. Nat Rev Neurosci. 2008, 9: 532-544.
Blass JP: Cerebrometabolic abnormalities in disease. Neurol Res. 2003, 25: 556-566. 10.1179/016164103101201995.
Eckert A, Hauptmann S, Scherping I, Rhein V, Muller-Spahn F, Gotz J, Muller WE: Soluble beta-amyloid leads to mitochondrial defects in amyloid precursor protein and tau transgenic mice. Neurodegener Dis. 2008, 5: 157-159. 10.1159/000113689.
Hauptmann S, Scherping I, Drose S, Brandt U, Schulz KL, Jendrach M, Leuner K, Eckert A, Muller WE: Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging. 2009, 30: 1574-1586. 10.1016/j.neurobiolaging.2007.12.005.
Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H: ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004, 304: 448-452. 10.1126/science.1091230.
Moreira PI, Santos MS, Oliveira CR: Alzheimer's disease: a lesson from mitochondrial dysfunction. Antioxid Redox Signal. 2007, 9: 1621-1630. 10.1089/ars.2007.1703.
Rhein V, Baysang G, Rao S, Meier F, Bonert A, Muller-Spahn F, Eckert A: Amyloid-beta leads to impaired cellular respiration, energy production and mitochondrial electron chain complex activities in human neuroblastoma cells. Cell Mol Neurobiol. 2009, 29: 1063-1071. 10.1007/s10571-009-9398-y.
Rhein V, Song X, Wiesner A, Ittner LM, Baysang G, Meier F, Ozmen L, Bluethmann H, Dröse S, Brandt U, Savaskan E, Czech C, Götz J, Eckert A: Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer's disease mice. Proc Natl Acad Sci USA. 2009, 106: 20057-20062.
Eckert A, Schulz KL, Rhein V, Gotz J: Convergence of amyloid-beta and tau pathologies on mitochondria in vivo. Mol Neurobiol. 2010, 41: 107-114. 10.1007/s12035-010-8109-5.
Mattson MP, Gleichmann M, Cheng A: Mitochondria in neuroplasticity and neurological disorders. Neuron. 2008, 60: 748-766. 10.1016/j.neuron.2008.10.010.
Scheffler IE: A century of mitochondrial research: achievements and perspectives. Mitochondrion. 2001, 1: 3-31. 10.1016/S1567-7249(00)00002-7.
Reddy PH: Mitochondrial dysfunction in aging and Alzheimer's disease: strategies to protect neurons. Antioxid Redox Signal. 2007, 9: 1647-1658. 10.1089/ars.2007.1754.
Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD: Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J. 2005, 19: 2040-2041.
Santos RX, Correia SC, Wang X, Perry G, Smith MA, Moreira PI, Zhu X: Alzheimer's disease: diverse aspects of mitochondrial malfunctioning. Int J Clin Exp Pathol. 2010, 3: 570-581.
Manczak M, Park BS, Jung Y, Reddy PH: Differential expression of oxidative phosphorylation genes in patients with Alzheimer's disease: implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med. 2004, 5: 147-162. 10.1385/NMM:5:2:147.
Mosconi L, Pupi A, De Leon MJ: Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer's disease. Ann N Y Acad Sci. 2008, 1147: 180-195. 10.1196/annals.1427.007.
Gibson GE, Sheu KF, Blass JP: Abnormalities of mitochondrial enzymes in Alzheimer disease. J Neural Transm. 1998, 105: 855-870. 10.1007/s007020050099.
Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK: Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006, 26: 9057-9068. 10.1523/JNEUROSCI.1469-06.2006.
Fernandez-Vizarra P, Fernandez AP, Castro-Blanco S, Serrano J, Bentura ML, Martinez-Murillo R, Martinez A, Rodrigo J: Intra- and extracellular Abeta and PHF in clinically evaluated cases of Alzheimer's disease. Histol Histopathol. 2004, 19: 823-844.
Pavlov PF, Hansson Petersen C, Glaser E, Ankarcrona M: Mitochondrial accumulation of APP and Abeta: significance for Alzheimer disease pathogenesis. J Cell Mol Med. 2009, 13: 4137-4145. 10.1111/j.1582-4934.2009.00892.x.
Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA: Mitochondrial abnormalities in Alzheimer's disease. J Neurosci. 2001, 21: 3017-3023.
Casley CS, Canevari L, Land JM, Clark JB, Sharpe MA: Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem. 2002, 80: 91-100.
Sorbi S, Bird ED, Blass JP: Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann Neurol. 1983, 13: 72-78. 10.1002/ana.410130116.
Cardoso SM, Proenca MT, Santos S, Santana I, Oliveira CR: Cytochrome c oxidase is decreased in Alzheimer's disease platelets. Neurobiol Aging. 2004, 25: 105-110. 10.1016/S0197-4580(03)00033-2.
Kish SJ, Bergeron C, Rajput A, Dozic S, Mastrogiacomo F, Chang LJ, Wilson JM, DiStefano LM, Nobrega JN: Brain cytochrome oxidase in Alzheimer's disease. J Neurochem. 1992, 59: 776-779. 10.1111/j.1471-4159.1992.tb09439.x.
Mutisya EM, Bowling AC, Beal MF: Cortical cytochrome oxidase activity is reduced in Alzheimer's disease. J Neurochem. 1994, 63: 2179-2184.
Aksenov MY, Tucker HM, Nair P, Aksenova MV, Butterfield DA, Estus S, Markesbery WR: The expression of key oxidative stress-handling genes in different brain regions in Alzheimer's disease. J Mol Neurosci. 1998, 11: 151-164. 10.1385/JMN:11:2:151.
Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Ramsey K, Caselli RJ, Kukull WA, McKeel D, Morris JC, Hulette CM, Schmechel D, Reiman EM, Rogers J, Stephan DA: Altered neuronal gene expression in brain regions differentially affected by Alzheimer's disease: a reference data set. Physiol Genomics. 2008, 33: 240-256. 10.1152/physiolgenomics.00242.2007.
Moreira PI, Santos MS, Oliveira CR, Shenk JC, Nunomura A, Smith MA, Zhu X, Perry G: Alzheimer disease and the role of free radicals in the pathogenesis of the disease. CNS Neurol Disord Drug Targets. 2008, 7: 3-10. 10.2174/187152708783885156.
Perry G, Nunomura A, Hirai K, Takeda A, Aliev G, Smith MA: Oxidative damage in Alzheimer's disease: the metabolic dimension. Int J Dev Neurosci. 2000, 18: 417-421. 10.1016/S0736-5748(00)00006-X.
Wang X, Su B, Fujioka H, Zhu X: Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer's disease patients. Am J Pathol. 2008, 173: 470-482. 10.2353/ajpath.2008.071208.
Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA: S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009, 324: 102-105. 10.1126/science.1171091.
Eckert A, Steiner B, Marques C, Leutz S, Romig H, Haass C, Muller WE: Elevated vulnerability to oxidative stress-induced cell death and activation of caspase-3 by the Swedish amyloid precursor protein mutation. J Neurosci Res. 2001, 64: 183-192. 10.1002/jnr.1064.
Leutz S, Steiner B, Marques CA, Haass C, Muller WE, Eckert A: Reduction of trophic support enhances apoptosis in PC12 cells expressing Alzheimer's APP mutation and sensitizes cells to staurosporine-induced cell death. J Mol Neurosci. 2002, 18: 189-201. 10.1385/JMN:18:3:189.
Marques CA, Keil U, Bonert A, Steiner B, Haass C, Muller WE, Eckert A: Neurotoxic mechanisms caused by the Alzheimer's disease-linked Swedish amyloid precursor protein mutation: oxidative stress, caspases, and the JNK pathway. J Biol Chem. 2003, 278: 28294-28302. 10.1074/jbc.M212265200.
Keil U, Bonert A, Marques CA, Scherping I, Weyermann J, Strosznajder JB, Müller-Spahn F, Haass C, Czech C, Pradier L, Müller WE, Eckert A: Amyloid beta-induced changes in nitric oxide production and mitochondrial activity lead to apoptosis. J Biol Chem. 2004, 279: 50310-50320. 10.1074/jbc.M405600200.
Pavlov PF, Wiehager B, Sakai J, Frykman S, Behbahani H, Winblad B, Ankarcrona M: Mitochondrial gamma-secretase participates in the metabolism of mitochondria-associated amyloid precursor protein. FASEB J. 2011, 25: 78-88. 10.1096/fj.10-157230.
Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X: Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci USA. 2008, 105: 19318-19323. 10.1073/pnas.0804871105.
Gotz J, Ittner LM, Lim YA: Common features between diabetes mellitus and Alzheimer's disease. Cell Mol Life Sci. 2009, 66: 1321-1325. 10.1007/s00018-009-9070-1.
Lim YA, Ittner LM, Lim YL, Gotz J: Human but not rat amylin shares neurotoxic properties with Abeta42 in long-term hippocampal and cortical cultures. FEBS Lett. 2008, 582: 2188-2194. 10.1016/j.febslet.2008.05.006.
Lim YA, Rhein V, Baysang G, Meier F, Poljak A, Raftery MJ, Guilhaus M, Ittner LM, Eckert A, Gotz J: Abeta and human amylin share a common toxicity pathway via mitochondrial dysfunction. Proteomics. 2010, 10: 1621-1633. 10.1002/pmic.200900651.
Gillardon F, Rist W, Kussmaul L, Vogel J, Berg M, Danzer K, Kraut N, Hengerer B: Proteomic and functional alterations in brain mitochondria from Tg2576 mice occur before amyloid plaque deposition. Proteomics. 2007, 7: 605-616. 10.1002/pmic.200600728.
Crouch PJ, Blake R, Duce JA, Ciccotosto GD, Li QX, Barnham KJ, Curtain CC, Cherny RA, Cappai R, Dyrks T, Masters CL, Trounce IA: Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1-42. J Neurosci. 2005, 25: 672-679. 10.1523/JNEUROSCI.4276-04.2005.
Richards JG, Higgins GA, Ouagazzal AM, Ozmen L, Kew JN, Bohrmann B, Malherbe P, Brockhaus M, Loetscher H, Czech C, Huber G, Bluethmann H, Jacobsen H, Kemp JA: PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J Neurosci. 2003, 23: 8989-9003.
Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS: Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc Natl Acad Sci USA. 2010, 107: 18670-18675. 10.1073/pnas.1006586107.
Muller WE, Eckert A, Kurz C, Eckert GP, Leuner K: Mitochondrial dysfunction: common final pathway in brain aging and Alzheimer's disease - therapeutic aspects. Mol Neurobiol. 2010, 41: 159-171. 10.1007/s12035-010-8141-5.
Yan SD, Fu J, Soto C, Chen X, Zhu H, Al-Mohanna F, Collison K, Zhu A, Stern E, Saido T, Tohyama M, Ogawa S, Roher A, Stern D: An intracellular protein that binds amyloid-beta peptide and mediates neurotoxicity in Alzheimer's disease. Nature. 1997, 389: 689-695. 10.1038/39522.
Yao J, Taylor M, Davey F, Ren Y, Aiton J, Coote P, Fang F, Chen JX, Yan SD, Gunn-Moore FJ: Interaction of amyloid binding alcohol dehydrogenase/Abeta mediates up-regulation of peroxiredoxin II in the brains of Alzheimer's disease patients and a transgenic Alzheimer's disease mouse model. Mol Cell Neurosci. 2007, 35: 377-382. 10.1016/j.mcn.2007.03.013.
Du H, Yan SS: Mitochondrial permeability transition pore in Alzheimer's disease: cyclophilin D and amyloid beta. Biochim Biophys Acta. 2010, 1802: 198-204.
Anandatheerthavarada HK, Devi L: Mitochondrial translocation of amyloid precursor protein and its cleaved products: relevance to mitochondrial dysfunction in Alzheimer's disease. Rev Neurosci. 2007, 18: 343-354.
Ebneth A, Drewes G, Mandelkow EM, Mandelkow E: Phosphorylation of MAP2c and MAP4 by MARK kinases leads to the destabilization of microtubules in cells. Cell Motil Cytoskeleton. 1999, 44: 209-224. 10.1002/(SICI)1097-0169(199911)44:3<209::AID-CM6>3.0.CO;2-4.
Gotz J, Ittner LM, Fandrich M, Schonrock N: Is tau aggregation toxic or protective: a sensible question in the absence of sensitive methods?. J Alzheimers Dis. 2008, 14: 423-429.
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J: Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010, 142: 387-397. 10.1016/j.cell.2010.06.036.
Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM: Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002, 156: 1051-1063. 10.1083/jcb.200108057.
Gotz J, Chen F, van Dorpe J, Nitsch RM: Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001, 293: 1491-1495. 10.1126/science.1062097.
David DC, Hauptmann S, Scherping I, Schuessel K, Keil U, Rizzu P, Ravid R, Dröse S, Brandt U, Muller WE, Eckert A, Götz J: Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J Biol Chem. 2005, 280: 23802-23814. 10.1074/jbc.M500356200.
David DC, Ittner LM, Gehrig P, Nergenau D, Shepherd C, Halliday G, Gotz J: Beta-amyloid treatment of two complementary P301L tau-expressing Alzheimer's disease models reveals similar deregulated cellular processes. Proteomics. 2006, 6: 6566-6577. 10.1002/pmic.200600634.
Melov S, Adlard PA, Morten K, Johnson F, Golden TR, Hinerfeld D, Schilling B, Mavros C, Masters CL, Volitakis I, Li QX, Laughton K, Hubbard A, Cherny RA, Gibson B, Bush AI: Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS One. 2007, 2: e536-10.1371/journal.pone.0000536.
Escobar-Khondiker M, Höllerhage M, Muriel MP, Champy P, Bach A, Depienne C, Respondek G, Yamada ES, Lannuzel A, Yagi T, Hirsch EC, Oertel WH, Jacob R, Michel PP, Ruberg M, Höglinger GU: Annonacin, a natural mitochondrial complex I inhibitor, causes tau pathology in cultured neurons. J Neurosci. 2007, 27: 7827-7837. 10.1523/JNEUROSCI.1644-07.2007.
Gotz J, Ittner LM, Kins S: Do axonal defects in tau and amyloid precursor protein transgenic animals model axonopathy in Alzheimer's disease?. J Neurochem. 2006, 98: 993-1006. 10.1111/j.1471-4159.2006.03955.x.
Bolmont T, Clavaguera F, Meyer-Luehmann M, Herzig MC, Radde R, Staufenbiel M, Lewis J, Hutton M, Tolnay M, Jucker M: Induction of tau pathology by intracerebral infusion of amyloid-beta-containing brain extract and by amyloid-beta deposition in APP × Tau transgenic mice. Am J Pathol. 2007, 171: 2012-2020. 10.2353/ajpath.2007.070403.
Gotz J, Schild A, Hoerndli F, Pennanen L: Amyloid-induced neurofibrillary tangle formation in Alzheimer's disease: insight from transgenic mouse and tissue-culture models. Int J Dev Neurosci. 2004, 22: 453-465. 10.1016/j.ijdevneu.2004.07.013.
Ittner LM, Gotz J: Amyloid-beta and tau - a toxic pas de deux in Alzheimer's disease. Nat Rev Neurosci. 2011, 12: 65-72.
Eckert A, Hauptmann S, Scherping I, Meinhardt J, Rhein V, Drose S, Brandt U, Fandrich M, Muller WE, Gotz J: Oligomeric and fibrillar species of betaamyloid (A beta 42) both impair mitochondrial function in P301L tau transgenic mice. J Mol Med. 2008, 86: 1255-1267. 10.1007/s00109-008-0391-6.
Crouch PJ, Hung LW, Adlard PA, Cortes M, Lal V, Filiz G, Perez KA, Nurjono M, Caragounis A, Du T, Laughton K, Volitakis I, Bush AI, Li QX, Masters CL, Cappai R, Cherny RA, Donnelly PS, White AR, Barnham KJ: Increasing Cu bioavailability inhibits Abeta oligomers and tau phosphorylation. Proc Natl Acad Sci USA. 2009, 106: 381-386. 10.1073/pnas.0809057106.
Hu M, Waring JF, Gopalakrishnan M, Li J: Role of GSK-3beta activation and alpha7 nAChRs in Abeta(1-42)-induced tau phosphorylation in PC12 cells. J Neurochem. 2008, 106: 1371-1377. 10.1111/j.1471-4159.2008.05483.x.
Li HL, Wang HH, Liu SJ, Deng YQ, Zhang YJ, Tian Q, Wang XC, Chen XQ, Yang Y, Zhang JY, Wang Q, Xu H, Liao FF, Wang JZ: Phosphorylation of tau antagonizes apoptosis by stabilizing beta-catenin, a mechanism involved in Alzheimer's neurodegeneration. Proc Natl Acad Sci USA. 2007, 104: 3591-3596. 10.1073/pnas.0609303104.
Wang ZF, Yin J, Zhang Y, Zhu LQ, Tian Q, Wang XC, Li HL, Wang JZ: Overexpression of tau proteins antagonizes amyloid-beta-potentiated apoptosis through mitochondria-caspase-3 pathway in N2a cells. J Alzheimers Dis. 2010, 20: 145-157.
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM: Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003, 39: 409-421. 10.1016/S0896-6273(03)00434-3.
Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD: Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2009, 106: 14670-14675. 10.1073/pnas.0903563106.
Hyun DH, Mughal MR, Yang H, Lee JH, Ko EJ, Hunt ND, de Cabo R, Mattson MP: The plasma membrane redox system is impaired by amyloid betapeptide and in the hippocampus and cerebral cortex of 3xTgAD mice. Exp Neurol. 2010, 225: 423-429. 10.1016/j.expneurol.2010.07.020.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Eckert, A., Schmitt, K. & Götz, J. Mitochondrial dysfunction - the beginning of the end in Alzheimer's disease? Separate and synergistic modes of tau and amyloid-β toxicity. Alz Res Therapy 3, 15 (2011). https://doi.org/10.1186/alzrt74
Published:
DOI: https://doi.org/10.1186/alzrt74