
Abstract
Alzheimer's disease (AD) is a progressive neurodegenerative disease characterised by extracellular amyloid-ß (Aß) and intraneuronal tau protein brain pathologies. The most significant risk factor for non-familial AD is the presence of the E4 isoform of the cholesterol transporter apolipoprotein E (apoE). Despite extensive basic research, the exact role of apoE in disease aetiology remains unclear. Correspondingly, therapeutic targeting of apoE in AD is at an early preclinical stage. In this review, I discuss the key interactions of apoE and Aß pathology, the current progress of preclinical animal models and the caveats of existing therapeutic approaches targeting apoE. Finally, novel Alzheimer's genetics and Aß-independent disease mechanisms are highlighted.
Similar content being viewed by others

Introduction
Alzheimer's disease (AD) is the most common cause of dementia in aged populations, being characterized by cerebrovascular and neuronal dysfunctions that induce a progressive decline in cognitive functions [1]. The occurrence of AD in individuals aged over 65 years is defined as late-onset AD (LOAD) - representing the majority of AD sufferers. Patients with early-onset AD (EOAD) represent approximately 1% of the overall population [2].
Symptomatic AD is diagnosed clinically using a battery of cognitive tests, with significant efforts ongoing to move diagnosis to earlier disease stages using the additional tools of genetic testing, blood and cerebrospinal fluid biomarkers and neuroimaging [3]. Previous to these advances, however, AD could only be definitively diagnosed as the cause of dementia by post-mortem detection of two major neuropathologies. These comprise senile plaques of aggregated Aß peptide, and neurofibrillary tangles of hyperphosphorylated, aggregated tau protein.
Amyloid-ß
Aß peptides are produced through sequential proteolysis of the amyloid precursor protein (APP) by ß-secretase/BACE and the γ-secretase complex (partly comprising the presenilin PS1 or PS2). Aß peptides vary in length from 39 to 43 amino acids with the predominant species being Aß40 and Aß42 [4]. Disease-modifying AD drug discovery research has focused on strategies targeting production or clearance of the Aß peptide. This 'amyloid hypothesis' has been driven by the fact that familial EOAD with autosomal dominant inheritance is caused by mutations in the APP, PS1 or PS2 genes. In simple terms, the net effect of these mutations is to increase either bulk Aß levels or the ratio of Aß42:Aß40 production [5]. An increase in brain Aß42 levels, whether absolute or ratiometric, is hence critical to the aetiology of familial EOAD.
In agreement with the amyloid hypothesis, studies in transgenic mouse models of AD imply a cascade of events in which abnormal forms of tau act as downstream mediators of Aß toxicity [6, 7]. Contrary to this proposed cascade, however, whilst neuronal loss and neurofibrillary tangle counts strongly predict cognitive status in LOAD patients, total Aß plaque load correlates weakly with cognitive impairment [8]. The prevalent explanation for this disparity is that it is diffusible Aß oligomers, rather than Aß plaques, that represent the actual toxic species. The E693Δ APP mutation, for example, causes Alzheimer's-type dementia through the toxicity of non-fibrillar, intracellular Aß oligomers [9]. Conversely, the 'Arctic' APP mutation (E693G) induces formation of large Aß oligomers known as protofibrils [10]. Experimental disagreement over the physicochemical nature of toxic oligomers in LOAD has hampered delineation of their exact role in disease [11].
Apolipoprotein E
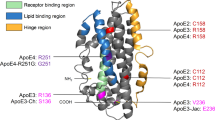
Apolipoprotein E (apoE) is the primary transporter of cholesterol in the central nervous system (CNS), being synthesised within the blood brain barrier (BBB) predominantly by astrocytes [12]. Three apoE polymorphic alleles (APOE2, APOE3, and APOE4) encode three protein isoforms (apoE2, apoE3 and apoE4) that differ by cysteine/arginine polymorphisms at position 112 or 158. The APOE4 allele, found in 15% of the population, remains the most significant genetic risk factor for LOAD [13].
In support of the amyloid hypothesis, APOE4 carrier status is associated with greater Aß plaque load in both AD patients and cognitively normal individuals [14, 15]. The APOE4 allele also correlates with increased cerebro-vascular Aß deposition [16] and, correspondingly, is a risk factor for cerebral amyloid angiopathy [17]. As a consequence, research into the mechanistic connection between apoE4 and LOAD has focused on delineating the interaction of apoE with Aß pathology (Figure 1). Experimental data now support a clear and necessary role for apoE in Aß toxicity.

Apolipoprotein E/amyloid-ß interaction pathways and therapeutic approaches. Amyloid-ß (Aß) and apolipoprotein E (apoE) are predominantly produced by neurons and astrocytes, respectively. ApoE regulates Aß oligomerisation, aggregation and receptor-mediated clearance, contributing to Aß pathology in LOAD. Potential therapeutic approaches are indicated in red. LDLR, LDL receptor; LRP, LDL receptor-related protein; LXR, lipid × receptor; RXR, retinoid × receptor; VLDLR, VLDL receptor.
Interactions of ApoE with Aß pathology
In vitro studies have demonstrated that apoE4 more than apoE3 interacts directly with Aß [18], enhancing Aß fibrillisation [19]. Interpretation of such data is complicated by the difficulties of replicating in vivo Aß conformation and apoE lipidation status. However, early Aß amyloidosis mouse model data also support a clear role for apoE in Aß pathology [20]. As a consequence of these findings, apoE/Aß interaction inhibitors are being developed as AD therapeutics. Small Aß-mimetic peptides intially demonstrated reductions in apoE-stimulated formation of neurotoxic Aß aggregates in vitro [21], with these data being subsequently confirmed in vivo using a mouse model of Aß brain amyloidosis [22].
ApoE proteins comprise an amino-terminal receptor-interacting domain and carboxy-terminal lipid-binding domain. Fluorescence lifetime imaging-fluorescence resonance energy transfer (FLIM-FRET) studies on human post-mortem tissue sections indicate that Aß is preferentially associated with the carboxyl terminus of apoE4 compared to that of apoE3, and that apoE4 undergoes greater amino-terminal degradation, prolonging Aß interaction [23]. This prolonged interaction may enhance formation and stabilisation of toxic Aß oligomers [24]. Analyses of AD brain samples have demonstrated a higher burden of oligomeric Aß in APOE4 carriers with increased amyloid plaque-associated synaptic loss. ApoE4 colocalises with oligomeric Aß at the synapse, indicating a key role as a co-factor in Aß toxicity [25].
The greater susceptibility of apoE4 to proteolytic cleavage, and the subsequent prolongation of Aß interactions, is thought to be a consequence of differential domain interaction. The C112R polymorphism in apoE4 results in a salt bridge between R61 and E255, which is lacking in apoE3 [26]. This brings the amino- and carboxy-terminal domains into closer proximity and exposes the hinge region of apoE4 to proteolysis [23]. Consequently, the development of small-molecule 'structure correctors' that shift apoE4 to an apoE3-like conformation has also been proposed as a therapeutic strategy for AD [27].
The main challenge for small molecule approaches aiming to disrupt apoE intradomain or apoE/Aß protein-protein interactions is to achieve a compound with sufficient potency, specificity and BBB permeability to be suitable for clinical trials.
ApoE mouse models of Aß amyloidosis
Multiple mouse models of Aß brain amyloidosis have been generated, predominantly comprising familial, EOAD APP and PS1/2 mutations either alone or in combination [28]. To varying degrees, these mice recapitulate brain parenchymal and cerebrovascular Aß deposition with cognitive behavioural disorder; however, neuronal loss is relatively lacking in most models. When considering the impact of apoE on Aß pathology in these mice it is important to consider that endogenous murine apoE is non-polymorphic and does not display domain interaction [29]. Consequently, mouse apoE behaves most similarly to human apoE3. In order to determine the effects of human apoE isoforms, Aß amyloidosis transgenics have now been combined with a variety of human apoE mouse models. These crosses display delayed onset of Aß pathology relative to their murine equivalents, emphasising the importance of interspecies differences [30].
Mice expressing mutant V717F APP in conjunction with human apoE isoform knock-ins (PDAPP/TRE mice) show isoform-dependent Aß deposition, with apoE4 showing the strongest effect followed by apoE3 and then apoE2 [31].
Gene dosage is critically important, with haplo-insufficiency of both human apoE3 and apoE4 knock-in isoforms causing marked reductions in Aß deposition in APP/PS1 mutant mice [32, 33]. This is a key point, as there is an ongoing debate regarding the potential therapeutic benefits of raising versus lowering apoE expression levels. Whilst the transgenic data indicate that reducing apoE levels would be more beneficial, small-molecule upregulation of apoE levels, particularly through agonism of the lipid × receptor (LXR) [34] or retinoid × receptor (RXR) [35], has been reported as a promising therapeutic approach. In vivo studies of such agonists, whilst successfully demonstrating reductions in Aß pathology, were carried out against a background of endogenous murine apoE. It remains a possibility, therefore, that increasing expression of human apoE4 may actually be deleterious to disease. It should also be noted that LXR/RXR agonism has side effects, such as hypertriglyceridaemia, and the relatively hydrophobic nature of ligands makes complicating interactions with the γ-secretase multispan membrane complex a possibility [36].
ApoE and Aß production
There is limited evidence for modulation of Aß production by apoE with in vitro studies using cultured cells co-overexpressing apoE and APP - a relatively unphysiological paradigm [37]. ApoE4-induced increases in Aß production could be mediated by a novel, apoE-interacting protein, TMC22, proposed to facilitate an interaction between APP and the γ-secretase complex [38].
ApoE and Aß aggregation
Neprilysin is the major protease mediating brain Aß degradation [39]. In vivo inhibition of neprilysin by thiorphan infusion induces apoE isoform-dependent aggregation of Aß, with apoE4 causing the greatest increase in aggregation [40]. It is possible that apoE acts to stabilise oligomeric Aß, causing enhanced toxicity and seeding deposition of larger aggregates [24].
ApoE and Aß clearance
Aß is cleared from the brain by proteolytic degradation [41], bulk flow along the perivascular interstitial fluid drainage pathway [42], or by receptor-mediated clearance across the BBB [43]. In addition, the 'peripheral sink' hypothesis postulates that clearance of Aß from the brain is accelerated by removal of Aß from the plasma via the liver and kidneys [44]. APOE4 carriers may display clearance deficits in both compartments as Aß removal from both the CNS and the plasma is reduced in human apoE4 knock-in mice [31, 45].
ApoE isoform status may influence CNS Aß degradation through indirect mechanisms such as regulation of cellular cholesterol - enhancing endocytosis and lysosomal degradation of Aß [46]. The major impact of apoE is, however, likely to be through interaction of Aß with cell-surface apoE receptors, including LDL receptor-related protein 1 (LRP1), the LDL receptor (LDLR) and the VLDL receptor (VLDLR) [47]. Receptor binding of Aß, alone or in complex with apoE, either delivers Aß to the lysosome or leads to transcytosis into the plasma via the BBB. LRP1 is perhaps the best characterised transporter acting in the latter instance [48]. ApoE isoforms (apoE4 > apoE3 > apoE2) may disrupt rapid, LRP1-mediated clearance of unbound Aß by diverting it to the VLDLR, which has a slower rate of endocytosis [49].
From a therapeutic perspective, peripheral administration of soluble fragments of LRP1 has been shown to reduce brain Aß load in K670N/M671L APP mice through plasma Aß binding - theoretically exploiting the peripheral sink hypothesis [50]. However, the primary investigation of this type of approach has been through enhancement of peripheral Aß clearance through anti-Aß immunisation strategies. These remain, despite early setbacks, one of the most promising current therapeutic avenues. Passive immunisation with the humanised anti-Aß antibody bapinuezumab demonstrated lower efficacy in APOE4 carriers with a corresponding increase in vasogenic oedema, suggestive of transient increases in vascular permeability [51, 52]. If phase III trials are positive, determination of APOE status is likely to become an important aspect of treatment.
In addition to LRP1, LDLR has also been implicated in Aß removal from the CNS. LDLR over-expression decreased Aß deposition and enhanced clearance in the K670N/M671L APP, ΔE9 PS1 amyloidosis mouse model [53]. LDLR knockout data are inconsistent, however, as whilst two studies reported increased Aß load [54, 55] a further analysis failed to show any effect [56]. Although LDLR-upregulating compounds have been reported [57], clinical usage of such drugs would be challenging due to specificity and toxicity concerns.
Aß-independent disease mechanisms
Collaborative large-scale genome-wide association studies have identified, in addition to apoE, novel LOAD risk genes. These include CLU (encoding apolipoprotein J), PICALM, CR1 and BIN1 [58]. Conversely, variants of APP and PS1/2, which increase Aß42 production in familial EOAD, were not hits in these studies. The genetic drivers of LOAD and EOAD are hence likely to be different. Whilst the novel LOAD risk genes may function in either Aß clearance [43, 59] or toxicity [60], there remains a possibility that key implicated pathways, such as lipid homeostasis and innate immunity, play Aß-independent roles in the aetiology of LOAD. ApoE is linked to autoimmune inflammation, diabetes and coronary heart disease - environmental risk factors for LOAD magnified by the APOE4 genotype [61]. The clinical failures of non-steroidal anti-inflammatories [62], a peroxisome proliferator-activated receptor (PPAR)γ agonist [63] and HMG-CoA reductase inhibitors [64] suggest, however, that targeting mid-life risk factors for LOAD in late stage disease is unlikely to be therapeutically successful. Such treatments, including apoE-based therapeutics, may need to be given earlier in the disease process. This places additional importance on early diagnosis of AD and/or preventative treatment in individuals at high risk of developing LOAD.
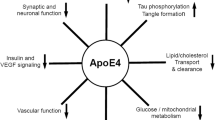
ApoE, and related cell signalling, is also purported to modulate synaptic plasticity, tau phosphorylation, and neuroinflammation [47]. The extent to which apoE drives the aetiology of LOAD through these mechanisms is unclear; however, apoE mimetic peptides designed to mediate putative, beneficial effects of apoE demonstrated both behavioural and pathological benefits in mutant APP mice [65]. The main challenge with such an approach will be to achieve a candidate molecule with appropriate physicochemical properties for clinical use.
Conclusions
Understanding of the interplay between APOE genotype and Aß pathology has progressed significantly in recent years, particularly with respect to human apoE knock-in animal models of Aß amyloidosis. These demonstrate an isoform-specific role for apoE4 in retarding Aß clearance from the CNS. By virtue of the nature of the target, however, apoE therapeutics are still at an early preclinical stage, with appreciable chemistry challenges facing small-molecule approaches. The most immediate impact of apoE on AD therapeutics will likely be the profiling of patients for APOE4 status to help determine dosing of anti-Aß immunotherapy treatments. ApoE has multiple systemic functions, some of which relate to novel LOAD risk genes, which may also affect the aetiology of AD independently of Aß. The understanding, and modelling, of these functions remain goals for future research.
Abbreviations
- Aß:
-
amyloid-ß peptide
- AD:
-
Alzheimer's disease
- apoE:
-
apolipoprotein E
- APP:
-
amyloid precursor protein
- BBB:
-
blood brain barrier
- CNS:
-
central nervous system
- EOAD:
-
early onset Alzheimer's disease
- LDL:
-
low-density lipoprotein
- LDLR:
-
LDL receptor
- LOAD:
-
late onset Alzheimer's disease
- LRP:
-
LDL receptor-related protein
- PS:
-
presenilin
- VLDLR:
-
VLDL receptor.
References
Amaducci LA, Rocca WA, Schoenberg BS: Origin of the distinction between Alzheimer's disease and senile dementia: how history can clarify nosology. Neurology. 1986, 36: 1497-1499. 10.1212/WNL.36.11.1497.
Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, Thomas-Anterion C, Michon A, Martin C, Charbonnier F, Raux G, Camuzat A, Penet C, Mesnage V, Martinez M, Clerget-Darpoux F, Brice A, Frebourg T: Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999, 65: 664-670. 10.1086/302553.
Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH: Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011, 7: 280-292. 10.1016/j.jalz.2011.03.003.
Zhang YW, Thompson R, Zhang H, Xu H: APP processing in Alzheimer's disease. Mol Brain. 2011, 4: 3-10.1186/1756-6606-4-3.
Weggen S, Beher D: Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal-dominant Alzheimer's disease. Alzheimer's Res Ther. 2012, 4: 9-10.1186/alzrt107.
Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM: Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006, 281: 39413-39423. 10.1074/jbc.M608485200.
Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L: Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007, 316: 750-754. 10.1126/science.1141736.
Giannakopoulos P, Herrmann FR, Bussière T, Bouras C, Kövari E, Perl DP, Morrison JH, Gold G, Hof PR: Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003, 60: 1495-1500. 10.1212/01.WNL.0000063311.58879.01.
Tomiyama T, Nagata T, Shimada H, Teraoka R, Fukushima A, Kanemitsu H, Takuma H, Kuwano R, Imagawa M, Ataka S, Wada Y, Yoshioka E, Nishizaki T, Watanabe Y, Mori H: A new amyloid beta variant favoring oligomerization in Alzheimer's-type dementia. Ann Neurol. 2008, 63: 377-387. 10.1002/ana.21321.
Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, Näslund J, Lannfelt L: The 'Arctic' APP mutation (E693G) causes Alzheimer's disease by enhanced Abeta protofibril formation. Nat Neurosci. 2001, 9: 887-893.
Benilova I, Karran E, De Strooper B: The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci. 2012, 15: 349-357. 10.1038/nn.3028.
Pitas RE, Boyles JK, Lee SH, Foss D, Mahley RW: Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim Biophys Acta. 1987, 917: 148-161. 10.1016/0005-2760(87)90295-5.
Myers RH, Schaefer EJ, Wilson PW, D'Agostino R, Ordovas JM, Espino A, Au R, White RF, Knoefel JE, Cobb JL, McNulty KA, Beiser A, Wolf PA: Apolipoprotein E epsilon4 association with dementia in a population-based study: The Framingham study. Neurology. 1996, 46: 673-677. 10.1212/WNL.46.3.673.
Drzezga A, Grimmer T, Henriksen G, Mühlau M, Perneczky R, Miederer I, Praus C, Sorg C, Wohlschläger A, Riemenschneider M, Wester HJ, Foerstl H, Schwaiger M, Kurz A: Effect of APOE genotype on amyloid plaque load and gray matter volume in Alzheimer disease. Neurology. 2009, 72: 1487-1494. 10.1212/WNL.0b013e3181a2e8d0.
Kantarci K, Lowe V, Przybelski SA, Weigand SD, Senjem ML, Ivnik RJ, Preboske GM, Roberts R, Geda YE, Boeve BF, Knopman DS, Petersen RC, Jack CR: APOE modifies the association between Aβ load and cognition in cognitively normal older adults. Neurology. 2012, 78: 232-240. 10.1212/WNL.0b013e31824365ab.
Peuralinna T, Tanskanen M, Mäkelä M, Polvikoski T, Paetau A, Kalimo H, Sulkava R, Hardy J, Lai SL, Arepalli S, Hernandez D, Traynor BJ, Singleton A, Tienari PJ, Myllykangas L: APOE and AβPP gene variation in cortical and cerebrovascular amyloid-β pathology and Alzheimer's disease: a population-based analysis. J Alzheimers Dis. 2011, 26: 377-385.
Keage HA, Carare RO, Friedland RP, Ince PG, Love S, Nicoll JA, Wharton SB, Weller RO, Brayne C: Population studies of sporadic cerebral amyloid angiopathy and dementia: a systematic review. BMC Neurol. 2009, 9: 3-10.1186/1471-2377-9-3.
Sanan DA, Weisgraber KH, Russell SJ, Mahley RW, Huang D, Saunders A, Schmechel D, Wisniewski T, Frangione B, Roses AD: Apolipoprotein E associates with beta amyloid peptide of Alzheimer's disease to form novel monofibrils. Isoform apoE4 associates more efficiently than apoE3. J Clin Invest. 1994, 94: 860-869. 10.1172/JCI117407.
Wisniewski T, Castaño EM, Golabek A, Vogel T, Frangione B: Acceleration of Alzheimer's fibril formation by apolipoprotein E in vitro. Am J Pathol. 1994, 145: 1030-1035.
Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, Fishman CE, DeLong CA, Piccardo P, Petegnief V, Ghetti B, Paul SM: Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 1999, 96: 15233-15238. 10.1073/pnas.96.26.15233.
Ma J, Brewer HB, Potter H: Alzheimer A beta neurotoxicity: promotion by antichymotrypsin, ApoE4; inhibition by Abeta-related peptides. Neurobiol Aging. 1996, 17: 773-780. 10.1016/0197-4580(96)00112-1.
Yang J, Ji Y, Mehta P, Bates KA, Sun Y, Wisniewski T: Blocking the apolipoprotein E/amyloid-β interaction reduces fibrillar vascular amyloid deposition and cerebral microhemorrhages in TgSwDI mice. J Alzheimers Dis. 2011, 24: 269-285.
Jones PB, Adams KW, Rozkalne A, Spires-Jones TL, Hshieh TT, Hashimoto T, von Armin CA, Mielke M, Bacskai BJ, Hyman BT: Apolipoprotein E: isoform specific differences in tertiary structure and interaction with amyloid-β in human Alzheimer brain. PLoS One. 2011, 6: e14586-10.1371/journal.pone.0014586.
Cerf E, Gustot A, Goormaghtigh E, Ruysschaert JM, Raussens V: High ability of apolipoprotein E4 to stabilize amyloid-β peptide oligomers, the pathological entities responsible for Alzheimer's disease. FASEB J. 2011, 25: 1585-1595. 10.1096/fj.10-175976.
Koffie RM, Hashimoto T, Tai HC, Kay KR, Serrano-Pozo A, Joyner D, Hou S, Kopeikina KJ, Frosch MP, Lee VM, Holtzman DM, Hyman BT, Spires-Jones TL: Apolipoprotein E4 effects in Alzheimer's disease are mediated by synaptotoxic oligomeric amyloid-β. Brain. 2012, 135: 2155-2168.
Mahley RW, Weisgraber KH, Huang Y: Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer's disease. Proc Natl Acad Sci USA. 2006, 103: 5644-5651. 10.1073/pnas.0600549103.
Chen HK, Liu Z, Meyer-Franke A, Brodbeck J, Miranda RD, McGuire JG, Pleiss MA, Ji ZS, Balestra ME, Walker DW, Xu Q, Jeong DE, Budamagunta MS, Voss JC, Freedman SB, Weisgraber KH, Huang Y, Mahley RW: Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J Biol Chem. 2012, 287: 5253-5266. 10.1074/jbc.M111.276162.
Van Dam D, De Deyn PP: Animal models in the drug discovery pipeline for Alzheimer's disease. Br J Pharmacol. 2011, 164: 1285-1300. 10.1111/j.1476-5381.2011.01299.x.
Raffai RL, Dong LM, Farese RV, Weisgraber KH: Introduction of human apolipoprotein E4 "domain interaction" into mouse apolipoprotein E. Proc Natl Acad Sci USA. 2001, 98: 11587-11591. 10.1073/pnas.201279298.
Tai LM, Youmans KL, Jungbauer L, Yu C, Ladu MJ: Introducing human APOE into Aβ transgenic mouse models. Int J Alzheimers Dis. 2011, 2011: 810981-
Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM: Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med. 2011, 3: 89ra57-10.1126/scitranslmed.3002156.
Kim J, Jiang H, Park S, Eltorai AE, Stewart FR, Yoon H, Basak JM, Finn MB, Holtzman DM: Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-β amyloidosis. J Neurosci. 2011, 31: 18007-18012. 10.1523/JNEUROSCI.3773-11.2011.
Bien-Ly N, Gillespie AK, Walker D, Yoon SY, Huang Y: Reducing human apolipoprotein E levels attenuates age-dependent Aβ accumulation in mutant human amyloid precursor protein transgenic mice. J Neurosci. 2012, 32: 4803-4811. 10.1523/JNEUROSCI.0033-12.2012.
Riddell DR, Zhou H, Comery TA, Kouranova E, Lo CF, Warwick HK, Ring RH, Kirksey Y, Aschmies S, Xu J, Kubek K, Hirst WD, Gonzales C, Chen Y, Murphy E, Leonard S, Vasylyev D, Oganesian A, Martone RL, Pangalos MN, Reinhart PH, Jacobsen JS: The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer's disease. Mol Cell Neurosci. 2007, 34: 621-628. 10.1016/j.mcn.2007.01.011.
Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE: ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science. 2012, 335: 1503-1506. 10.1126/science.1217697.
Czech C, Burns MP, Vardanian L, Augustin A, Jacobsen H, Baumann K, Rebeck GW: Cholesterol independent effect of LXR agonist TO-901317 on gammasecretase. J Neurochem. 2007, 101: 929-936. 10.1111/j.1471-4159.2007.04467.x.
Ye S, Huang Y, Müllendorff K, Dong L, Giedt G, Meng EC, Cohen FE, Kuntz ID, Weisgraber KH, Mahley RW: Apolipoprotein (apo) E4 enhances amyloid beta peptide production in cultured neuronal cells: apoE structure as a potential therapeutic target. Proc Natl Acad Sci USA. 2005, 102: 18700-18705. 10.1073/pnas.0508693102.
Hopkins PC, Sáinz-Fuertes R, Lovestone S: The impact of a novel apolipoprotein E and amyloid-β protein precursor-interacting protein on the production of amyloid-β. J Alzheimers Dis. 2011, 26: 239-253.
Marr RA, Guan H, Rockenstein E, Kindy M, Gage FH, Verma I, Masliah E, Hersh LB: Neprilysin regulates amyloid Beta peptide levels. J Mol Neurosci. 2004, 22: 5-11. 10.1385/JMN:22:1-2:5.
Zepa L, Frenkel M, Belinson H, Kariv-Inbal Z, Kayed R, Masliah E, Michaelson DM: ApoE4-driven accumulation of intraneuronal oligomerized Aβ42 following activation of the amyloid cascade in vivo is mediated by a gain of function. Int J Alzheimers Dis. 2011, 792070-
Saido T, Leissring MA: Proteolytic degradation of amyloid β-protein. Cold Spring Harb Perspect Med. 2012, 2: a006379-
Weller RO, Subash M, Preston SD, Mazanti I, Carare RO: Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol. 2008, 18: 253-266.
Deane R, Bell RD, Sagare A, Zlokovic BV: Clearance of amyloid-beta peptide across the blood-brain barrier: implication for therapies in Alzheimer's disease. CNS Neurol Disord Drug Targets. 2009, 8: 16-30. 10.2174/187152709787601867.
Matsuoka Y, Saito M, LaFrancois J, Saito M, Gaynor K, Olm V, Wang L, Casey E, Lu Y, Shiratori C, Lemere C, Duff K: Novel therapeutic approach for the treatment of Alzheimer's disease by peripheral administration of agents with an affinity to beta-amyloid. J Neurosci. 2003, 23: 29-33.
Sharman MJ, Morici M, Hone E, Berger T, Taddei K, Martins IJ, Lim WL, Singh S, Wenk MR, Ghiso J, Buxbaum JD, Gandy S, Martins RN: APOE genotype results in differential effects on the peripheral clearance of amyloid-beta42 in APOE knock-in and knock-out mice. J Alzheimers Dis. 2010, 21: 403-409.
Lee CY, Tse W, Smith JD, Landreth GE: Apolipoprotein E promotes β-amyloid trafficking and degradation by modulating microglial cholesterol levels. J Biol Chem. 2012, 287: 2032-2044. 10.1074/jbc.M111.295451.
Holtzman DM, Herz J, Bu G: Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med. 2012, 2: a006312-
Zlokovic BV, Deane R, Sagare AP, Bell RD, Winkler EA: Low-density lipoprotein receptor-related protein-1: a serial clearance homeostatic mechanism controlling Alzheimer's amyloid β-peptide elimination from the brain. J Neurochem. 2010, 115: 1077-1089. 10.1111/j.1471-4159.2010.07002.x.
Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, Holtzman DM, Zlokovic BV: ApoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 2008, 118: 4002-4013. 10.1172/JCI36663.
Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R, Marky A, Lenting PJ, Wu Z, Zarcone T, Goate A, Mayo K, Perlmutter D, Coma M, Zhong Z, Zlokovic BV: Clearance of amyloid-beta by circulating lipoprotein receptors. Nat Med. 2007, 13: 1029-1031. 10.1038/nm1635.
Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, Sabbagh M, Honig LS, Doody R, van Dyck CH, Mulnard R, Barakos J, Gregg KM, Liu E, Lieberburg I, Schenk D, Black R, Grundman M, Bapineuzumab 201 Clinical Trial Investigators: A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009, 73: 2061-2070. 10.1212/WNL.0b013e3181c67808.
Roher AE, Maarouf CL, Daugs ID, Kokjohn TA, Hunter JM, Sabbagh MN, Beach TG: Neuropathology and amyloid-β spectrum in a bapineuzumab immunotherapy recipient. J Alzheimers Dis. 2011, 24: 315-325.
Kim J, Castellano JM, Jiang H, Basak JM, Parsadanian M, Pham V, Mason SM, Paul SM, Holtzman DM: Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular Aβ clearance. Neuron. 2009, 64: 632-644. 10.1016/j.neuron.2009.11.013.
Katsouri L, Georgopoulos S: Lack of LDL receptor enhances amyloid deposition and decreases glial response in an Alzheimer's disease mouse model. PLoS One. 2011, 6: e21880-10.1371/journal.pone.0021880.
Cao D, Fukuchi K, Wan H, Kim H, Li L: Lack of LDL receptor aggravates learning deficits and amyloid deposits in Alzheimer transgenic mice. Neurobiol Aging. 2006, 27: 1632-1643. 10.1016/j.neurobiolaging.2005.09.011.
Fryer JD, Demattos RB, McCormick LM, O'Dell MA, Spinner ML, Bales KR, Paul SM, Sullivan PM, Parsadanian M, Bu G, Holtzman DM: The low density lipoprotein receptor regulates the level of central nervous system human and murine apolipoprotein E but does not modify amyloid plaque pathology in PDAPP mice. J Biol Chem. 2005, 280: 25754-25759. 10.1074/jbc.M502143200.
Murakami S, Nitanai I, Uchida S, Kondo-Ohta Y, Asami Y, Kondo K, Sato M, Kawashima A, Hara H, Tomisawa K, Mei HB, Xiang CZ: Up-regulation of low density lipoprotein receptor by a novel isobenzofranone derivative, MD-700. Atherosclerosis. 1999, 146: 281-290. 10.1016/S0021-9150(99)00148-3.
Hollingworth P, Harold D, Jones L, Owen MJ, Williams J: Alzheimer's disease genetics: current knowledge and future challenges. Int J Geriatr Psychiatry. 2011, 26: 793-802. 10.1002/gps.2628.
Biffi A, Shulman JM, Jagiella JM, Cortellini L, Ayres AM, Schwab K, Brown DL, Silliman SL, Selim M, Worrall BB, Meschia JF, Slowik A, De Jager PL, Greenberg SM, Schneider JA, Bennett DA, Rosand J: Genetic variation at CR1 increases risk of cerebral amyloid angiopathy. Neurology. 2012, 78: 334-341. 10.1212/WNL.0b013e3182452b40.
Treusch S, Hamamichi S, Goodman JL, Matlack KE, Chung CY, Baru V, Shulman JM, Parrado A, Bevis BJ, Valastyan JS, Han H, Lindhagen-Persson M, Reiman EM, Evans DA, Bennett DA, Olofsson A, DeJager PL, Tanzi RE, Caldwell KA, Caldwell GA, Lindquist S: Functional links between Aβ toxicity, endocytic trafficking, and Alzheimer's disease risk factors in yeast. Science. 2011, 334: 1241-1245. 10.1126/science.1213210.
Kivipelto M, Rovio S, Ngandu T, Kåreholt I, Eskelinen M, Winblad B, Hachinski V, Cedazo-Minguez A, Soininen H, Tuomilehto J, Nissinen A: Apolipoprotein E epsilon4 magnifies lifestyle risks for dementia: a population-based study. J Cell Mol Med. 2008, 12: 2762-2771. 10.1111/j.1582-4934.2008.00296.x.
ADAPT Research Group, Martin BK, Szekely C, Brandt J, Piantadosi S, Breitner JC, Craft S, Evans D, Green R, Mullan M: Cognitive function over time in the Alzheimer's Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol. 2008, 65: 896-905.
Harrington C, Sawchak S, Chiang C, Davies J, Donovan C, Saunders AM, Irizarry M, Jeter B, Zvartau-Hind M, van Dyck CH, Gold M: Rosiglitazone does not improve cognition or global function when used as adjunctive therapy to AChE inhibitors in mild-to-moderate Alzheimer's disease: two phase 3 studies. Curr Alzheimer Res. 2011, 8: 592-606. 10.2174/156720511796391935.
Feldman HH, Doody RS, Kivipelto M, Sparks DL, Waters DD, Jones RW, Schwam E, Schindler R, Hey-Hadavi J, DeMicco DA, Breazna A, LEADe Investigators: Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology. 2010, 74: 956-964. 10.1212/WNL.0b013e3181d6476a.
Vitek MP, Christensen DJ, Wilcock D, Davis J, Van Nostrand WE, Li FQ, Colton CA: APOE-mimetic peptides reduce behavioral deficits, plaques and tangles in Alzheimer's disease transgenics. Neurodegenerative Dis. 2012, 10: 122-126. 10.1159/000334914.
Acknowledgements
The author thanks Eisai Limited for approving this review for publication.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
Adam Kline was in the past 5 years an employee of Eisai Limited and received a fixed salary. Adam Kline was not an Eisai employee at the time of publication.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Kline, A. Apolipoprotein E, amyloid-ß clearance and therapeutic opportunities in Alzheimer's disease. Alz Res Therapy 4, 32 (2012). https://doi.org/10.1186/alzrt135
Published:
DOI: https://doi.org/10.1186/alzrt135