
Abstract
An important pathological feature of Alzheimer's disease (AD) is the presence of extracellular senile plaques in the brain. Senile plaques are composed of aggregations of small peptides called β-amyloid (Aβ). Multiple lines of evidence demonstrate that overproduction/aggregation of Aβ in the brain is a primary cause of AD and inhibition of Aβ generation has become a hot topic in AD research. Aβ is generated from β-amyloid precursor protein (APP) through sequential cleavages first by β-secretase and then by γ-secretase complex. Alternatively, APP can be cleaved by α-secretase within the Aβ domain to release soluble APPα and preclude Aβ generation. Cleavage of APP by caspases may also contribute to AD pathologies. Therefore, understanding the metabolism/processing of APP is crucial for AD therapeutics. Here we review current knowledge of APP processing regulation as well as the patho/physiological functions of APP and its metabolites.
Similar content being viewed by others

Background
Alzheimer's disease (AD) is the most prevalent neurodegenerative disorder, afflicting 10% of the population over the age of 65 and 50% of the population over the age of 85. A small subset (<10%) of AD cases result from an inherited autosomal dominant gene mutation and have an early-onset (the fourth to sixth decade). The majority of these familial AD (FAD) mutations are in the genes encoding β-amyloid precursor protein (APP) and presenilins (PS1 and PS2) [1–3]. Significant efforts have gone into understanding the mechanisms underlying the genes tied to FAD as the clinicopathological features are indistinguishable from regular onset AD.
AD is characterized in patients by an inexorably progressing dementia. In vulnerable brain regions, such as the hippocampus and cortex, there is an accumulation of extracellular neuritic plaques and intracellular neurofibrillary tangles. The neurofibrillary tangles (NFTs) consist largely of hyperphosphorylated twisted filaments of the microtubule-associated protein tau [4, 5]. Extracellular neuritic plaques are deposits of differently sized small peptides called β-amyloid (Aβ) that are derived via sequential proteolytic cleavages of the β-amyloid precursor protein (APP) [6].
APP and Its Function
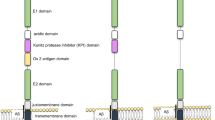
The APP gene is located on chromosome 21 in humans with three major isoforms arising from alternative splicing [3]. These are APP695, APP751 and APP770 (containing 695, 751, and 770 amino acids, respectively). APP751 and APP770 are expressed in most tissues and contain a 56 amino acid Kunitz Protease Inhibitor (KPI) domain within their extracellular regions. APP695 is predominantly expressed in neurons and lacks the KPI domain [7, 8]. There are reports showing that the protein and mRNA levels of KPI-containing APP isoforms are elevated in AD brain and associated with increased Aβ deposition [9]; and prolonged activation of extrasynaptic NMDA receptor in neurons can shift APP expression from APP695 to KPI-containing APP isoforms, accompanied with increased production of Aβ [10]. These findings may suggest that a dysregulated splicing of APP RNA contributes to disease pathogenesis.
APP belongs to a protein family that includes APP-like protein 1 (APLP1) and 2 (APLP2) in mammals [11–13], all are type-I transmembrane proteins and are processed in a similar fashion. The Aβ domain is unique to the APP protein, though the family shares several other conserved domains such as the E1 and E2 domains in the extracellular sequence. Studies with APP knockout mice suggest some functional redundancy between these APP homologs that appears to be exerted by motifs other than Aβ. APP knockout mice are viable and fertile, showing a relatively subtle abnormal phenotype [14, 15]. APLP1 and APLP2 knockout mice are also viable and fertile, though APP/APLP2 and APLP1/APLP2 double null mice and APP/APLP1/APLP2 triple null mice show early postnatal lethality [16–18]. Interestingly, the APP/APLP1 double null mice are viable [17], suggesting that APLP2 is crucial when either APP or APLP1 is absent.
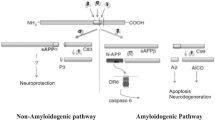
Although APP has been the subject of much study since its identification, its physiological function remains largely undetermined. A role for APP has been suggested in neurite outgrowth and synaptogenesis, neuronal protein trafficking along the axon, transmembrane signal transduction, cell adhesion, calcium metabolism, etc, all requiring additional in vivo evidence (reviewed in [19]). APP is proteolyzed into various fragments (see Figure 1) during its intracellular trafficking and these APP metabolites mediate various and sometimes adverse functions. Therefore, the net effect of full-length APP on cellular activity may be a combination of its metabolites' functions, temporospatially depending on the proportion of levels of each APP metabolite. Here we list several possible functions of full-length APP per se.

Schematic diagram of APP processing (not drawn in proportion). It is not clear whether caspases cleave membrane-associated APP forms or released AICD.
The similarity in topology and proteolytic processing between APP and Notch suggest that APP may function as a membrane receptor like Notch. Indeed, several APP ligands have been identified, such as Aβ [20], F-spondin [21] and nectrin-1 [22]. However, while binding of APP by these ligands can affect APP processing, the exact downstream signaling events triggered by such binding remains to be clarified and a bona fide membrane receptor function for APP remains speculative.
There is evidence linking APP to cell adhesion. APP was found to colocalize with β1 intergrins in neural cells [23]. An X-ray analysis showed that the E2 domain of APP can form antiparallel dimers [24]. Indeed, further study in cell cultures demonstrated that APP can form homodimers and heterodimers in a trans-dimerization manner with other APP family members and that such dimerization promotes intercellular adhesion [25].
APP undergoes rapid anterograde transport in neurons. During its transport, APP was found to interact with kinesin-I and functions as a kinesin-I membrane receptor to mediate axonal transport of β-secretase (BACE1) and PS1 [26, 27]. However, another study failed to verify the interaction between APP and kinesin-I and the co-transport of BACE1 and PS1 with APP [28]. We recently found that APP and its derived membrane-associated form, CTFs, can regulate cell surface delivery of PS1/γ-secretase but not BACE1 [29]. In addition, APP was found to be a major component of herpes simplex viral particles and likely mediates fast anterograde transport of these particles [30, 31]. Another study showed that increased doses of APP markedly decreased retrograde transport of nerve growth factor and resulted in degeneration of forebrain cholinergic neurons in a mouse model of Down's Syndrome [32]. APP was also found to interact with high-affinity choline transporter (CHT) through the C-terminal domain and APP deficiency affected CHT endocytosis [33]. Overall, most studies suggest that APP plays some role in regulating protein trafficking.
APP Processing
Full-length APP is a type I transmembrane protein. APP is synthesized in the endoplasmic reticulum (ER) and then transported through the Golgi apparatus to the trans-Golgi-network (TGN) where the highest concentration of APP is found in neurons at steady state [34–36]. Aβ is generated in the ER and Golgi/TGN [36]. From the TGN, APP can be transported in TGN-derived secretory vesicles to the cell surface where it is either cleaved by α-secretase to produce a soluble molecule, sAPPα [37], or re-internalized via an endosomal/lysosomal degradation pathway [38, 39]. It has been proposed that Aβ can also be generated in the endosomal/lysosomal system [40, 41]. While Aβ is neurotoxic, studies suggest that sAPPα is neuroprotective, making the subcellular distribution of APP an important factor in neurodegeneration [42–44]. Delineation of the mechanisms involved in APP trafficking are thus relevant and crucial to understanding the pathogenesis of AD.
α-secretase and α-processing
Cleavage of APP by α-secretase precludes Aβ generation as the cleavage site is within the Aβ domain (at the Lys16-Leu17 bond), and releases a large soluble ectodomain of APP called sAPPα. The generation of sAPPα is a constitutive event but can also be regulated by various reagents. Early studies suggested that α-secretase is a membrane-bound endoprotease which cleaves APP primarily at the plasma membrane [37]. Using proteinase inhibitor profiling, it was determined that α-secretase is a zinc metalloproteinase [45]. Several members of the ADAM (a disintegrin and metalloproteinase) family possess α-secretase-like activity and three of them have been suggested as the α-secretase: ADAM9, ADAM10, and ADAM17. Like APP, they are also type-I transmembrane proteins.
ADAM17 (also called tumor necrosis factor-α converting enzyme, TACE) can be proteolytically cleaved to release its extracellular domain as soluble TGF-α [46]. Manipulation of ADAM17 can alter α-cleavage of APP and Aβ generation, with regulated α-cleavage abolished in ADAM17-deficient cells, suggesting that ADAM17 is likely the α-secretase responsible for regulated APP cleavage [47]. Additionally, an ADAM17 inhibitor prevented regulated α-secretase activity in human neurons [48], whereas RNAi downregulation of ADAM10 had no effect on α-cleavage of APP [49]. Various other studies confirm that ADAM17 likely affects regulated, but not constitutive, α-cleavage in various cell lines [50].
Co-expression of ADAM9 with APP promoted sAPPα production upon phorbol ester treatment, suggesting that ADAM9 possesses α-secretase activity [51]. However, RNAi of ADAM9 had no effect on sAPPα generation [49], implying that ADAM9 is involved only in regulated α-cleavage.
Overexpression of ADAM10 increases α-cleavage, whereas a dominant-negative form of ADAM10 and RNAi of ADAM10 inhibit endogenous α-cleavage activity in several cell lines, including murine primary neurons [49, 52, 53]. Significantly, sAPPα generation was nearly abolished in the neurons of mice with neural ADAM10 conditionally knocked-out [54]. A dramatically reduced ADAM10 protein level in the platelets of sporadic AD patients was also found to correlate with the significantly decreased sAPPα levels found in their platlets and cerebrospinal fluid [55] and the reduced α-secretase activity in the temporal cortex homogenates of AD patients [56]. These studies strongly suggest that ADAM10 is the constitutive α-secretase that is active at the cell surface, though there may be some functional redundancy in α-cleavage among the ADAM family.
In contrast to Aβ, sAPPα has an important role in neuronal plasticity/survival and is protective against excitotoxicity [42, 43]. sAPPα also regulates neural stem cell proliferation and is important for early CNS development [57, 58]. We and others have also found that sAPPα can inhibit stress-induced CDK5 activation and participate in various neuroprotective reagent-mediated excitoprotection [44, 59–61]. Interestingly, expression of sAPPα alone is able to rescue the abnormalities of APP deficient mice [62], implying that most of APP's physiological function is mediated by sAPPα.
β-secretase and β-processing
The first step in Aβ generation is cleavage of APP by the β-secretase. In 1999-2000, several groups concomitantly identified BACE1 (also called Asp2 or memapsin 2) as the major β-secretase [63–66]. BACE1, the most common name for the protease, is a membrane-bound aspartyl protease with a characteristic type I transmembrane domain near the C-terminus [63, 64]. Overexpression or downregulation of BACE1 induces or inhibits cleavage of APP at the known β-site locations, Asp1 and Glu11, respectively. In vitro studies with synthetic APP peptides confirm cleavage by BACE1. These results provide convincing evidence that BACE1 is the β-secretase involved in APP metabolism [63–67]; and BACE1 activity is thought to be the rate-limiting factor in Aβ generation from APP.
A larger precursor, pro-BACE1, is modified by glycosylation, phosphorylation and cleaved by a furin-like endoprotease to produce mature BACE1 [68, 69]. BACE1 requires an acidic environment for optimal activity and, as expected, overexpressed BACE1 in various pre-mitotic cell lines is mainly found in the early Golgi, late Golgi/early endosomes, and endosomes that provide an acidic environment. In addition, BACE1 can be found at the cell surface [64, 70–72]. The mechanisms regulating BACE1 trafficking and activity have not been fully elucidated. Some studies found that BACE1 can interact with reticulon/Nogo proteins, whose increased expression can block BACE1 in the ER with a neutral pH environment and thus inhibit BACE1 activity in Aβ generation [73–75]. On the other hand, Golgi-localized γ-ear-containing ARF-binding (GGA) proteins have been found to interact with BACE1 and regulate its trafficking between the late Golgi and early endosomes; and depletion of GGA proteins increases the accumulation of BACE1 in acidic early endosomes for enhanced BACE1 stability and cleavage of APP [76–78].
The viability of BACE1 as a therapeutic target has been investigated by a number of studies. An early study suggested that BACE1 knockout mice do not produce detectable levels of Aβ and have no severe phenotypic abnormalities [79]. BACE1 deficiency in AD model mice have been shown to rescue cholinergic dysfunction, neuronal loss and memory deficits, correlating with a dramatic reduction in Aβ40/42 levels [79–81]. Several studies have found that BACE1 protein and activity levels are elevated in the regions of the brain affected by AD [82, 83]. Together these results suggest BACE1 as a good therapeutic target for AD. However, more recent studies have found several phenotypic abnormalities in BACE1 KO mice. Dominguez et al. [84] observed a variable but significant number of BACE1 null mice died in the first weeks after birth. The BACE1 null mice that survive were smaller than their littermates, presented with hyperactive behavior, and had subtle electrophysiological alterations in the steady-state inactivation of their voltage-gated sodium channels. They also were affected by hypomyelination of peripheral nerves and had altered neurological behaviors such as reduced grip strength and elevated pain sensitivity, likely due to the deficiency of neuregulin processing in the absence of BACE1, as neuregulin 1 is another substrate of BACE1 [85, 86]. Furthermore, additional BACE1 substrates have been identified, including the voltage-gated sodium channel (Nav1) β2 subunit, Golgi-localized membrane-bound α2,6-sialyltransferase, P-selectin glycoprotein ligand -1, etc. (reviewed in [87]). Therefore, BACE1 is likely not as safe a drug target as first assumed.
BACE2 is a homolog of BACE1 that maps to 21q22.3 [88], the region critical for Down's syndrome (DS). As DS also results in Aβ accumulation, the genes location suggests a link between BACE2 and APP processing. Indeed, BACE2 cleaves β-secretase substrates such as wild-type and Swedish mutant APP, similar to BACE1, in enzymatic In vitro assays [89]. However, BACE2 expression in neurons is substantially lower than BACE1 [90] and cellular BACE2 cleaves APP near the α-secretase site much more efficiently than at the β-secretase site [91]. These results suggest that BACE1 is the primary β-secretase but do not exclude a potential contribution of BACE2 towards AD pathogenesis. While BACE2 knockout mice are healthy overall, a deficiency of both BACE1 and BACE2 enhanced the BACE1 KO lethality phenotype, suggesting a slight functional redundancy [84].
In addition to BACE1 and BACE2, cathepsin B has been proposed as an additional β-secretase. Inhibition of cathepsin B has been found to reduce Aβ production both in vivo and in vitro[92, 93]. However, whether cathepsin B really exerts physiological β-secretase activity requires further validation.
Upon β-cleavage, the ectodomain of APP is also released as soluble APPβ (sAPPβ). Although sAPPβ only differs from sAPPα by lacking the Aβ1-16 region at its carboxyl-terminus, sAPPβ was reported to function as a death receptor 6 ligand and mediate axonal pruning and neuronal cell death [94]. A recent report found that sAPPβ can rescue gene expression of transthyretin and Klotho, which is decreased in APP/APLP2 deficient mice, but cannot rescue the lethality and neuromuscular synapse defects of these mice, suggesting a gene expression regulation function for sAPPβ that is independent of developmental APP functions [95].
After α- and β-cleavage, the carboxyl terminal fragments (CTFs) of APP, known as αCTF and βCTF, respectively, remain membrane-associated and will be further cleaved by γ-secretase. Since these APP CTFs are intermediate products, their functions have been less characterized. However, overexpression of APP βCTF was found to be cytotoxic and cause neuronal degeneration, perhaps by perturbing APP signal transduction [96, 97]. It is also possible that APP βCTF's cytotoxic effect is actually mediated by the end products of γ- and/or caspase-cleavage including APP intracellular domain (AICD), C31 and Jcasp which are cytotoxic (see below). We recently found that APP βCTF can regulate cell surface delivery of γ-sceretase, perhaps through the direct binding of enzyme-substrate [29]. It is possible that APP αCTF possesses a similar effect since it is also the substrate of γ-secretase.
γ-secretase and γ-processing
APP αCTF and βCTF are further cleaved by γ-secretase to generate p83 and Aβ, respectively. The p83 fragment is rapidly degraded and widely believed to possess no important function, if any. γ-secretase-mediated cleavage is unique in that the cleavage takes place within the transmembrane domain, though the exact site can vary. γ-cleavage can yield both Aβ40, the majority species, and Aβ42, the more amyloidogenic species, as well as release the intracellular domain of APP (AICD). Recent data has shown that PS/γ-secretase also mediates ζ-site cleavage (Aβ46) [98, 99] and ε-site cleavage (Aβ49) [100, 101], suggesting a sequential cleavage model where cleavage at the ε-site is followed by the ζ-site and γ-site.
Multiple lines of biochemical evidence have shown γ-secretase activity to reside in a high molecular weight complex consisting of at least four components: presenilin (PS, PS1 or PS2), Nicastrin, anterior pharynx-defective-1 (APH-1), and presenilin enhancer-2 (PEN-2) [102, 103]. In mammals there are two presenilin homologs, PS1 and PS2 [1, 2]. Mutations in these two genes, particularly PS1, are causative in the majority of familial AD (FAD) cases. PSs are multi-transmembrane proteins with an unclear number of transmembrane domains [104]. Nascent PSs undergo endroproteolytic cleavage with the resulting amino-terminal fragment (NTF) and carboxyl-terminal fragment (CTF) forming a functional PS heterodimer [105]. PSs possess two highly conserved aspartate residues indispensable for γ-secretase activity. The PS1 NTF/CTF heterodimers are bound by transition-state analogue γ-secretase inhibitors [102, 106], suggesting that PSs are the crucial catalytic components of γ-secretase. This notion has recently been confirmed by in vitro assays [107]. Nicastrin, the first identified cofactor of PS, is a type I transmembrane glycoprotein that is considered the scaffolding protein within the γ-secretase complex. One study showed that the ectodomain of Nicastrin binds to APP and Notch and can recruit them into the γ-secretase complex, suggesting that Nicastrin may act as the γ-secretase receptor [108]. Another two components, APH1 and PEN2, were identified through genetic screening of Caenorhabditis elegans[109, 110]. APH-1 interacts with Nicastrin to form a stable intermediate in an early assembly stage of the γ-secretase complex [102]. PEN-2 regulates PS endoproteolysis [107, 108]. Each of these four γ-secretase components has been found necessary for the enzymatic activity of the complex with deficiency in any of them dramatically impairing γ-secretase activity. Coexpression of the four components in the yeast Saccharomyces cerevisiae has been found to be necessary and sufficient to reconstitute γ-secretase activity, which is not endogenous to yeast [111, 112].
In addition to the four critical components, several other factors have been proposed as additional γ-secretase components. However, these factors play a modulatory role and are not essential for γ-secretase activity: CD147 is a transmembrane glycoprotein and interacts with all four essential γ-secretase components. Downregulation of CD147 increases Aβ production but its overexpression has no effect on Aβ generation [113]. TMP21/p23 binds to the γ-secretase complex and regulates γ-cleavage, but not ε-cleavage, through its transmembrane domain [114, 115]. However, another study failed to confirm the binding of TMP23/p21 to γ-secretase, but rather suggested that TMP21/p23, which belongs to the p24 cargo family involved in vesicular trafficking regulation, influences APP trafficking and thus Aβ generation [116]. Recently, a novel γ-secretase activating protein (GSAP) was identified and GSAP was found to selectively increase Aβ production through interaction with both γ-secretase and the APP CTF substrate [117]. Additional validation and investigation of the role of these proteins in the γ-secretase complex is required.
Strong evidence suggests that the γ-secretase complex resides primarily in the ER, Golgi/TGN, endocytic and intermediate compartments--most of which (except the TGN) are not major subcellular localizations for APP [118, 119]. In addition to cleaving APP CTFs, γ-secretase cleaves a series of functionally important transmembrane proteins, including Notch [120], cadherin [114], tyrosinase [121], ErbB4 [79], CD44 [70], etc.) (see review [122]). The cleavage of various substrates appears to be dependent on the subcellular compartment; APP is mainly cleaved in the TGN and early endosomal domains whereas Notch is primarily cleaved at the plasma membrane [34, 36, 123]. Thus a disturbance in the localization of the γ-secretase complex may play some role in abnormal Aβ generation and AD pathogenesis.
γ-cleavage can release the intracellular domains (ICDs) of the substrates. Notch intracellular domain (NICD) is well-known to translocate into the nucleus and regulate genes critical to development [124, 125]. The other intracellular domains may be of comparable importance. For example, the ErbB4 ICD has been found to bind to astrocytic gene promoters to suppress their expression [126]. In a similar fashion, released AICD has been shown to possess transactivation activity and can regulate transcription of multiple genes including APP, GSK-3β, KAI1, neprilysin, BACE1, p53, EGFR, and LRP1[127–132]. In addition, free AICD can induce apoptosis and may play a role in sensitizing neurons to toxic stimuli [133, 134]. However, as the intracellular domain of APP, one important function of AICD is to facilitate the interaction of APP with various cytosolic factors that regulate APP's intracellular trafficking and/or signal transduction function. Interestingly, it seems that AICD-mediated APP interaction with different factors is controlled by the phosphorylation state of AICD [135].
Caspase processing
In addition to secretases, caspases (predominantly caspase-3) can directly cleave APP at position Asp664 (based on the APP695 sequence) within the cytoplasmic tail during apoptosis to release a fragment containing the last 31 amino acids of APP (called C31). Additional γ-cleavage further generates the fragment (called Jcasp) containing the region between γ- and caspase-cleavage sites [136–138]. Although original data found that caspase cleavage affects amyloidogenic processing of APP [137], further study suggests not [139]. However, during Aβ-induced neurotoxicity, activated caspases cleave APP to generate C31 and Jcasp, which are also neurotoxic, therefore initiating a detrimental cascade [140]. One possible mechanism for C31's toxicity is that C31 complexes with APP to recruit the interacting partners that initiate the signals related to cellular toxicity [136]. Compared to C31, Jcasp appears to play a minor role in cytotoxicity [136]. Importantly, caspase cleavage of APP seems to be crucial for Aβ-mediated neurotoxicity, as an APP mutation at position Asp664 to inhibit the caspase-cleavage in transgenic mice negated the synapse, electrophysiology, and behavioral abnormalities, even though Aβ plaques were still abundant in the brain [141].
Aβ Function
The neurotoxic effect of Aβ has been well-established and will not be specifically emphasized here. Multiple lines of evidence demonstrate that overproduction of Aβ results in a neurodegenerative cascade leading to synaptic dysfunction, formation of intraneuronal fibrillary tangles and eventually neuron loss in affected areas of the brain [6, 142]. There are two main toxic species, Aβ40 and Aβ42, with Aβ42 more hydrophobic and more prone to fibril formation while only making up about 10% of the Aβ peptide produced [143]. Studies done on familial AD (FAD) mutations consistently show increases in the ratio of Aβ42/40 [105, 144], suggesting that elevated levels of Aβ42 relative to Aβ40 is critical for AD pathogenesis, probably by providing the core for Aβ assembly into oligomers, fibrils and amyloidogenic plaques [145, 146].
Although the majority of Aβ is secreted out of the cell, Aβ can be generated in several subcellular compartments within the cell, such as the ER, Golgi/TGN, and endosome/lysosome. In addition, extracellular Aβ can be internalized by the cell for degradation. The intracellular existence of Aβ implies that Aβ may accumulate within neurons and contribute to disease pathogenesis. Confirming this, intraneuronal Aβ immunoreactivity has been found in the hippocampal and entorhinal cortical regions which are prone to early AD pathology in patients with mild cognitive impairment (MCI) [147]. In Down Syndrome (DS) patients, the accumulation of intracellular Aβ precedes extracellular plaque formation [148] and the level of intraneuronal Aβ decreases as the extracellular Aβ plaques accumulate [149]. Studies with transgenic mouse models consistently confirm these results, revealing intracellular Aβ accumulation as an early event in the neuropathological phenotype with decreasing intraneuronal levels of Aβ as extracellular plaques build up [150–152]. Intraneuronal Aβ can also impair amygdala-dependent emotional responses by affecting the ERK/MAPK signaling pathway [153]. Inhibition of dynamin-mediated but not clathrin-mediated Aβ internalization was also found to reduce Aβ-induced neurotoxicity [154]. One recent study suggests that internalized Aβ can aggregate within the cell and disrupt the vesicular membrane, thus contributing to its pathological effect [155].
Aβ was originally regarded as an abnormal and toxic species restricted to the brains of aged or demented humans. The discovery of soluble Aβ species in the bodily fluids of various species [156] and in the conditioned medium of cultured cells [157] has refuted this concept and implied a physiological function for Aβ. Although excessive Aβ causes synaptic dysfunction and synapse loss [142], low levels of Aβ increase hippocampal long-term potentiation and enhances memory, indicating a novel positive, modulatory role on neurotransmission and memory [158, 159]. Picomolar levels of Aβ can also rescue neuronal cell death induced by inhibition of Aβ generation (by exposure to inhibitors of β- or γ-scretases) [160], possibly through regulating the potassium ion channel expression, hence affecting neuronal excitability [161]. One study using a transgenic Caenorpabditis elegans model found that intracellular Aβ aggregation in muscle cells may trap excess free copper to reduce copper-mediated cytotoxic effects [162]. However, whether Aβ can form intracellular aggregates in human peripheral cells to exert a physiologically protective function remains to be determined.
Regulation of APP Processing at the Trafficking Level
Alterations in APP intracellular trafficking and localization directly impact Aβ production as APP is processed by two mutually exclusive pathways. The available evidence has shown that intracellular trafficking of APP is regulated by a number of factors.
Trafficking factors
Intracellular trafficking of proteins requires the involvement of a series of cytosolic factors. An increasing number of proteins that interact with APP or act as trafficking factors are being implicated in the regulation of Aβ generation and APP trafficking. For example, the APP C-terminus has been found to interact with all three mint (X11) family members (mint1, mint2, and mint3) involved in trafficking regulation [163–165]. APP interaction with mint proteins has been shown to affect APP processing by stabilizing cellular APP, altering both sAPPα and Aβ generation and secretion [166]. Rab6, a member of the GTP-binding protein family of membrane trafficking regulators, is implicated in protein transport along biosynthetic and endocytic pathways and has also been found to affect APP processing. Moreover, internalization of APP from the cell surface for endosomal/lysosomal degradation can be mediated by clathrin. Clathrin-modulated endocytosis is tightly controlled, requiring the participation of AP-2, dynamin I, and many other factors [167–169]. When the endocytic pathway is inhibited by overexpression of a dominant-negative form of dynamin I, APP processing is also affected [170, 171]. It is conceivable that other important vesicular transport factors may also affect APP processing through regulation of general protein trafficking.
In addition to general trafficking modulators, several other proteins have been found to regulate APP trafficking in a more specific manner, possibly through their direct binding to APP. PS1 is the catalytic component of the γ-secretase complex but has also been demonstrated to regulate the intracellular trafficking of several membrane proteins, including the other γ-secretase components (nicastrin, APH-1 and PEN-2), TrkB, and ICAM-5/telecephalin [122]. We and others have shown that PS1 can also regulate the intracellular trafficking of APP. Expression of a loss of function PS1 variant, or the absence of PS1, results in increased budding/generation of vesicles from both the ER and TGN containing APP along with a concomitant increase in complex glycosylation and APP localization at the cell surface. In contrast, the FAD-linked PS1 mutant variants significantly reduce budding from the ER and TGN and result in decreased delivery of APP to the cell surface [172]. These results suggest the possibility that FAD-linked PS1 variants increase Aβ production by decreasing intracellular transport of APP, prolonging the availability of APP for cleavage by β- and γ-secretases within the TGN. PS1 may regulate protein trafficking through its interaction with several cytosolic factors involved in the regulation of vesicular transport such as Rab11, Rab6 and Rab GDI [173–175]. We have also found that PS1 interacts with phospholipase D1 (PLD1), a phospholipid-modifying enzyme regulating membrane trafficking events. This PS1-PLD1 interaction recruits PLD1 to the Golgi/TGN and thus potentially alters APP trafficking as PLD1 overexpression promotes budding of vesicles from the TGN containing APP and increases cell surface levels of APP [176, 177].
SorLA/LR11 is a type I membrane protein expressed in neurons and reduced in the brains of AD patients [178, 179]. Although the function of SorLA/LR11 is not known, its homology with sorting receptors that are involved with transport between the plasma membrane, endosomes and the Golgi suggests a protein trafficking function [180, 181]. Recently it was found that SorLA/LR11 overexpression redistributed APP to the Golgi, decreasing Aβ generation, while SorLA/LR11 knockout mice have increased levels of Aβ, as found in AD patients [182]. Additionally, some inherited variants of the SorLA/LR11 gene were found to associate with late-onset AD [183].
Low-density lipoprotein receptor-related protein (LRP) is a SorLA/LR11-related protein that binds to APP through Fe65, a cytoplasmic adaptor protein [184]. LRP has been shown to bind, directly or indirectly, with Aβ to mediate its clearance [185, 186]. Antagonizing the extracellular interaction between cell-surface APP and LRP increased the level of cell surface APP while decreasing Aβ generation [187]. Using an AD mouse model, expression of a functional LRP minireceptor in neurons resulted in increased memory deficits and higher Aβ levels in the aged mice [188]. An LRP-related protein 1B (LRP1B) has a similar effect, binding APP at the plasma membrane, preventing APP internalization, and leading to decreased Aβ generation and increased sAPPα secretion [189].
Signal transduction
Epidemiological evidence suggests that post-menopausal women receiving replacement therapy of the sex hormone estrogen have a reduced risk and delayed onset of AD while elderly women with reduced levels of circulating estrogen have an increased incidence of AD [190–192]. The primary mechanism of estrogen's protection against AD development is still unclear. Several potential mechanisms have been proposed: (1) estrogen may act on interlukin 6 to antagonize inflammation [193]; (2) the phenolic structure of estrogen may contribute to its antioxidant effect in cells [194]; (3) estrogen may reduce the level of appolipoprotein E (ApoE), with the isoform ApoE4 being a strong risk factor for AD development [195]; and (4) gonadal steroids may reduce the protein level of PS1 and thus γ-secretase activity [196].
We have found that estrogen may reduce Aβ levels by stimulating the α-secretase pathway and thereby inhibit Aβ generation. Estrogen can stimulate the formation of APP-containing vesicles from the TGN in cell-free systems derived from both neuroblastomas and primary neurons [197–199]. Interestingly, the stimulation of sAPPα secretion by estrogen can be blocked by a PKC inhibitor, suggesting the involvement of a PKC-dependent pathway [200]. Indeed, phorbol ester's effect on sAPPα secretion and Aβ generation though activation of protein kinase C (PKC) has been known for a long time [201–203]. PKC stimulates sAPPα secretion, reducing Aβ levels, even when the phosphorylation sites on APP are mutated or the entire cytoplasmic domain is deleted [204]. While PKC can directly phosphorylate APP Ser655 [205], it appears to affect APP metabolism by phosphorylating a different target. One potential target is a TGN phosphoprotein, resulting in transport of APP from the TGN to the cell surface. Our studies have shown that PKC increases the formation of APP-containing secretory vesicles from the TGN in a cell-free system [206]. In support of this, protein kinase A (PKA) has similar effects on reducing Aβ generation and stimulating the budding of APP-containing vesicles from the TGN [207]. The effects of PKC and PKA are additive, suggesting that while they both appear to act through stimulating vesicle formation from the TGN, the regulatory mechanisms involved are independent [207]. Additionally, estrogen has been found to facilitate binding of Rab11 to the TGN membrane and a dominant negative Rab11 mutant abolishes the estrogen-regulated change in APP trafficking, leading to increased Aβ formation [197].
The sex hormone testosterone decreases with age in older men and postmenopausal women. Animal model studies of testosterone treatments show neuroprotective and neuroexcitatory benefits along with improved cognitive performance [208]. Some studies have suggested that testosterone exerts its beneficial effect through an aromatase-mediated conversion into estrogen [209, 210]. However, a recent study blocking the conversion of testosterone to estrogen found an estrogen-independent improvement in cognitive function and lowering of plaque formation along with a decrease in BACE1 mRNA, protein level, and activity [211]. In addition, testosterone may also reduce the protein level of PS1 [196].
Conclusion
The overproduction and accumulation of Aβ in the brain are key pathogenic events in AD progression. In addition to Aβ, APP can be proteolyzed by different secretases and caspases. In this review we have discussed APP processing regulation and the physio/pathological functions of various APP metabolites. Further elucidation of APP metabolism will be important for identifying new potential therapies to reduce Aβ accumulation and combat AD.
References
Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, et al: Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995, 269: 973-977. 10.1126/science.7638622.
Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, et al: Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995, 375: 754-760. 10.1038/375754a0.
Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al: Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991, 349: 704-706. 10.1038/349704a0.
Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR: Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000, 33: 95-130. 10.1016/S0165-0173(00)00019-9.
Gendron TF, Petrucelli L: The role of tau in neurodegeneration. Mol Neurodegener. 2009, 4: 13-10.1186/1750-1326-4-13.
Selkoe DJ: The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol. 1998, 8: 447-453. 10.1016/S0962-8924(98)01363-4.
Rohan de Silva HA, Jen A, Wickenden C, Jen LS, Wilkinson SL, Patel AJ: Cell-specific expression of beta-amyloid precursor protein isoform mRNAs and proteins in neurons and astrocytes. Brain Res Mol Brain Res. 1997, 47: 147-156. 10.1016/S0169-328X(97)00045-4.
Kang J, Muller-Hill B: Differential splicing of Alzheimer's disease amyloid A4 precursor RNA in rat tissues: PreA4(695) mRNA is predominantly produced in rat and human brain. Biochem Biophys Res Commun. 1990, 166: 1192-1200. 10.1016/0006-291X(90)90992-V.
Menendez-Gonzalez M, Perez-Pinera P, Martinez-Rivera M, Calatayud MT, Blazquez Menes B: APP processing and the APP-KPI domain involvement in the amyloid cascade. Neurodegener Dis. 2005, 2: 277-283. 10.1159/000092315.
Bordji K, Becerril-Ortega J, Nicole O, Buisson A: Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid-ss production. J Neurosci. 2010, 30: 15927-15942. 10.1523/JNEUROSCI.3021-10.2010.
Wasco W, Bupp K, Magendantz M, Gusella JF, Tanzi RE, Solomon F: Identification of a mouse brain cDNA that encodes a protein related to the Alzheimer disease-associated amyloid beta protein precursor. Proc Natl Acad Sci USA. 1992, 89: 10758-10762. 10.1073/pnas.89.22.10758.
Wasco W, Gurubhagavatula S, Paradis MD, Romano DM, Sisodia SS, Hyman BT, Neve RL, Tanzi RE: Isolation and characterization of APLP2 encoding a homologue of the Alzheimer's associated amyloid beta protein precursor. Nat Genet. 1993, 5: 95-100. 10.1038/ng0993-95.
Coulson EJ, Paliga K, Beyreuther K, Masters CL: What the evolution of the amyloid protein precursor supergene family tells us about its function. Neurochem Int. 2000, 36: 175-184. 10.1016/S0197-0186(99)00125-4.
Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, Heavens RP, Dawson GR, Boyce S, Conner MW, et al: beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell. 1995, 81: 525-531. 10.1016/0092-8674(95)90073-X.
Dawson GR, Seabrook GR, Zheng H, Smith DW, Graham S, O'Dowd G, Bowery BJ, Boyce S, Trumbauer ME, Chen HY, et al: Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the beta-amyloid precursor protein. Neuroscience. 1999, 90: 1-13. 10.1016/S0306-4522(98)00410-2.
von Koch CS, Zheng H, Chen H, Trumbauer M, Thinakaran G, van der Ploeg LH, Price DL, Sisodia SS: Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol Aging. 1997, 18: 661-669. 10.1016/S0197-4580(97)00151-6.
Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rulicke T, von Kretzschmar H, von Koch C, Sisodia S, Tremml P, et al: Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci. 2000, 20: 7951-7963.
Herms J, Anliker B, Heber S, Ring S, Fuhrmann M, Kretzschmar H, Sisodia S, Muller U: Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. Embo J. 2004, 23: 4106-4115. 10.1038/sj.emboj.7600390.
Zheng H, Koo EH: The amyloid precursor protein: beyond amyloid. Mol Neurodegener. 2006, 1: 5-10.1186/1750-1326-1-5.
Lorenzo A, Yuan M, Zhang Z, Paganetti PA, Sturchler-Pierrat C, Staufenbiel M, Mautino J, Vigo FS, Sommer B, Yankner BA: Amyloid beta interacts with the amyloid precursor protein: a potential toxic mechanism in Alzheimer's disease. Nat Neurosci. 2000, 3: 460-464. 10.1038/74833.
Ho A, Sudhof TC: Binding of F-spondin to amyloid-beta precursor protein: a candidate amyloid-beta precursor protein ligand that modulates amyloid-beta precursor protein cleavage. Proc Natl Acad Sci USA. 2004, 101: 2548-2553. 10.1073/pnas.0308655100.
Lourenco FC, Galvan V, Fombonne J, Corset V, Llambi F, Muller U, Bredesen DE, Mehlen P: Netrin-1 interacts with amyloid precursor protein and regulates amyloid-beta production. Cell Death Differ. 2009, 16: 655-663. 10.1038/cdd.2008.191.
Yamazaki T, Koo EH, Selkoe DJ: Cell surface amyloid beta-protein precursor colocalizes with beta 1 integrins at substrate contact sites in neural cells. J Neurosci. 1997, 17: 1004-1010.
Wang Y, Ha Y: The X-ray structure of an antiparallel dimer of the human amyloid precursor protein E2 domain. Mol Cell. 2004, 15: 343-353. 10.1016/j.molcel.2004.06.037.
Soba P, Eggert S, Wagner K, Zentgraf H, Siehl K, Kreger S, Lower A, Langer A, Merdes G, Paro R, et al: Homo- and heterodimerization of APP family members promotes intercellular adhesion. Embo J. 2005, 24: 3624-3634. 10.1038/sj.emboj.7600824.
Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS: Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron. 2000, 28: 449-459. 10.1016/S0896-6273(00)00124-0.
Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS: Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature. 2001, 414: 643-648. 10.1038/414643a.
Lazarov O, Morfini GA, Lee EB, Farah MH, Szodorai A, DeBoer SR, Koliatsos VE, Kins S, Lee VM, Wong PC, et al: Axonal transport, amyloid precursor protein, kinesin-1, and the processing apparatus: revisited. J Neurosci. 2005, 25: 2386-2395. 10.1523/JNEUROSCI.3089-04.2005.
Liu Y, Zhang YW, Wang X, Zhang H, You X, Liao FF, Xu H: Intracellular trafficking of presenilin 1 is regulated by beta-amyloid precursor protein and phospholipase D1. J Biol Chem. 2009, 284: 12145-12152. 10.1074/jbc.M808497200.
Satpute-Krishnan P, DeGiorgis JA, Bearer EL: Fast anterograde transport of herpes simplex virus: role for the amyloid precursor protein of alzheimer's disease. Aging Cell. 2003, 2: 305-318. 10.1046/j.1474-9728.2003.00069.x.
Satpute-Krishnan P, DeGiorgis JA, Conley MP, Jang M, Bearer EL: A peptide zipcode sufficient for anterograde transport within amyloid precursor protein. Proc Natl Acad Sci USA. 2006, 103: 16532-16537. 10.1073/pnas.0607527103.
Salehi A, Delcroix JD, Belichenko PV, Zhan K, Wu C, Valletta JS, Takimoto-Kimura R, Kleschevnikov AM, Sambamurti K, Chung PP, et al: Increased App expression in a mouse model of Down's syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006, 51: 29-42. 10.1016/j.neuron.2006.05.022.
Wang B, Yang L, Wang Z, Zheng H: Amyolid precursor protein mediates presynaptic localization and activity of the high-affinity choline transporter. Proc Natl Acad Sci USA. 2007, 104: 14140-14145. 10.1073/pnas.0704070104.
Xu H, Sweeney D, Wang R, Thinakaran G, Lo AC, Sisodia SS, Greengard P, Gandy S: Generation of Alzheimer beta-amyloid protein in the trans-Golgi network in the apparent absence of vesicle formation. Proc Natl Acad Sci USA. 1997, 94: 3748-3752. 10.1073/pnas.94.8.3748.
Hartmann T, Bieger SC, Bruhl B, Tienari PJ, Ida N, Allsop D, Roberts GW, Masters CL, Dotti CG, Unsicker K, et al: Distinct sites of intracellular production for Alzheimer's disease A beta40/42 amyloid peptides. Nat Med. 1997, 3: 1016-1020. 10.1038/nm0997-1016.
Greenfield JP, Tsai J, Gouras GK, Hai B, Thinakaran G, Checler F, Sisodia SS, Greengard P, Xu H: Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc Natl Acad Sci USA. 1999, 96: 742-747. 10.1073/pnas.96.2.742.
Sisodia SS: Beta-amyloid precursor protein cleavage by a membrane-bound protease. Proc Natl Acad Sci USA. 1992, 89: 6075-6079. 10.1073/pnas.89.13.6075.
Nordstedt C, Caporaso GL, Thyberg J, Gandy SE, Greengard P: Identification of the Alzheimer beta/A4 amyloid precursor protein in clathrin-coated vesicles purified from PC12 cells. J Biol Chem. 1993, 268: 608-612.
Caporaso GL, Takei K, Gandy SE, Matteoli M, Mundigl O, Greengard P, De Camilli P: Morphologic and biochemical analysis of the intracellular trafficking of the Alzheimer beta/A4 amyloid precursor protein. J Neurosci. 1994, 14: 3122-3138.
Haass C, Hung AY, Schlossmacher MG, Teplow DB, Selkoe DJ: beta-Amyloid peptide and a 3-kDa fragment are derived by distinct cellular mechanisms. J Biol Chem. 1993, 268: 3021-3024.
Haass C, Hung AY, Schlossmacher MG, Oltersdorf T, Teplow DB, Selkoe DJ: Normal cellular processing of the beta-amyloid precursor protein results in the secretion of the amyloid beta peptide and related molecules. Ann N Y Acad Sci. 1993, 695: 109-116. 10.1111/j.1749-6632.1993.tb23037.x.
Furukawa K, Sopher BL, Rydel RE, Begley JG, Pham DG, Martin GM, Fox M, Mattson MP: Increased activity-regulating and neuroprotective efficacy of alpha-secretase-derived secreted amyloid precursor protein conferred by a C-terminal heparin-binding domain. J Neurochem. 1996, 67: 1882-1896. 10.1046/j.1471-4159.1996.67051882.x.
Mattson MP: Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997, 77: 1081-1132.
Han P, Dou F, Li F, Zhang X, Zhang YW, Zheng H, Lipton SA, Xu H, Liao FF: Suppression of cyclin-dependent kinase 5 activation by amyloid precursor protein: a novel excitoprotective mechanism involving modulation of tau phosphorylation. J Neurosci. 2005, 25: 11542-11552. 10.1523/JNEUROSCI.3831-05.2005.
Roberts SB, Ripellino JA, Ingalls KM, Robakis NK, Felsenstein KM: Non-amyloidogenic cleavage of the beta-amyloid precursor protein by an integral membrane metalloendopeptidase. J Biol Chem. 1994, 269: 3111-3116.
Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, et al: A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997, 385: 729-733. 10.1038/385729a0.
Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA: Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998, 273: 27765-27767. 10.1074/jbc.273.43.27765.
Blacker M, Noe MC, Carty TJ, Goodyer CG, LeBlanc AC: Effect of tumor necrosis factor-alpha converting enzyme (TACE) and metalloprotease inhibitor on amyloid precursor protein metabolism in human neurons. J Neurochem. 2002, 83: 1349-1357. 10.1046/j.1471-4159.2002.01228.x.
Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW, Kremmer E, Rossner S, Lichtenthaler SF: ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. Embo J. 2010, 29: 3020-3032. 10.1038/emboj.2010.167.
Merlos-Suarez A, Ruiz-Paz S, Baselga J, Arribas J: Metalloprotease-dependent protransforming growth factor-alpha ectodomain shedding in the absence of tumor necrosis factor-alpha-converting enzyme. J Biol Chem. 2001, 276: 48510-48517.
Koike H, Tomioka S, Sorimachi H, Saido TC, Maruyama K, Okuyama A, Fujisawa-Sehara A, Ohno S, Suzuki K, Ishiura S: Membrane-anchored metalloprotease MDC9 has an alpha-secretase activity responsible for processing the amyloid precursor protein. Biochem J. 1999, 343 (Pt 2): 371-375. 10.1042/0264-6021:3430371.
Lopez-Perez E, Zhang Y, Frank SJ, Creemers J, Seidah N, Checler F: Constitutive alpha-secretase cleavage of the beta-amyloid precursor protein in the furin-deficient LoVo cell line: involvement of the pro-hormone convertase 7 and the disintegrin metalloprotease ADAM10. J Neurochem. 2001, 76: 1532-1539. 10.1046/j.1471-4159.2001.00180.x.
Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F: Constitutive and regulated alpha-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA. 1999, 96: 3922-3927. 10.1073/pnas.96.7.3922.
Jorissen E, Prox J, Bernreuther C, Weber S, Schwanbeck R, Serneels L, Snellinx A, Craessaerts K, Thathiah A, Tesseur I, et al: The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J Neurosci. 2010, 30: 4833-4844. 10.1523/JNEUROSCI.5221-09.2010.
Colciaghi F, Borroni B, Pastorino L, Marcello E, Zimmermann M, Cattabeni F, Padovani A, Di Luca M: [alpha]-Secretase ADAM10 as well as [alpha]APPs is reduced in platelets and CSF of Alzheimer disease patients. Mol Med. 2002, 8: 67-74.
Tyler SJ, Dawbarn D, Wilcock GK, Allen SJ: alpha- and beta-secretase: profound changes in Alzheimer's disease. Biochem Biophys Res Commun. 2002, 299: 373-376. 10.1016/S0006-291X(02)02635-9.
Caille I, Allinquant B, Dupont E, Bouillot C, Langer A, Muller U, Prochiantz A: Soluble form of amyloid precursor protein regulates proliferation of progenitors in the adult subventricular zone. Development. 2004, 131: 2173-2181. 10.1242/dev.01103.
Ohsawa I, Takamura C, Morimoto T, Ishiguro M, Kohsaka S: Amino-terminal region of secreted form of amyloid precursor protein stimulates proliferation of neural stem cells. Eur J Neurosci. 1999, 11: 1907-1913. 10.1046/j.1460-9568.1999.00601.x.
Ma T, Zhao Y, Kwak YD, Yang Z, Thompson R, Luo Z, Xu H, Liao FF: Statin's excitoprotection is mediated by sAPP and the subsequent attenuation of calpain-induced truncation events, likely via rho-ROCK signaling. J Neurosci. 2009, 29: 11226-11236. 10.1523/JNEUROSCI.6150-08.2009.
Marcade M, Bourdin J, Loiseau N, Peillon H, Rayer A, Drouin D, Schweighoffer F, Desire L: Etazolate, a neuroprotective drug linking GABA(A) receptor pharmacology to amyloid precursor protein processing. J Neurochem. 2008, 106: 392-404. 10.1111/j.1471-4159.2008.05396.x.
Levites Y, Amit T, Mandel S, Youdim MB: Neuroprotection and neurorescue against Abeta toxicity and PKC-dependent release of nonamyloidogenic soluble precursor protein by green tea polyphenol (-)-epigallocatechin-3-gallate. Faseb J. 2003, 17: 952-954.
Ring S, Weyer SW, Kilian SB, Waldron E, Pietrzik CU, Filippov MA, Herms J, Buchholz C, Eckman CB, Korte M, et al: The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J Neurosci. 2007, 27: 7817-7826. 10.1523/JNEUROSCI.1026-07.2007.
Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, et al: Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999, 402: 537-540. 10.1038/990114.
Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, et al: Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999, 286: 735-741. 10.1126/science.286.5440.735.
Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, et al: Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature. 1999, 402: 533-537. 10.1038/990107.
Lau KF, McLoughlin DM, Standen C, Miller CC: X11 alpha and x11 beta interact with presenilin-1 via their PDZ domains. Mol Cell Neurosci. 2000, 16: 557-565. 10.1006/mcne.2000.0898.
Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC: BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci. 2001, 4: 233-234. 10.1038/85064.
Creemers JW, Ines Dominguez D, Plets E, Serneels L, Taylor NA, Multhaup G, Craessaerts K, Annaert W, De Strooper B: Processing of beta-secretase by furin and other members of the proprotein convertase family. J Biol Chem. 2001, 276: 4211-4217. 10.1074/jbc.M006947200.
Bennett BD, Denis P, Haniu M, Teplow DB, Kahn S, Louis JC, Citron M, Vassar R: A furin-like convertase mediates propeptide cleavage of BACE, the Alzheimer's beta-secretase. J Biol Chem. 2000, 275: 37712-37717. 10.1074/jbc.M005339200.
Walter J, Fluhrer R, Hartung B, Willem M, Kaether C, Capell A, Lammich S, Multhaup G, Haass C: Phosphorylation regulates intracellular trafficking of beta-secretase. J Biol Chem. 2001, 276: 14634-14641. 10.1074/jbc.M011116200.
Huse JT, Pijak DS, Leslie GJ, Lee VM, Doms RW: Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme. The Alzheimer's disease beta-secretase. J Biol Chem. 2000, 275: 33729-33737. 10.1074/jbc.M004175200.
Huse JT, Liu K, Pijak DS, Carlin D, Lee VM, Doms RW: Beta-secretase processing in the trans-Golgi network preferentially generates truncated amyloid species that accumulate in Alzheimer's disease brain. J Biol Chem. 2002, 277: 16278-16284. 10.1074/jbc.M111141200.
He W, Lu Y, Qahwash I, Hu XY, Chang A, Yan R: Reticulon family members modulate BACE1 activity and amyloid-beta peptide generation. Nat Med. 2004, 10: 959-965. 10.1038/nm1088.
Murayama KS, Kametani F, Saito S, Kume H, Akiyama H, Araki W: Reticulons RTN3 and RTN4-B/C interact with BACE1 and inhibit its ability to produce amyloid beta-protein. Eur J Neurosci. 2006, 24: 1237-1244. 10.1111/j.1460-9568.2006.05005.x.
Shi Q, Prior M, He W, Tang X, Hu X, Yan R: Reduced amyloid deposition in mice overexpressing RTN3 is adversely affected by preformed dystrophic neurites. J Neurosci. 2009, 29: 9163-9173. 10.1523/JNEUROSCI.5741-08.2009.
He X, Li F, Chang WP, Tang J: GGA proteins mediate the recycling pathway of memapsin 2 (BACE). J Biol Chem. 2005, 280: 11696-11703. 10.1074/jbc.M411296200.
Tesco G, Koh YH, Kang EL, Cameron AN, Das S, Sena-Esteves M, Hiltunen M, Yang SH, Zhong Z, Shen Y, et al: Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron. 2007, 54: 721-737. 10.1016/j.neuron.2007.05.012.
Wahle T, Prager K, Raffler N, Haass C, Famulok M, Walter J: GGA proteins regulate retrograde transport of BACE1 from endosomes to the trans-Golgi network. Mol Cell Neurosci. 2005, 29: 453-461. 10.1016/j.mcn.2005.03.014.
Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, et al: Mice deficient in BACE1, the Alzheimer's beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci. 2001, 4: 231-232. 10.1038/85059.
Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF: BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron. 2004, 41: 27-33. 10.1016/S0896-6273(03)00810-9.
Ohno M, Cole SL, Yasvoina M, Zhao J, Citron M, Berry R, Disterhoft JF, Vassar R: BACE1 gene deletion prevents neuron loss and memory deficits in 5XFAD APP/PS1 transgenic mice. Neurobiol Dis. 2007, 26: 134-145. 10.1016/j.nbd.2006.12.008.
Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, et al: Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003, 9: 3-4. 10.1038/nm0103-3.
Johnston JA, Liu WW, Todd SA, Coulson DT, Murphy S, Irvine GB, Passmore AP: Expression and activity of beta-site amyloid precursor protein cleaving enzyme in Alzheimer's disease. Biochem Soc Trans. 2005, 33: 1096-1100. 10.1042/BST20051096.
Dominguez D, Tournoy J, Hartmann D, Huth T, Cryns K, Deforce S, Serneels L, Camacho IE, Marjaux E, Craessaerts K, et al: Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J Biol Chem. 2005, 280: 30797-30806. 10.1074/jbc.M505249200.
Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, Yan R: Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006, 9: 1520-1525. 10.1038/nn1797.
Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooper B, Saftig P, Birchmeier C, Haass C: Control of peripheral nerve myelination by the beta-secretase BACE1. Science. 2006, 314: 664-666. 10.1126/science.1132341.
Vassar R, Kovacs DM, Yan R, Wong PC: The beta-secretase enzyme BACE in health and Alzheimer's disease: regulation, cell biology, function, and therapeutic potential. J Neurosci. 2009, 29: 12787-12794. 10.1523/JNEUROSCI.3657-09.2009.
Solans A, Estivill X, de La Luna S: A new aspartyl protease on 21q22.3, BACE2, is highly similar to Alzheimer's amyloid precursor protein beta-secretase. Cytogenet Cell Genet. 2000, 89: 177-184. 10.1159/000015608.
Hussain I, Powell DJ, Howlett DR, Chapman GA, Gilmour L, Murdock PR, Tew DG, Meek TD, Chapman C, Schneider K, et al: ASP1 (BACE2) cleaves the amyloid precursor protein at the beta-secretase site. Mol Cell Neurosci. 2000, 16: 609-619. 10.1006/mcne.2000.0884.
Bennett BD, Babu-Khan S, Loeloff R, Louis JC, Curran E, Citron M, Vassar R: Expression analysis of BACE2 in brain and peripheral tissues. J Biol Chem. 2000, 275: 20647-20651. 10.1074/jbc.M002688200.
Yan R, Munzner JB, Shuck ME, Bienkowski MJ: BACE2 functions as an alternative alpha-secretase in cells. J Biol Chem. 2001, 276: 34019-34027. 10.1074/jbc.M105583200.
Hook V, Toneff T, Bogyo M, Greenbaum D, Medzihradszky KF, Neveu J, Lane W, Hook G, Reisine T: Inhibition of cathepsin B reduces beta-amyloid production in regulated secretory vesicles of neuronal chromaffin cells: evidence for cathepsin B as a candidate beta-secretase of Alzheimer's disease. Biol Chem. 2005, 386: 931-940. 10.1515/BC.2005.108.
Hook VY, Kindy M, Reinheckel T, Peters C, Hook G: Genetic cathepsin B deficiency reduces beta-amyloid in transgenic mice expressing human wild-type amyloid precursor protein. Biochem Biophys Res Commun. 2009, 386: 284-288. 10.1016/j.bbrc.2009.05.131.
Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M: APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009, 457: 981-989. 10.1038/nature07767.
Li H, Wang B, Wang Z, Guo Q, Tabuchi K, Hammer RE, Sudhof TC, Zheng H: Soluble amyloid precursor protein (APP) regulates transthyretin and Klotho gene expression without rescuing the essential function of APP. Proc Natl Acad Sci USA. 2010, 107: 17362-17367. 10.1073/pnas.1012568107.
Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL: Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science. 1989, 245: 417-420. 10.1126/science.2474201.
Oster-Granite ML, McPhie DL, Greenan J, Neve RL: Age-dependent neuronal and synaptic degeneration in mice transgenic for the C terminus of the amyloid precursor protein. J Neurosci. 1996, 16: 6732-6741.
Zhao G, Mao G, Tan J, Dong Y, Cui MZ, Kim SH, Xu X: Identification of a new presenilin-dependent zeta-cleavage site within the transmembrane domain of amyloid precursor protein. J Biol Chem. 2004, 279: 50647-50650. 10.1074/jbc.C400473200.
Zhao G, Tan J, Mao G, Cui MZ, Xu X: The same gamma-secretase accounts for the multiple intramembrane cleavages of APP. J Neurochem. 2007, 100: 1234-1246. 10.1111/j.1471-4159.2006.04302.x.
Sastre M, Steiner H, Fuchs K, Capell A, Multhaup G, Condron MM, Teplow DB, Haass C: Presenilin-dependent gamma-secretase processing of beta-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep. 2001, 2: 835-841. 10.1093/embo-reports/kve180.
Weidemann A, Eggert S, Reinhard FB, Vogel M, Paliga K, Baier G, Masters CL, Beyreuther K, Evin G: A novel epsilon-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with Notch processing. Biochemistry. 2002, 41: 2825-2835. 10.1021/bi015794o.
Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ: Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc Natl Acad Sci USA. 2003, 100: 6382-6387. 10.1073/pnas.1037392100.
Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T: The role of presenilin cofactors in the gamma-secretase complex. Nature. 2003, 422: 438-441. 10.1038/nature01506.
Kim J, Schekman R: The ins and outs of presenilin 1 membrane topology. Proc Natl Acad Sci USA. 2004, 101: 905-906. 10.1073/pnas.0307297101.
Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, et al: Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron. 1996, 17: 1005-1013. 10.1016/S0896-6273(00)80230-5.
Li YM, Lai MT, Xu M, Huang Q, DiMuzio-Mower J, Sardana MK, Shi XP, Yin KC, Shafer JA, Gardell SJ: Presenilin 1 is linked with gamma-secretase activity in the detergent solubilized state. Proc Natl Acad Sci USA. 2000, 97: 6138-6143. 10.1073/pnas.110126897.
Ahn K, Shelton CC, Tian Y, Zhang X, Gilchrist ML, Sisodia SS, Li YM: Activation and intrinsic {gamma}-secretase activity of presenilin 1. Proc Natl Acad Sci USA. 2010
Luo WJ, Wang H, Li H, Kim BS, Shah S, Lee HJ, Thinakaran G, Kim TW, Yu G, Xu H: PEN-2 and APH-1 coordinately regulate proteolytic processing of presenilin 1. J Biol Chem. 2003, 278: 7850-7854. 10.1074/jbc.C200648200.
Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC, et al: aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev Cell. 2002, 3: 85-97. 10.1016/S1534-5807(02)00189-2.
Goutte C, Tsunozaki M, Hale VA, Priess JR: APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc Natl Acad Sci USA. 2002, 99: 775-779. 10.1073/pnas.022523499.
Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C: Reconstitution of gamma-secretase activity. Nat Cell Biol. 2003, 5: 486-488. 10.1038/ncb960.
Greenfield JP, Xu H, Greengard P, Gandy S, Seeger M: Generation of the amyloid-beta peptide N terminus in Saccharomyces cerevisiae expressing human Alzheimer's amyloid-beta precursor protein. J Biol Chem. 1999, 274: 33843-33846. 10.1074/jbc.274.48.33843.
Zhou S, Zhou H, Walian PJ, Jap BK: CD147 is a regulatory subunit of the gamma-secretase complex in Alzheimer's disease amyloid beta-peptide production. Proc Natl Acad Sci USA. 2005, 102: 7499-7504. 10.1073/pnas.0502768102.
Chen F, Hasegawa H, Schmitt-Ulms G, Kawarai T, Bohm C, Katayama T, Gu Y, Sanjo N, Glista M, Rogaeva E, et al: TMP21 is a presenilin complex component that modulates gamma-secretase but not epsilon-secretase activity. Nature. 2006, 440: 1208-1212. 10.1038/nature04667.
Pardossi-Piquard R, Bohm C, Chen F, Kanemoto S, Checler F, Schmitt-Ulms G, St George-Hyslop P, Fraser PE: TMP21 transmembrane domain regulates gamma-secretase cleavage. J Biol Chem. 2009, 284: 28634-28641. 10.1074/jbc.M109.059345.
Vetrivel KS, Gong P, Bowen JW, Cheng H, Chen Y, Carter M, Nguyen PD, Placanica L, Wieland FT, Li YM, et al: Dual roles of the transmembrane protein p23/TMP21 in the modulation of amyloid precursor protein metabolism. Mol Neurodegener. 2007, 2: 4-10.1186/1750-1326-2-4.
He G, Luo W, Li P, Remmers C, Netzer WJ, Hendrick J, Bettayeb K, Flajolet M, Gorelick F, Wennogle LP, et al: Gamma-secretase activating protein is a therapeutic target for Alzheimer's disease. Nature. 2010, 467: 95-98. 10.1038/nature09325.
Cupers P, Bentahir M, Craessaerts K, Orlans I, Vanderstichele H, Saftig P, De Strooper B, Annaert W: The discrepancy between presenilin subcellular localization and gamma-secretase processing of amyloid precursor protein. J Cell Biol. 2001, 154: 731-740. 10.1083/jcb.200104045.
Kovacs DM, Fausett HJ, Page KJ, Kim TW, Moir RD, Merriam DE, Hollister RD, Hallmark OG, Mancini R, Felsenstein KM, et al: Alzheimer-associated presenilins 1 and 2: neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat Med. 1996, 2: 224-229. 10.1038/nm0296-224.
Haass C, De Strooper B: The presenilins in Alzheimer's disease--proteolysis holds the key. Science. 1999, 286: 916-919. 10.1126/science.286.5441.916.
Wang R, Tang P, Wang P, Boissy RE, Zheng H: Regulation of tyrosinase trafficking and processing by presenilins: partial loss of function by familial Alzheimer's disease mutation. Proc Natl Acad Sci USA. 2006, 103: 353-358. 10.1073/pnas.0509822102.
Vetrivel KS, Zhang YW, Xu H, Thinakaran G: Pathological and physiological functions of presenilins. Mol Neurodegener. 2006, 1: 4-10.1186/1750-1326-1-4.
Tarassishin L, Yin YI, Bassit B, Li YM: Processing of Notch and amyloid precursor protein by gamma-secretase is spatially distinct. Proc Natl Acad Sci USA. 2004, 101: 17050-17055. 10.1073/pnas.0408007101.
Kopan R, Schroeter EH, Weintraub H, Nye JS: Signal transduction by activated mNotch: importance of proteolytic processing and its regulation by the extracellular domain. Proc Natl Acad Sci USA. 1996, 93: 1683-1688. 10.1073/pnas.93.4.1683.
Schroeter EH, Kisslinger JA, Kopan R: Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998, 393: 382-386. 10.1038/30756.
Sardi SP, Murtie J, Koirala S, Patten BA, Corfas G: Presenilin-dependent ErbB4 nuclear signaling regulates the timing of astrogenesis in the developing brain. Cell. 2006, 127: 185-197. 10.1016/j.cell.2006.07.037.
Baek SH, Ohgi KA, Rose DW, Koo EH, Glass CK, Rosenfeld MG: Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-kappaB and beta-amyloid precursor protein. Cell. 2002, 110: 55-67. 10.1016/S0092-8674(02)00809-7.
Kim HS, Kim EM, Lee JP, Park CH, Kim S, Seo JH, Chang KA, Yu E, Jeong SJ, Chong YH, et al: C-terminal fragments of amyloid precursor protein exert neurotoxicity by inducing glycogen synthase kinase-3beta expression. Faseb J. 2003, 17: 1951-1953.
Pardossi-Piquard R, Petit A, Kawarai T, Sunyach C, Alves da Costa C, Vincent B, Ring S, D'Adamio L, Shen J, Muller U, et al: Presenilin-dependent transcriptional control of the Abeta-degrading enzyme neprilysin by intracellular domains of betaAPP and APLP. Neuron. 2005, 46: 541-554. 10.1016/j.neuron.2005.04.008.
von Rotz RC, Kohli BM, Bosset J, Meier M, Suzuki T, Nitsch RM, Konietzko U: The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J Cell Sci. 2004, 117: 4435-4448. 10.1242/jcs.01323.
Zhang YW, Wang R, Liu Q, Zhang H, Liao FF, Xu H: Presenilin/gamma-secretase-dependent processing of beta-amyloid precursor protein regulates EGF receptor expression. Proc Natl Acad Sci USA. 2007, 104: 10613-10618. 10.1073/pnas.0703903104.
Liu Q, Zerbinatti CV, Zhang J, Hoe HS, Wang B, Cole SL, Herz J, Muglia L, Bu G: Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron. 2007, 56: 66-78. 10.1016/j.neuron.2007.08.008.
Giliberto L, Zhou D, Weldon R, Tamagno E, De Luca P, Tabaton M, D'Adamio L: Evidence that the Amyloid beta Precursor Protein-intracellular domain lowers the stress threshold of neurons and has a "regulated" transcriptional role. Mol Neurodegener. 2008, 3: 12-10.1186/1750-1326-3-12.
Kinoshita A, Whelan CM, Berezovska O, Hyman BT: The gamma secretase-generated carboxyl-terminal domain of the amyloid precursor protein induces apoptosis via Tip60 in H4 cells. J Biol Chem. 2002, 277: 28530-28536. 10.1074/jbc.M203372200.
Tamayev R, Zhou D, D'Adamio L: The interactome of the amyloid beta precursor protein family members is shaped by phosphorylation of their intracellular domains. Mol Neurodegener. 2009, 4: 28-10.1186/1750-1326-4-28.
Park SA, Shaked GM, Bredesen DE, Koo EH: Mechanism of cytotoxicity mediated by the C31 fragment of the amyloid precursor protein. Biochem Biophys Res Commun. 2009, 388: 450-455. 10.1016/j.bbrc.2009.08.042.
Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, et al: Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999, 97: 395-406. 10.1016/S0092-8674(00)80748-5.
Bredesen DE: Neurodegeneration in Alzheimer's disease: caspases and synaptic element interdependence. Mol Neurodegener. 2009, 4: 27-10.1186/1750-1326-4-27.
Soriano S, Lu DC, Chandra S, Pietrzik CU, Koo EH: The amyloidogenic pathway of amyloid precursor protein (APP) is independent of its cleavage by caspases. J Biol Chem. 2001, 276: 29045-29050. 10.1074/jbc.M102456200.
Lu DC, Soriano S, Bredesen DE, Koo EH: Caspase cleavage of the amyloid precursor protein modulates amyloid beta-protein toxicity. J Neurochem. 2003, 87: 733-741. 10.1046/j.1471-4159.2003.02059.x.
Galvan V, Gorostiza OF, Banwait S, Ataie M, Logvinova AV, Sitaraman S, Carlson E, Sagi SA, Chevallier N, Jin K, et al: Reversal of Alzheimer's-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc Natl Acad Sci USA. 2006, 103: 7130-7135. 10.1073/pnas.0509695103.
Shankar GM, Walsh DM: Alzheimer's disease: synaptic dysfunction and Abeta. Mol Neurodegener. 2009, 4: 48-10.1186/1750-1326-4-48.
Burdick D, Soreghan B, Kwon M, Kosmoski J, Knauer M, Henschen A, Yates J, Cotman C, Glabe C: Assembly and aggregation properties of synthetic Alzheimer's A4/beta amyloid peptide analogs. J Biol Chem. 1992, 267: 546-554.
Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, et al: Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996, 2: 864-870. 10.1038/nm0896-864.
Jarrett JT, Berger EP, Lansbury PT: The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry. 1993, 32: 4693-4697. 10.1021/bi00069a001.
Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y: Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron. 1994, 13: 45-53. 10.1016/0896-6273(94)90458-8.
Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, et al: Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000, 156: 15-20.
Gyure KA, Durham R, Stewart WF, Smialek JE, Troncoso JC: Intraneuronal abeta-amyloid precedes development of amyloid plaques in Down syndrome. Arch Pathol Lab Med. 2001, 125: 489-492.
Mori C, Spooner ET, Wisniewsk KE, Wisniewski TM, Yamaguch H, Saido TC, Tolan DR, Selkoe DJ, Lemere CA: Intraneuronal Abeta42 accumulation in Down syndrome brain. Amyloid. 2002, 9: 88-102.
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM: Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003, 39: 409-421. 10.1016/S0896-6273(03)00434-3.
Oddo S, Caccamo A, Smith IF, Green KN, LaFerla FM: A dynamic relationship between intracellular and extracellular pools of Abeta. Am J Pathol. 2006, 168: 184-194. 10.2353/ajpath.2006.050593.
Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, et al: Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006, 26: 10129-10140. 10.1523/JNEUROSCI.1202-06.2006.
Espana J, Gimenez-Llort L, Valero J, Minano A, Rabano A, Rodriguez-Alvarez J, LaFerla FM, Saura CA: Intraneuronal beta-amyloid accumulation in the amygdala enhances fear and anxiety in Alzheimer's disease transgenic mice. Biol Psychiatry. 2010, 67: 513-521. 10.1016/j.biopsych.2009.06.015.
Yu C, Nwabuisi-Heath E, Laxton K, Ladu MJ: Endocytic pathways mediating oligomeric Abeta42 neurotoxicity. Mol Neurodegener. 2010, 5: 19-10.1186/1750-1326-5-19.
Friedrich RP, Tepper K, Ronicke R, Soom M, Westermann M, Reymann K, Kaether C, Fandrich M: Mechanism of amyloid plaque formation suggests an intracellular basis of Abeta pathogenicity. Proc Natl Acad Sci USA. 2010, 107: 1942-1947. 10.1073/pnas.0904532106.
Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C, et al: Isolation and quantification of soluble Alzheimer's beta-peptide from biological fluids. Nature. 1992, 359: 325-327. 10.1038/359325a0.
Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB, et al: Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature. 1992, 359: 322-325. 10.1038/359322a0.
Puzzo D, Privitera L, Leznik E, Fa M, Staniszewski A, Palmeri A, Arancio O: Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008, 28: 14537-14545. 10.1523/JNEUROSCI.2692-08.2008.
Morley JE, Farr SA, Banks WA, Johnson SN, Yamada KA, Xu L: A physiological role for amyloid-beta protein:enhancement of learning and memory. J Alzheimers Dis. 2010, 19: 441-449.
Plant LD, Boyle JP, Smith IF, Peers C, Pearson HA: The production of amyloid beta peptide is a critical requirement for the viability of central neurons. J Neurosci. 2003, 23: 5531-5535.
Plant LD, Webster NJ, Boyle JP, Ramsden M, Freir DB, Peers C, Pearson HA: Amyloid beta peptide as a physiological modulator of neuronal 'A'-type K+ current. Neurobiol Aging. 2006, 27: 1673-1683. 10.1016/j.neurobiolaging.2005.09.038.
Minniti AN, Rebolledo DL, Grez PM, Fadic R, Aldunate R, Volitakis I, Cherny RA, Opazo C, Masters C, Bush AI, et al: Intracellular amyloid formation in muscle cells of Abeta-transgenic Caenorhabditis elegans: determinants and physiological role in copper detoxification. Mol Neurodegener. 2009, 4: 2-10.1186/1750-1326-4-2.
King GD, Scott Turner R: Adaptor protein interactions: modulators of amyloid precursor protein metabolism and Alzheimer's disease risk?. Exp Neurol. 2004, 185: 208-219. 10.1016/j.expneurol.2003.10.011.
Rogelj B, Mitchell JC, Miller CC, McLoughlin DM: The X11/Mint family of adaptor proteins. Brain Res Rev. 2006, 52: 305-315. 10.1016/j.brainresrev.2006.04.005.
Borg JP, Ooi J, Levy E, Margolis B: The phosphotyrosine interaction domains of X11 and FE65 bind to distinct sites on the YENPTY motif of amyloid precursor protein. Mol Cell Biol. 1996, 16: 6229-6241.
Borg JP, Yang Y, De Taddeo-Borg M, Margolis B, Turner RS: The X11alpha protein slows cellular amyloid precursor protein processing and reduces Abeta40 and Abeta42 secretion. J Biol Chem. 1998, 273: 14761-14766. 10.1074/jbc.273.24.14761.
Salcini AE, Chen H, Iannolo G, De Camilli P, Di Fiore PP: Epidermal growth factor pathway substrate 15, Eps15. Int J Biochem Cell Biol. 1999, 31: 805-809. 10.1016/S1357-2725(99)00042-4.
Sorkin A: Cargo recognition during clathrin-mediated endocytosis: a team effort. Curr Opin Cell Biol. 2004, 16: 392-399. 10.1016/j.ceb.2004.06.001.
Maldonado-Baez L, Wendland B: Endocytic adaptors: recruiters, coordinators and regulators. Trends Cell Biol. 2006, 16: 505-513. 10.1016/j.tcb.2006.08.001.
Chyung JH, Selkoe DJ: Inhibition of receptor-mediated endocytosis demonstrates generation of amyloid beta-protein at the cell surface. J Biol Chem. 2003, 278: 51035-51043. 10.1074/jbc.M304989200.
Carey RM, Balcz BA, Lopez-Coviella I, Slack BE: Inhibition of dynamin-dependent endocytosis increases shedding of the amyloid precursor protein ectodomain and reduces generation of amyloid beta protein. BMC Cell Biol. 2005, 6: 30-10.1186/1471-2121-6-30.
Cai D, Leem JY, Greenfield JP, Wang P, Kim BS, Wang R, Lopes KO, Kim SH, Zheng H, Greengard P, et al: Presenilin-1 regulates intracellular trafficking and cell surface delivery of beta-amyloid precursor protein. J Biol Chem. 2003, 278: 3446-3454. 10.1074/jbc.M209065200.
Dumanchin C, Czech C, Campion D, Cuif MH, Poyot T, Martin C, Charbonnier F, Goud B, Pradier L, Frebourg T: Presenilins interact with Rab11, a small GTPase involved in the regulation of vesicular transport. Hum Mol Genet. 1999, 8: 1263-1269. 10.1093/hmg/8.7.1263.
Scheper W, Zwart R, Sluijs P, Annaert W, Gool WA, Baas F: Alzheimer's presenilin 1 is a putative membrane receptor for rab GDP dissociation inhibitor. Hum Mol Genet. 2000, 9: 303-310. 10.1093/hmg/9.2.303.
Scheper W, Zwart R, Baas F: Rab6 membrane association is dependent of Presenilin 1 and cellular phosphorylation events. Brain Res Mol Brain Res. 2004, 122: 17-23. 10.1016/j.molbrainres.2003.11.013.
Cai D, Netzer WJ, Zhong M, Lin Y, Du G, Frohman M, Foster DA, Sisodia SS, Xu H, Gorelick FS, et al: Presenilin-1 uses phospholipase D1 as a negative regulator of beta-amyloid formation. Proc Natl Acad Sci USA. 2006, 103: 1941-1946. 10.1073/pnas.0510708103.
Cai D, Zhong M, Wang R, Netzer WJ, Shields D, Zheng H, Sisodia SS, Foster DA, Gorelick FS, Xu H, et al: Phospholipase D1 corrects impaired betaAPP trafficking and neurite outgrowth in familial Alzheimer's disease-linked presenilin-1 mutant neurons. Proc Natl Acad Sci USA. 2006, 103: 1936-1940. 10.1073/pnas.0510710103.
Scherzer CR, Offe K, Gearing M, Rees HD, Fang G, Heilman CJ, Schaller C, Bujo H, Levey AI, Lah JJ: Loss of apolipoprotein E receptor LR11 in Alzheimer disease. Arch Neurol. 2004, 61: 1200-1205. 10.1001/archneur.61.8.1200.
Grear KE, Ling IF, Simpson JF, Furman JL, Simmons CR, Peterson SL, Schmitt FA, Markesbery WR, Liu Q, Crook JE, et al: Expression of SORL1 and a novel SORL1 splice variant in normal and Alzheimers disease brain. Mol Neurodegener. 2009, 4: 46-10.1186/1750-1326-4-46.
Yamazaki H, Bujo H, Kusunoki J, Seimiya K, Kanaki T, Morisaki N, Schneider WJ, Saito Y: Elements of neural adhesion molecules and a yeast vacuolar protein sorting receptor are present in a novel mammalian low density lipoprotein receptor family member. J Biol Chem. 1996, 271: 24761-24768. 10.1074/jbc.271.11.6483.
Jacobsen L, Madsen P, Moestrup SK, Lund AH, Tommerup N, Nykjaer A, Sottrup-Jensen L, Gliemann J, Petersen CM: Molecular characterization of a novel human hybrid-type receptor that binds the alpha2-macroglobulin receptor-associated protein. J Biol Chem. 1996, 271: 31379-31383. 10.1074/jbc.271.49.31379.
Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, von Arnim CA, Breiderhoff T, Jansen P, Wu X, et al: Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci USA. 2005, 102: 13461-13466. 10.1073/pnas.0503689102.
Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, Katayama T, Baldwin CT, Cheng R, Hasegawa H, et al: The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007, 39: 168-177. 10.1038/ng1943.
Trommsdorff M, Borg JP, Margolis B, Herz J: Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and the amyloid precursor protein. J Biol Chem. 1998, 273: 33556-33560. 10.1074/jbc.273.50.33556.
Narita M, Holtzman DM, Schwartz AL, Bu G: Alpha2-macroglobulin complexes with and mediates the endocytosis of beta-amyloid peptide via cell surface low-density lipoprotein receptor-related protein. J Neurochem. 1997, 69: 1904-1911. 10.1046/j.1471-4159.1997.69051904.x.
Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, et al: LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004, 43: 333-344. 10.1016/j.neuron.2004.07.017.
Ulery PG, Beers J, Mikhailenko I, Tanzi RE, Rebeck GW, Hyman BT, Strickland DK: Modulation of beta-amyloid precursor protein processing by the low density lipoprotein receptor-related protein (LRP). Evidence that LRP contributes to the pathogenesis of Alzheimer's disease. J Biol Chem. 2000, 275: 7410-7415. 10.1074/jbc.275.10.7410.
Zerbinatti CV, Wozniak DF, Cirrito J, Cam JA, Osaka H, Bales KR, Zhuo M, Paul SM, Holtzman DM, Bu G: Increased soluble amyloid-beta peptide and memory deficits in amyloid model mice overexpressing the low-density lipoprotein receptor-related protein. Proc Natl Acad Sci USA. 2004, 101: 1075-1080. 10.1073/pnas.0305803101.
Cam JA, Zerbinatti CV, Knisely JM, Hecimovic S, Li Y, Bu G: The low density lipoprotein receptor-related protein 1B retains beta-amyloid precursor protein at the cell surface and reduces amyloid-beta peptide production. J Biol Chem. 2004, 279: 29639-29646. 10.1074/jbc.M313893200.
Tang MX, Jacobs D, Stern Y, Marder K, Schofield P, Gurland B, Andrews H, Mayeux R: Effect of oestrogen during menopause on risk and age at onset of Alzheimer's disease. Lancet. 1996, 348: 429-432. 10.1016/S0140-6736(96)03356-9.
Waring SC, Rocca WA, Petersen RC, O'Brien PC, Tangalos EG, Kokmen E: Postmenopausal estrogen replacement therapy and risk of AD: a population-based study. Neurology. 1999, 52: 965-970.
Baldereschi M, Di Carlo A, Lepore V, Bracco L, Maggi S, Grigoletto F, Scarlato G, Amaducci L: Estrogen-replacement therapy and Alzheimer's disease in the Italian Longitudinal Study on Aging. Neurology. 1998, 50: 996-1002.
Paganini-Hill A: Estrogen replacement therapy--something to smile about. Compend Contin Educ Dent Suppl. 1998, S4-8.
Wise PM, Dubal DB, Wilson ME, Rau SW, Bottner M, Rosewell KL: Estradiol is a protective factor in the adult and aging brain: understanding of mechanisms derived from in vivo and in vitro studies. Brain Res Brain Res Rev. 2001, 37: 313-319. 10.1016/S0165-0173(01)00136-9.
Colton CA, Brown CM, Vitek MP: Sex steroids, APOE genotype and the innate immune system. Neurobiol Aging. 2005, 26: 363-372. 10.1016/j.neurobiolaging.2004.08.001.
Ghosh S, Thakur MK: PS1 expression is downregulated by gonadal steroids in adult mouse brain. Neurochem Res. 2008, 33: 365-369. 10.1007/s11064-007-9424-8.
Greenfield JP, Leung LW, Cai D, Kaasik K, Gross RS, Rodriguez-Boulan E, Greengard P, Xu H: Estrogen lowers Alzheimer beta-amyloid generation by stimulating trans-Golgi network vesicle biogenesis. J Biol Chem. 2002, 277: 12128-12136. 10.1074/jbc.M110009200.
Xu H, Gouras GK, Greenfield JP, Vincent B, Naslund J, Mazzarelli L, Fried G, Jovanovic JN, Seeger M, Relkin NR, et al: Estrogen reduces neuronal generation of Alzheimer beta-amyloid peptides. Nat Med. 1998, 4: 447-451. 10.1038/nm0498-447.
Xu H, Wang R, Zhang YW, Zhang X: Estrogen, beta-amyloid metabolism/trafficking, and Alzheimer's disease. Ann N Y Acad Sci. 2006, 1089: 324-342. 10.1196/annals.1386.036.
Zhang S, Huang Y, Zhu YC, Yao T: Estrogen stimulates release of secreted amyloid precursor protein from primary rat cortical neurons via protein kinase C pathway. Acta Pharmacol Sin. 2005, 26: 171-176. 10.1111/j.1745-7254.2005.00538.x.
Buxbaum JD, Gandy SE, Cicchetti P, Ehrlich ME, Czernik AJ, Fracasso RP, Ramabhadran TV, Unterbeck AJ, Greengard P: Processing of Alzheimer beta/A4 amyloid precursor protein: modulation by agents that regulate protein phosphorylation. Proc Natl Acad Sci USA. 1990, 87: 6003-6006. 10.1073/pnas.87.15.6003.
Caporaso GL, Gandy SE, Buxbaum JD, Ramabhadran TV, Greengard P: Protein phosphorylation regulates secretion of Alzheimer beta/A4 amyloid precursor protein. Proc Natl Acad Sci USA. 1992, 89: 3055-3059. 10.1073/pnas.89.7.3055.
Gillespie SL, Golde TE, Younkin SG: Secretory processing of the Alzheimer amyloid beta/A4 protein precursor is increased by protein phosphorylation. Biochem Biophys Res Commun. 1992, 187: 1285-1290. 10.1016/0006-291X(92)90442-N.
Hung AY, Haass C, Nitsch RM, Qiu WQ, Citron M, Wurtman RJ, Growdon JH, Selkoe DJ: Activation of protein kinase C inhibits cellular production of the amyloid beta-protein. J Biol Chem. 1993, 268: 22959-22962.
Gandy S, Czernik AJ, Greengard P: Phosphorylation of Alzheimer disease amyloid precursor peptide by protein kinase C and Ca2+/calmodulin-dependent protein kinase II. Proc Natl Acad Sci USA. 1988, 85: 6218-6221. 10.1073/pnas.85.16.6218.
Xu H, Greengard P, Gandy S: Regulated formation of Golgi secretory vesicles containing Alzheimer beta-amyloid precursor protein. J Biol Chem. 1995, 270: 23243-23245. 10.1074/jbc.270.40.23243.
Xu H, Sweeney D, Greengard P, Gandy S: Metabolism of Alzheimer beta-amyloid precursor protein: regulation by protein kinase A in intact cells and in a cell-free system. Proc Natl Acad Sci USA. 1996, 93: 4081-4084. 10.1073/pnas.93.9.4081.
Driscoll I, Resnick SM: Testosterone and cognition in normal aging and Alzheimer's disease: an update. Curr Alzheimer Res. 2007, 4: 33-45. 10.2174/156720507779939878.
Goodenough S, Engert S, Behl C: Testosterone stimulates rapid secretory amyloid precursor protein release from rat hypothalamic cells via the activation of the mitogen-activated protein kinase pathway. Neurosci Lett. 2000, 296: 49-52. 10.1016/S0304-3940(00)01622-0.
Gouras GK, Xu H, Gross RS, Greenfield JP, Hai B, Wang R, Greengard P: Testosterone reduces neuronal secretion of Alzheimer's beta-amyloid peptides. Proc Natl Acad Sci USA. 2000, 97: 1202-1205. 10.1073/pnas.97.3.1202.
McAllister C, Long J, Bowers A, Walker A, Cao P, Honda S, Harada N, Staufenbiel M, Shen Y, Li R: Genetic targeting aromatase in male amyloid precursor protein transgenic mice down-regulates beta-secretase (BACE1) and prevents Alzheimer-like pathology and cognitive impairment. J Neurosci. 2010, 30: 7326-7334. 10.1523/JNEUROSCI.1180-10.2010.
Acknowledgements
This work was supported in part by National Institutes of Health grants (R01AG021173, R01NS046673, R01AG030197 and R03AG034366 to H.X.), and grants from the Alzheimer's Association (to H.X. and Y.-w.Z.), the American Health Assistance Foundation (to H.X.), National Natural Science Foundation of China (30973150 to Y.-w.Z.), National S&T Major Project of China (2009ZX09103-731 to Y.-w.Z.), the 973 Prophase Project (2010CB535004 to Y.-w.Z.), and Natural Science Funds for Distinguished Young Scholar of Fujian Province (2009J06022 to Y.-w.Z.). Y.-w.Z. is supported by the Program for New Century Excellent Talents in Universities (NCET), the Fundamental Research Funds for the Central Universities, and Fok Ying Tung Education Foundation.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
YWZ and RT have written the manuscript. YWZ and HX have conceived and conceptualized the manuscript. HZ has contributed to the figure. All authors have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zhang, Yw., Thompson, R., Zhang, H. et al. APP processing in Alzheimer's disease. Mol Brain 4, 3 (2011). https://doi.org/10.1186/1756-6606-4-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-6606-4-3