
Abstract
Background
Host-associated microbiota are integral to the ecology of their host and may help wildlife species cope with rapid environmental change. Urbanization is a globally replicated form of severe environmental change which we can leverage to better understand wildlife microbiomes. Does the colonization of separate cities result in parallel changes in the intestinal microbiome of wildlife, and if so, does within-city habitat heterogeneity matter? Using 16S rRNA gene amplicon sequencing, we quantified the effect of urbanization (across three cities) on the microbiome of eastern grey squirrels (Sciurus carolinensis). Grey squirrels are ubiquitous in rural and urban environments throughout their native range, across which they display an apparent coat colour polymorphism (agouti, black, intermediate).
Results
Grey squirrel microbiomes differed between rural and city environments; however, comparable variation was explained by habitat heterogeneity within cities. Our analyses suggest that operational taxonomic unit (OTU) community structure was more strongly influenced by local environmental conditions (rural and city forests versus human built habitats) than urbanization of the broader landscape (city versus rural). The bacterial genera characterizing the microbiomes of built-environment squirrels are thought to specialize on host-derived products and have been linked in previous research to low fibre diets. However, despite an effect of urbanization at fine spatial scales, phylogenetic patterns in the microbiome were coat colour phenotype dependent. City and built-environment agouti squirrels displayed greater phylogenetic beta-dispersion than those in rural or forest environments, and null modelling results indicated that the phylogenetic structure of urban agouti squirrels did not differ greatly from stochastic expectations.
Conclusions
Squirrel microbiomes differed between city and rural environments, but differences of comparable magnitude were observed between land classes at a within-city scale. We did not observe strong evidence that inter-environmental differences were the result of disparate selective pressures. Rather, our results suggest that microbiota dispersal and ecological drift are integral to shaping the inter-environmental differences we observed. However, these processes were partly mediated by squirrel coat colour phenotype. Given a well-known urban cline in squirrel coat colour melanism, grey squirrels provide a useful free-living system with which to study how host genetics mediate environment x microbiome interactions.
Similar content being viewed by others

Background
Recognition that host-associated microbial communities (microbiomes) affect host health [1, 2], phenotypes [3, 4], and fitness [5, 6] has sparked interest in understanding the causes, consequences, and eco-evolutionary relevance of microbiome variation in nature [7,8,9,10]. The microbiome’s greater mutability (relative to the host genome) undergirds theorization that the microbiome plays an important evolutionary role in vertebrates [10,11,12]. Namely, it is hypothesized that the microbiome may help to buffer the adverse effects of novel environmental change by extending a host’s phenotypic range. In doing so, the microbiome might facilitate adaptive stop-gap solutions during host colonization to a new ecological niche. The gut microbiome is commonly invoked in such theorization, since the semi-permeable intestinal epithelium provides an intimate host-microbe interface through which microbiota can shape host behaviour [13], immunity [14], homeostasis [15], digestion [16], and dietary detoxification [17]—traits which ultimately define a species fundamental niche. For example, dietary detoxification by intestinal microbes may facilitate persistence of woodrat (Neotoma lepida) populations at the southern extent of their range, despite the northward expansion of creosote bush (Larrea tridentata) which produces a toxic resin and an armament of plant secondary compounds [17, 18]. Central to the hypothesis that microbiomes facilitate host population persistence is the prediction that among host populations, there exists genetic variation in traits that shape microbiome plasticity [11]. Many free-living study systems are intractable for empirically testing these predictions, partly because it is difficult to define what constitutes a truly ‘novel’ environmental change.
Urban ecosystems have achieved recognition in the field of evolutionary biology as opportunistic, mass-replicated, free-living ecological and evolutionary experiments [19, 20]—experiments which are useful for probing the host-microbiome relationship. Cities are among the fastest growing terrestrial ecosystems in the world [21] and expose wildlife to combinations of stressors and stimuli unlike anything observed in nature [22]. The novelty of urban environmental selective pressures is such that, globally, rates of phenotypic change among plants and animals show a distinct urban signature [23]. However, for many of these phenotypic changes, an underlying host genetic basis has not be strongly demonstrated [24]. The rapidity of phenotypic change of plants and animals in response to urbanization, and the nascence of cities on an evolutionary timescale, make urban environments a useful testing-ground for hypotheses that the microbiome can facilitate wildlife or—from a holobiont perspective—drive adaptation to a new ecological niche [11, 25, 26].
Despite widespread evidence for an effect of urbanization on the microbiomes of wild plants and animals, reported patterns are inconsistent across host species [27,28,29,30,31,32,33,34,35]. Inconsistencies of urban effects are likely attributable to; 1) city-specific variation (most studies to-date have focused on populations residing within a single city); 2) differences in the operational definition of urbanization; 3) differences in the how host species interact with the urban environment. For example, among vertebrates, consumption of human food resources is hypothesized to be the primary driver of gut microbiome variation [30, 32, 36, 37]. Dietary variation is likely an important contributor to urban gut microbiome variation for some wildlife, but not all species rely on human food subsidies to the same extent [38]. Alongside diet, patterns of intra- and inter-specific microbiota dispersal between hosts may help shape microbiome differences between environments [39,40,41,42]. Likewise, the physiological responses of hosts to environmental stimuli may be more important than the details of the stimuli itself [35, 43]. The capacity of the microbiome to facilitate wildlife expansion to a new ecological niche may therefore strongly depend on the outcome of gene x environment x microbiome interactions [44].
Building on past research, we adopt a multi-city perspective in studying the effect of urbanization on the faecal microbiome of eastern grey squirrels (Sciurus carolinensis)—a species with a prominent coat colour polymorphism which is both linked to urbanization and pleiotropically connected to gene x environment interactions among squirrels [45, 46]. Specifically, this colour polymorphism is the result of an incompletely dominant mutation in the gene which encodes the melanocortin-1 receptor of the proopiomelanortin system [47]; agouti: homozygous for the wild type allele, black: homozygous for the mutant allele, intermediate: heterozygous for the wild-type and mutant alleles (Fig. 1). Along replicate rural-urban gradients, the melanic phenotype has been reported to increase in frequency relative to the agouti wild-type [48, 49]. Fur and feather colour polymorphisms are frequently pleiotropically linked to differences in vertebrate physiology and behaviour, such as immunity, stress physiology, social behaviour, and grooming [50]. Variation in each of these traits is known to affect the host-associated microbiomes [35, 51, 52]. If dimensions of eastern grey squirrel physiology are pleiotropically linked to coat colour, then we predict microbiome differences between colour morphs, or colour morph differences in the microbiome’s response to urbanization, if colour pleiotropy linked traits are subject to gene x environment interactions.

Representative photos of the (left) agouti (zero copies of the melanism causing MC1R-Δ24 mutant allele), (centre) intermediate (one copy of the MC1R-Δ24 mutant allele), and (right) black (two copies of the MC1R-Δ24 mutant allele) eastern grey squirrel (Sciurus carolinensis) coat colour phenotypes
Using a 16S rRNA gene amplicon sequencing approach, we characterized the faecal bacterial microbiome of 195 individual adult eastern grey squirrels spanning three cities (Guelph, Waterloo, Windsor) and three rural forests in southern Ontario (Canada). A single rural forest site was located a short distance outside each of the three focal cities, to mitigate the effect that spatial biases could have on environmental comparisons. To test for an effect of habitat heterogeneity within a single city (Guelph), we also sampled squirrels from three land classes presumed to possess disparate biotic and abiotic characteristics: 1) an urban university campus, 2) two suburban parks, and 3) two urban forest fragments.
We sought to test the prediction that the colonization of cities by eastern grey squirrels drives convergence in the bacterial microbiome, and to identify the spatial scale at which this convergence occurs. To inferentially parse what (non-mutually exclusive) ecological processes (e.g., selection, dispersal, drift) might underlay inter-environmental microbiome variation, we used a phylogenetic null modelling approach [53,54,55]. We predicted that homogenizing selection should act on the microbiome within environments, but that evidence for disparate selective pressures acting on the microbiome would be observed between environments. To evaluate competing hypotheses that local (forest versus human built-environment) [27, 29] versus landscape (city versus rural) [31] level environmental variation is more important in shaping the microbiome, we contrasted variation in the microbiome explained by between- versus within-city habitat classifications (university campuses, suburban parks, urban forests, rural forests). Finally, to test for an effect of intra-specific physiological variation, we tested for interactions between coat colour phenotype and the bacterial microbiome’s response to urbanization.
Results
α-Diversity
We observed 1256 operational taxonomic units (OTUs) across 195 squirrel faecal samples, with an average of 379 OTUs ±4 SE per squirrel. The eastern grey squirrel microbiome was comprised primarily of Lachnospiraceae (37% ± 1% SE), Ruminococcaceae (19% ± 0.6% SE), Muribaculaceae (10% ± 0.5% SE), Prevotellaceae (8% ± 0.5% SE), Lactobacillaceae (7% ± 0.8% SE), and Bacteroidaceae (5% ± 0.5% SE). The community membership patterns we observed are consistent with previously published microbiome characterizations of eastern grey squirrels [35], and other tree squirrel and ground squirrel species [56, 57].
No difference in α-diversity was observed between sexes (β = 2.11 ± 8.12 SE, t = 0.26, p = 0.80). Agouti squirrels from city sites trended towards having fewer OTUs than agouti squirrels from rural sites, albeit this effect was marginally non-significant (β = 59.88 ± 31.17 SE, t = − 1.92, p = 0.06). However, we observed a colour phenotype x environment interaction, whereby α-diversity decreased with increases in MC1R mutation number (agouti: 0, intermediate: 1, black: 2) at rural sites (β = − 29.39 ± 12.26 SE, t = − 2.40, p = 0.02) but increased with melanism mutation number at city sites (β = 37.27 ± 13.86 SE, t = 2.69, p < 0.01; Fig. 2). Qualitatively similar results were observed with respect to Shannon diversity estimates (box-cox-transformed, λ = 4), wherein no effect of sex was observed (β = 6.83 ± 4.59 SE, t = 1.49, p = 0.14) and city agouti squirrel microbiomes had marginally lower Shannon diversity than agouti squirrels at rural sites (β = − 37.36 ± 18.50 SE, t = − 2.02, p = 0.05). A marginally non-significant negative correlation between melanism and Shannon evenness among rural sites (β = − 12.59 ± 7.00 SE, t = − 1.80, p = 0.07), but a positive correlation between Shannon diversity and melanism existed among city sites (β = 18.32 ± 7.90 SE, t = 2.32, p = 0.02).

Bacterial α-diversity in the squirrel microbiome is negatively correlated with melanism at rural sites (p = 0.02), but positively correlated with melanism among squirrels within city sites (p < 0.01). Solid horizontal lines within violins denote the interquartile range
β-Diversity
OTU composition (Euclidean distance of centre log-transformed OTU counts) of the microbiome was correlated with environment (city versus rural forest; F = 6.96, R2 = 0.03, p < 0.01), within-city land class (urban campus, suburban park, urban forest, rural forest; F = 4.95, R2 = 0.05, p < 0.01), trapping site (F = 2.78, R2 = 0.08, p < 0.01; Supplementary Information: Fig. S1), and sex (F = 1.78, R2 = 0.01, p < 0.01; Supplementary Information: Fig. S2) based on PERMANOVA testing. Conversely, we observed no effect of coat colour phenotype (F = 1.01, R2 = 0.01, p = 0.38) and no interaction between environment and coat colour (F = 1.07, R2 = 0.01, p = 0.21). Based on ordination of the bacterial microbiome (Fig. 3) and hierarchical clustering analysis (Supplementary Information: Fig. S3), squirrels from suburban parks within the city of Guelph clustered most closely with those from university campuses (built-environment sites). Conversely, squirrels from urban forests in Guelph clustered most closely with squirrels from distant rural forests (forest sites).

The first three axes of a principal coordinate analysis ordination of Euclidean distances (centred log-ratio transformed OTU dataset) separating eastern grey squirrel microbiomes. Points coloured by land class and shaped by local environment type (forest versus built-environment). Solid lines denote 95% confidence ellipses around environment type
Like OTU composition, phylogenetic structure of the squirrel microbiome (weighted UniFrac distance) was correlated with environment (F = 4.24, R2 = 0.02, p < 0.01), within-city land class (F = 5.05, R2 = 0.05, p < 0.01), trapping site (F = 3.26, R2 = 0.09, p < 0.01), and sex (F = 2.47, R2 = 0.01, p < 0.01). Coat colour phenotype was marginally non-significant (F = 1.33, R2 = 0.01, p = 0.06), and interestingly, we observed a significant interaction between environment type and coat colour phenotype (F = 1.49, R2 = 0.01, p = 0.02).
Visualization of weighted UniFrac ordination plots suggest that the environment x phenotype interaction we observed might be partly the result of differences in β-dispersion between phenotypes across environments, rather than solely mean community dissimilarities (Fig. 4). This interpretation was supported by post-hoc permutation tests of multivariate homogeneity for phenotype-environment grouping beta-dispersion. Specifically, city agouti squirrels harboured microbiomes which were more phylogenetically variable than rural agouti squirrels (p = 0.05). No other phenotype-environment groups differed in beta-dispersion, but city intermediate squirrels trended towards displaying greater phylogenetic variability than rural agouti squirrels (p = 0.07).

Weighted UniFrac principal coordinate analysis ordination of the eastern grey squirrel microbiome separated by environment and coloured and shaped by phenotype with 95% confidence ellipses. Agouti squirrels display greater beta-dispersion (p = 0.05) within cities (right panel) than in rural environments (left panel)
Null-modelling inference
Conventional β-diversity analyses are used to test whether community structure differs between groups but provide little evidence with which to infer why communities may differ. Further, patterns in β-diversity can be influenced by imbalances in α-diversity between communities [58]. Phylogeny-independent and phylogeny-weighted null modelling methods can be used to determine whether community composition (or between community dissimilarities) deviate from random expectations, given community α-diversity and pre-defined pool of γ-diversity (see Methods) [53, 55].
We tested for a phylogenetic signal among the bacteria observed in the squirrel microbiome with respect to their average relative abundance within forest and built-environment squirrel populations. We observed a significant positive signal over short phylogenetic distances (Supplementary Information: Fig. S4). This suggests that closely related bacterial OTUs tended to occupy a similar ecological niche, a result which supports the use of mean nearest taxon distance (MNTD), a measure of phylogenetic clustering/dispersion within communities [54].
Effect size standardized and box-cox transformed (λ = 0.4) absolute MNTDses values were closer to stochastic expectations (i.e., 0) among city agouti squirrels compared to rural agouti squirrels (β = − 1.28 ± 0.58 SE, t = − 2.22, p = 0.03); however, we observed an interaction whereby—among city squirrels—|MNTDses| increased with melanism mutant allele number (β = 0.51 ± 0.24 SE, t = 2.15, p = 0.03; Fig. 5A). Conversely, we observed no effect of sex (β = 0.17 ± 0.14 SE, t = 1.20, p = 0.23) or melanism among rural squirrels (β = − 0.29 ± 0.21 SE, t = − 1.38, p = 0.17).

An interaction between coat colour phenotype and environment affected (A) |mean nearest taxon distances| (box-cox transformed) (city versus rural), and (B) β mean nearest taxon distance (βMNTDses) (built-environment versus forest), such that city and built-environment agouti squirrels had values closer to stochastic expectations. Dotted horizontal lines denote values beyond two standard deviations from the null distribution. Solid horizontal lines within violins denote the interquartile range. Horizontal brackets and ‘*’ denote significant differences in βMNTDses variance homogeneity using permutation tests (p < 0.05)
The same principles which underlie null modeling of within community phylogenetic structure can be applied to between community contrasts. Using this approach, phylogenetic distances between all taxa in one community are measured to their nearest related taxon in another community [53]. When effect-size is standardized by the standard deviation of the corresponding null distribution, these values provide an indication of whether any two communities are more (βMNTDses > 2), less (βMNTDses < 2), or no more (|βMNTDses| < 0) phylogenetically disparate than expected by chance [53]. If taxon niche-space is shallowly phylogenetically conserved (an assumption supported by the positive phylogenetic signal described above), deviations from null expectations are one indication that similar (or disparate) selective pressures act between communities. Based on a PERMANOVA test, microbiome βMNTDses values were not correlated with environment (F = 1.65, R2 = 0.01, p = 0.10), within-city land class (F = 0.42, R2 < 0.01, p = 0.96), trapping site (F = 1.27, R2 = 0.04, p = 0.16), sex (F = 1.16, R2 = 0.01, p = 0.33), or coat colour phenotype (F = 1.38, R2 = 0.01, p = 0.17); however, we observed an interaction between environment and coat colour phenotype (F = 1.57, R2 = 0.02, p = 0.05). Post-hoc permutation tests of multivariate homogeneity indicated that city agouti squirrels had smaller βMNTDses variances than either rural agouti squirrels (p = 0.04) or city black squirrels (p = 0.02). Similarly, city squirrels of the intermediate morph had smaller variances than city black squirrels (p = 0.02). Notably, the magnitude of βMNTDses values among city agouti squirrels and city intermediate morph squirrels, were more consistent with stochastic expectations than they were homogenizing selection. Conversely, black squirrels maintained comparably large βMNTDses values, regardless of their environment.
Because squirrels from rural and urban forests clustered together in Euclidean-based β-diversity analyses, we additionally considered a PERMANOVA in which squirrel faecal samples from city forests were grouped with those from rural forests to test for an effect of urbanization at a finer spatial scale (forest versus built-environment). Again, we observed no effect of within city land class (F = 0.00, R2 = 0, p = 0.99), trapping site (F = 1.27, R2 = 0.04, p = 0.10), sex (F = 1.17, R2 = 0.01, p = 0.36), or coat melanism (F = 1.39, R2 = 0.01, p = 0.14); however, we did observe a significant effect of urbanization at this finer spatial scale (human built environment versus forest; F = 2.73, R2 = 0.01, p < 0.01) and an interaction between habitat-type and coat colour phenotype (F = 1.86, R2 = 0.02, p = 0.01). As above, post-hoc permutation tests of multivariate homogeneity indicated that built-environment agouti squirrels had smaller βMNTDses variances than forest agouti squirrels (p < 0.01), forest black squirrels (p < 0.01), or built-environment black squirrels (p < 0.01; Fig. 5B) and trended towards smaller variances than forest intermediate morph squirrels (p = 0.06). Built-environment intermediate squirrels likewise had smaller βMNTDses values than forest black squirrels (p = 0.04), and although marginally non-significant, trended towards having smaller values than either forest agouti squirrels (p = 0.07) and built-environment black squirrels (p = 0.09).
Raup-Crick bray (RCbray) values which—in contrast to βMNTDses measurements—indicate the magnitude of OTU turnover, independent of taxa phylogenetic relatedness. In the absence of βMNTDses deviations from stochastic expectations (i.e., |βMNTDses| < 2), RCbray values which are higher (> 0.95) or lower (< − 0.95) are suggestive of dispersal limitation and homogenizing dispersal, respectively. Conversely, values between these extremes are considered indicative of ecological drift. Of 18,943 pairwise comparisons between squirrels, 14,143 (75%) had |βMNTDses| < 2, and of these pairs, 13,851 (98%) had corresponding values of RCbray > 0.95. Environment (city versus rural; F = 1.21, R2 = 0.01, p < 0.01), within-city land class (F = 1.78, R2 = 0.02, p < 0.01), trapping site (F = 1.34, R2 = 0.04, p < 0.01), and sex (F = 1.28, R2 = 0.01, p < 0.01) were all correlated with RCbray values in a PERMANOVA test. But we observed no effect of phenotype (F = 1.05, R2 = 0.01, p = 0.20), and only a marginally significant interaction between environment and phenotype (F = 1.10, R2 = 0.01, p = 0.06). However, the latter result might be expected, given the significant interaction of environment and phenotype on βMNTDses values.
Post-hoc Kruskal-Wallis tests indicated that RCbray values were higher among comparisons made within trapping sites than between trapping sites (χ2 = 530.97, p < 0.01). Interestingly, RCbray values within city sites were smaller than those observed within rural sites (χ2 = 19.77, p < 0.01; Supplementary Information: Fig. S5A). RCbray values also differed between city and rural environments with respect to between site comparisons (χ2 = 19.99, p < 0.01). Specifically, RCbray values were smaller between city sites than between rural sites (p < 0.01) or between city and rural sites (p < 0.01). RCbray values also differed by sex both within sites (χ2 = 7.83, p = 0.02; Supplementary Information: Fig. S5B) and between sites (χ2 = 15.41, p < 0.01). Within sites, post-hoc dunn tests indicated that female-female pairs had lower RCbray values than male-male pairs (p = 0.02) or female-male pairs (p < 0.01). In comparisons made between sites, female-male pairs had larger RCbray values than either female-female (p = 0.01) or male-male pairs (p < 0.01).
Differential abundance testing
To generate a robust understanding of which taxa drove the apparent divide in the microbiome between squirrels in forest versus built-environments, we used two different, but complimentary, statistical approaches. The first, selection balance analysis, accounts for the compositional nature of most 16S amplicon datasets by seeking the most parsimonious log-ratio of taxa that delineate two groups [59]. Results of selection balance analyses are therefore derived from parsimony and discriminative sensitivity, rather than test statistics typical of frequentist approaches. The second approach, analysis of compositions of microbiomes with bias correction (ANCOM-BC), normalizes sequence counts by a process similar to centred-log ratio transformations and applies corrections to control for false discovery rates [60].
Selection balance cross-validation identified that the apparent divide in the microbiome between squirrels in the human built-environment (university campuses and suburban parks) versus forests (urban forest and rural forests) could be most parsimoniously discriminated by a log-ratio balance of 12 genera. Squirrels from the built-environment had a greater abundance of Odoribacter, Oscillospira, Sulfurimonas, Ruminiclostridium (sub-group 5), Coprococcus (sub-group 3), and unclassified genera within the class Mollicutes RF39, relative to Alistipes, Ruminiclostridium (sub-group 1), Rikenellaceae (sub-group RC9), Lachnospiraceae (sub-group UCG-010), Clostridiales (sub-group vadinBB60), and unclassified genera of Lachnospiraceae, when compared to forest squirrels (Supplementary Information: Fig. S6A). This balance was able to discriminate between built-environment and forest samples with high sensitivity (AUC-ROC = 0.975). Similarly, at the family level, a log-ratio of Mollicutes RF39: Rikenellaceae most parsimoniously delineated built-environment and forest squirrels (AUC-ROC = 0.808; Supplementary Information: Fig. S6B).
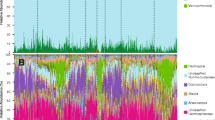
The results of ANCOM-BC analyses were qualitatively like those of the selection balance analysis, with 17 genera observed to differ between forest and human built-environments. Squirrels from the built-environment had greater relative abundances of Mollicutes (sub-group RF39), Ruminiclostridium (sub-group 5), Prevotella, Parasutterella, Oxalobacter, unclassified genera of Eggerthellaceae, as well as genera of Ruminococcaceae (sub-group UCG-014, sub-group UCG-008) and Lachnospiraceae (Lachnoclostridium, Blautia, Eisenbergiella, Coprococcus sub-group 3). Conversely, forest squirrels had greater abundances of Alistipes, Clostridiales (sub-group vadinBB60), Lachnospiraceae (sub-group FCS020), Lachnospiraceae (sub-group NK4A136), and a collection of unclassified Lachnospiraceae genera (Supplementary Information: Table S1). Notably, these environmental differences appeared driven primarily by the agouti morph (Fig. 6). After repeating ANCOM-BC analyses upon datasets subset by colour phenotype, we observed that between forest and human built-environments, 14 genera differed among agouti squirrels, 4 genera differed among the intermediate morph, and 0 genera differed among black coated squirrels (Supplementary Information: Fig. S7).

A stacked barplot of bacterial genera indicated by ANCOM-BC analyses to significantly differ in relative abundance between forest and built-environments in the full dataset or among at least one coat colour phenotype. Facetted by coat colour phenotype and environment type
Discussion
Urbanization clearly affected the eastern grey squirrel microbiome. This result is consistent with findings from birds [27, 29,30,31,32], reptiles [36], humans [61, 62], insects [63], plants [64], and wild mammals [35]. We further demonstrate that convergence occurs across cities, but also that substantive variation exists both between and within cities. For example, the variation explained by city-scale urbanization was comparable to the variation explained by land class heterogeneity within a single city (university campuses, suburban parks, urban forests). This is consistent with reports that fine-scale environmental variation can play a substantive role in shaping the microbiome [65, 66] and suggests the need for caution when trying to quantify an individual’s environment with simplified univariate terms, like percent impermeable landcover in the context of urban landscapes. For example, our urban forest and suburban park sites were characterised by similarly sized urban greenspaces, but squirrels from urban forests tended to cluster with squirrels from rural forests in ordination and hierarchical clustering plots, while those from suburban parks were more alike those from university campuses. Broad-scale urbanization (city versus rural) had no effect on βMNTDses, however, βMNTDses values were affected by whether squirrels were from local habitats characterized as forest or human built. These phylogenetic patterns in the microbiome suggest that environmental selective pressures act on the microbiome (perhaps indirectly through host physiology) at fine spatial scales. Therefore, although we report an effect of urbanization on the microbiome, habitat heterogeneity within cities makes a representative ‘urban microbiome’ for host species unlikely.
Anthropogenic food subsidies in cities are hypothesized to underlie microbiome variation among urban mammals [30, 37], as well as reports of obesity and hyperglycaemia [67,68,69]. Western diet-induced obesity in mice and pigs maintained on diets low in indigestible fibre and starch (as might be expected of a western diet) are strongly characterized by blooms of Mollicutes which appear to specialize on fermenting simple sugars [70,71,72]. Tellingly, we observed Mollicutes to be one of the strongest discriminating features separating built-environment squirrels from forest squirrels in both selection balance and ANCOM-BC testing.
ANCOM-BC tests further identified that built-environment squirrels harboured greater relative abundances of Parasutterella—sister genus to the very ecologically similar Sutterella [73]. Sutterella were observed in one study to be a strong feature characterizing the microbiome of wild red squirrels supplemented with peanut butter, a food higher in sugar and fat than natural components of the squirrel’s diet [56]. Rather than dietary fats, Parasutterella and Sutterella appear to specialize on the bile acids, which hosts produce to solubilize dietary fats [74].
These results are exemplative of a broader pattern, whereby city and built-environment squirrel microbiomes were generally characterized by a shift towards taxa which show evidence of metabolizing fats and host-derived products, especially bile acids. For example, a large collection of unclassified genera of Lachnospiraceae—a family comprised of primarily fibrolytic specialists and plant fibre fermenters [75]—was less abundant among built-environment squirrels compared to forest squirrels. Despite this, a handful of Lachnospiraceae genera were found in ANCOM-BC analyses to be more abundant in squirrels from the built-environment, but like Parasutterella and Sutterella, these exceptions are known to metabolize animal host derived products (Lachnoclostridium [76,77,78,79], Blautia [80,81,82], Eisenbergiella [83]). Genera within the family Eggerthellaceae and the genus Odoribacter were likewise built-environment associated—in ANCOM-BC and selection balance analyses, respectively—and likewise specialize on bile acids and other host derived products [76, 84,85,86,87]. In addition to metabolizing host products, many of these genera have been implicated in western diet-related metabolic and gastro-intestinal diseases in humans (Lachnoclostridium [76, 88], Blautia [89,90,91,92], Eisenbergiella [88], Eggerthellaceae [76, 86, 88, 93, 94], Odoribacter [87, 92, 95,96,97]). While diet may be the ultimate cause of the built-environment versus forest squirrel microbiome divide, host physiological responses to their diet may be a complimentary, if not more proximate, mechanism. This distinction is important, since a genetic basis to host physiological responses to their diet allows for evolution in the diet x microbiome relationship [35].
The need to consider proximate host physiological mechanisms was exemplified by our observation that although urbanization had an apparent effect on the squirrel microbiome, these effects were partly mediated by squirrel coat colour phenotype. Coat melanism was negatively correlated with OTU richness in rural environments but positively correlated with OTU richness and Shannon diversity in the city. Black squirrels harboured microbiomes which were more phylogenetically dissimilar than predicted by null expectations but did not differ systematically between environments. When parsed by phenotype, no genera differed in abundance between black squirrels sampled in built-environments and forests, whereas 14 genera differed among agouti squirrels between habitats. Agouti squirrels also displayed greater phylogenetic variability in the microbiome within cities than black squirrels, and null modelling results suggest that this might be due to stochastic (rather than deterministic) processes (|MNTDses| and βMNTDses values closer to 0). If diet is indeed an ultimate cause for the effects of urbanization on the grey squirrel microbiome, then dietary effects might be mediated by physiological differences between squirrel colour morphs.
Fur and feather melanism—like that observed in grey squirrels—is often pleiotropically linked through the pro-opiomelanocortin system to myriad physiological pathways [50]. These pathways include baseline hypothalamic-pituitary-adrenal (HPA) physiology, HPA axis reactivity to stressors, and the immune system [98,99,100,101,102]—each of which is a dimension of host physiology known to affect wildlife microbiomes [35, 43, 103]. In eastern grey squirrels, the melanism-causing MC1R mutation [104] has been connected to behavioural and physiological differences [105, 106], most notably thermogenic physiology [45, 46]. Specifically, melanic squirrels show greater plasticity in their ability to adaptively lower their basal metabolic rate when exposed to sub-zero ambient temperatures. This gene x environment interaction may be the result of MC1R linked pleiotropy, however, more recent evidence suggests that the MC1R mutation in grey squirrels might have originated from introgression with eastern fox squirrels (Sciurus niger) [107]. Therefore, the MC1R mutation—and the greater physiological plasticity with which it appears correlated—might reflect more substantive underlying genetic differences between colour morphs.
Greater physiological plasticity among black squirrels might paradoxically explain why this morph tended to maintain similar microbiomes regardless of their external environment. Conversely, if agouti squirrels are physiologically inflexible, they may be incapable of acclimating to novel urban conditions or environmental stressors, and thereby lose, or relax, control of their resident microbiota. The resultant homeostatic disruption might explain the greater evidence for ecological drift within the city and built-environment squirrel microbiome. A physiological basis to the colour phenotype patterns we observed seems likely given past research [45, 46, 48, 50]. In testing the hypothesis that microbiome plasticity is adaptive for hosts in novel environments [11], we caution that it is important to parse whether observed variance in the microbiome is the result of stochastic processes (perhaps signalling a loss of host homeostatic control) or deterministic processes (possibly mediated by plasticity in host physiology); the outcome for host health and fitness may be very different depending on ecological process responsible for driving beta-dispersion in the microbiome [2].
Despite the emphasis we have placed on selection and ecological drift, patterns in bacterial dispersal likely also strongly contribute to the inter-environmental variation that we observed [41]. For example, bacterial dispersal limitation undoubtedly occurs between sampling locations—an interpretation supported by the smaller RCbray values observed within sites than between sites. This is unsurprising as spatial structure and social structure within [39, 108,109,110,111] and between [40, 65, 112, 113] mammalian populations has been demonstrated to affect the microbiome. More surprising, was our observation that RCbray values tended to be smaller within city sites than within rural sites—despite no effect of environment on βMNTDses values. This suggests that bacterial dispersal limitation might be stronger between squirrels within rural habitats versus between squirrels within city environments. Squirrel populations persist at greater densities on urban landscapes [114], which could facilitate greater microbial exchange via more frequent interactions with conspecifics [108]. Conspecific interactions are further catalyzed by spatial clustering of anthropogenic food sources (bird feeders, garbage cans, picnic areas etc.) which are known to increase rates of pathogen transmission in wildlife [115]; the same process could as easily facilitate greater exchange of commensal or mutualistic bacteria. Further, bacterial dispersal is not restricted to among hosts of the same species, but rather, are partly shaped by trophic interactions [40]. Urban food webs tend to have fewer species, and more interactions per species (i.e. greater connectivity [116]), which might help to promote the exchange of microbiota between the microbiomes of co-occurring colonizing wildlife. Greater connectivity and spatial overlap between con- and hetero-specific hosts within urban environments could facilitate more frequent microbial dispersal when compared to rural sites.
Environment dependent patterns of microbiota dispersal may likewise partly underlie beta-diversity differences in the microbiome between environments. We observed that RCbray values were smaller among pairwise comparisons made between city sites than comparisons made between rural sites or between city and rural sites. Substantive differences in abiotic and biotic factors between these environments ensure that urban and rural squirrels are very likely exposed to different pools of bacterial γ-diversity. For example, urbanization has been connected to predictable biodiversity loss and landscape homogenization [117], effects which might extend to the microbial communities. Furthermore, urban biological communities are strongly shaped by human socio-cultural factors [118]. Since the cities included in our study were built in a very similar socio-cultural context, the plant and animal species within these cities (and therefore the microbiota to which squirrels are exposed) might be more similar between cities than between rural forests. Furthermore, even when plant or animal species are found in both city and rural environments, the microbiota they transmit to squirrels may differ. For example, the phyllosphere microbiota of trees—with which squirrels closely associate—are themselves affected by urbanization [34]. Finally, the near-constant exchange of people and resources between cities facilitates gene flow and prevent the genetic isolation of urban wildlife populations [119, 120]. A similar mechanism might allow for greater bacterial dispersal between cities than between isolated rural forest fragments. Although speculative, it is important to consider an organism’s broader microbial milieu when studying host-microbe symbioses, rather than lay causality solely at the feet of host diet and physiology.
Lastly, we unexpectedly observed evidence that some of the sex effects among squirrels might derive partially from patterns in microbiota dispersal. Namely, RCbray values were smaller between females than between sexes or between males, despite no effect of sex on βMNTDses values. These differences could derive from behavioural differences in bacterial transmission, or physiological differences which affect colonization success, as suggested by researchers who characterized a similar pattern of female-biased bacterial transmission among co-housed common marmosets (Callithrix jacchus [121];). Among North American red squirrels, inter-individual bacterial dispersal appears to occur primarily through the maternal line [56]; therefore, a pattern of lower OTU turnover might have been observed between females because bacterial dispersal occurs both from a female’s parents and to a female’s offspring. By contrast, males may not directly contribute microbiota to their offspring. These familial-structured bacterial dispersal patterns are likely continually reinforced among related female grey squirrels, which show a greater propensity for social grooming and nest sharing when compared to males [122]. These results are consistent with patterns in the microbiome of black howler monkeys (Alouatta pigra) in which social bonds are strongest among female-female dyads [123]. Interestingly, the opposite patterns are observed among semi-feral welsh ponies (Equus ferus caballus), in which males show greater centrality in both social and (inferred) microbiota dispersal networks [109]. Similarly, bacterial transmission within social networks of wild wood mice (Apodemus sylvaticus) is most strongly driven by males, despite no difference in social association strength between sexes [52]. Therefore, while dietary and physiological differences between hosts affect microbiota colonization success and abundances, organismal behaviour and variation in social structure shape microbiota metacommunities, and determine which microbiota are given the opportunity to colonize a new host [13, 41, 110].
Conclusions
Urbanization affected the eastern grey squirrel microbiome at landscape and intra-city spatial scales. Characteristics of the urban squirrel microbiome echo those reported in humans and laboratory mice maintained on high-fat, low-fibre, westernized diets; however, the response of the squirrel microbiome to urbanization was coat colour phenotype dependent. Namely, the inter-environmental differences we observed were driven primarily by the agouti morph, however, we did not observe evidence that this pattern was the result of divergent selective pressures. Rather, the city agouti squirrel microbiome displayed phylogenetic patterns more consistent with stochastic processes than black squirrels. Given the putative importance of a westernized diet in shaping the urban wildlife microbiome, colour polymorphic grey squirrels provide an interesting free-living system with which to study the gene x diet x microbiome interactions. Further research and a full holo-omics approach (integrated host and microbiome genomics, transcriptomics, proteomics, and metabolomics [26]) are required to probe the causes and fitness consequences of metagenomic plasticity and understand its importance for host species as they endeavour to colonize a new ecological niche.
Methods
Study sites
Sampling was divided among three site pairs throughout southern Ontario. Site pairs were comprised of one city and one nearby rural deciduous forest outside of city limits. Each city and rural site within a pair were trapped consecutively to limit the effect of seasonal confounds. Cities are inherently environmentally heterogeneous, therefore, to standardize our efforts, we limited sampling to urban university campuses as our representative city sites. Those sites were the University of Guelph (43°31′52.45″N 80°13′36.70″W, n = 29 squirrels), University of Waterloo (43°28′17.68″N 80°32′36.03″W, n = 30), and University of Windsor (42°18′23.77″N 83°4′1.92″W, n = 15), which were paired with forests at Starkey Hill Conservation Area (43°32′37.99″N 80°9′15.82″W, n = 20), the rare Charitable Research Reserve (43°22′57.72″N 80°20′54.43″W, n = 26), and the Kopegaron Woods Conservation Area (42° 4′38.52″N 82°29′34.04″W, n = 2), respectively. To understand how habitat heterogeneity within cities can affect the microbiome, we also sampled other urban land classes within the City of Guelph: two suburban parks (Exhibition Park, n = 29; Royal City Park, n = 13) and two urban forests (University of Guelph Dairy Bush, n = 11; Arboretum, n = 10)—small forest fragments entirely enveloped by an urban landscape.
Capture protocol
All trapping occurred in southern Ontario between May 9th and September 12th using tomahawk Model 102 traps (Tomahawk Live Trap Co., WI, USA) baited with peanuts. Traps were set between 6:00–16:00 and checked in 1 h intervals. Upon capture, we transferred squirrels to a cloth bag and checked in 15 min intervals for faecal pellets. Using single-use sterilized toothpicks, we collected fresh faecal pellets which we stored on ice in the field before transfer to -20 °C for storage within 4 h of collection. After faecal pellet collection, we transferred squirrels to a handling bag, recorded sex, and marked squirrels with unique alpha-numeric ear-tags before release to allow for identification of recaptures and prevent resampling of individuals.
Sequencing and bioinformatics
Using QIAamp DNA Stool Mini Kits (Qiagen, Hilden Germany), we extracted bacterial DNA from 0.2 g subsamples of faecal samples, which in other rodents are indicative of bacterial communities in the large intestines [124]. Extracts underwent triplicate PCR amplification of the 16S rRNA gene v4 region (515F-806R modified primers; [125]) at MetaGenomBio Inc. (Toronto Canada) alongside PCR negative controls. Triplicate PCR products were then pooled prior to sequencing on an Illumina MiSeq using v2 chemistry (250 bp read). We then processed pair-end reads in mothur using a standardized amplicon processing pipeline [126], aligned sequences to the silva v132 reference database [127], and classified operational taxonomic units (OTUs) clustered using OptiClust [128] based on a 97% similarity threshold. To remove potential contaminants or sequencing errors, we discarded all non-bacterial OTUs and OTUs which were not represented by at least 1 read in 5% of the samples prior to analysis [113]. In total, we generated 3,826,931 merged bacterial amplicon sequences, with an average sequencing depth of ~ 20,000 reads/sample (min = 9724, max = 60,891), after assembly, quality control, and filtering. Samples were rarefied to a depth of 9724 reads (Supplementary Information: Fig. S5), except for ANCOM-BC analysis and analyses pertaining to Euclidean measures of β-diversity which were calculated from raw count # centred log ratio transformed OTU datasets [129]. A relaxed neighbour-joining method was used to construct a phylogenetic tree using the mothur implementation of clearcut [126, 130]. We sequenced extraction kit negative controls which contained swabs of the bags which were used to cover the tomahawk traps. However, negative controls showed nominal amplification and most negative control OTUs were removed during the filtering procedures described above. The remaining OTUs were present at low abundance in negative controls but were abundant among biological samples and therefore likely the result of index hopping.
Diversity testing
We tested whether microbiome α-diversity (# of OTUs and Shannon diversity) differed with sex, coat melanism mutation number (agouti = 0, intermediate = 1, black = 2), environment (city versus rural), and an interaction between coat melanism and environment using linear mixed effects models in the R package ‘lmerTest’ [131], treating sampling location as a random effect. To determine whether bacterial microbiome β-diversity differed between city and rural eastern grey squirrel populations, we used permutational multivariate analyses of variance (PERMANOVA) tests of β-diversity in Euclidean and weighted UniFrac space. PERMANOVAs were parameterized by environment type (city versus rural), land class (campus, suburban park, urban forest, rural forest), sampling location, sex, coat colour phenotype, and an interaction between phenotype and environment type, in that order. To test for differences in environment-phenotype groupings, we used permutation tests of multivariate homogeneity, with β-dispersion calculated relative to the group centroid with a bias adjustment for small sample sizes.
Community null modeling
To determine whether observed patterns of microbiome diversity deviated from stochastic expectations, we used a null modelling approach [53, 54, 132]. These methods are predicated on species’ ecological niche-spaces being non-independent of their phylogeny [55], a notion broadly supported via analysis of functional traits derived from publicly archived microbial genome sequences [133, 134]. We tested for a positive phylogenetic signal in urban associated niche-space using the phyloCorrelogram() function from the R package ‘phylosignal’ [135]. OTU niche space was estimated as the deviation of an OTU from a 1:1 line of mean OTU relative abundance in city versus rural habitats (Supplementary Information: Fig. S4).
In brief, we calculated mean nearest taxon distance (MNTD) and β mean nearest taxon distance (βMNTD), which are among community and between community measures of phylogenetic dissimilarity, respectively. For all analyses, these measures were effect size standardized by the mean and standard deviation of MNTD or βMNTD null distributions created through stochastically assembled communities which possessed the same α-diversity as the focal observed communities [53, 54]. Null distributions were created by randomly shuffling taxa names and relative abundances across the system’s γ-diversity phylogenetic tree (MNTD: 9999 iterations, βMNTD: 999 iterations) using the function ses.mntd() from the package ‘picante’ [136] and ses.comdistnt() from the package ‘MicEco’, respectively [137]. Additionally, we calculated RCbray values, a phylogeny-independent measure of between community OTU turnover [54, 58]. In brief, observed Bray-Curtis values were compared to 9999 probabilistically assembled community pairs of the same α-diversity as observed community pairs [54].
A linear mixed effects model was used to test for an effect of sex, environment, and colour phenotype, as well as a phenotype x environment interaction on |MNTD|, which we used as a measure of among community deviation from stochastic expectations. Again, sampling location was included in the mixed model as a random effect. To determine whether the phylogenetic structure between communities were more similar (βMNTDses < 0) or more disparate (βMNTDses > 0) than expected by chance alone (|βMNTDses| > 2), we used a PERMANOVA (parameterized as in the β-diversity analyses described above). As above, post-hoc permutation tests of multivariate homogeneity were used to parse significant relationships. Dispersion in βMNTDses values were calculated relative to the group centroid with a bias adjustment for small sample sizes [138], using the ‘vegan’ function betadisper() [139].
RCbray values were analyzed using a PERMANOVA, as described above. To make within and between group comparisons, we used post-hoc Kruskal-Wallis and dunn tests to further parse results into within versus between trapping sites. We used an ultrametric phylogenetic tree transformed using the chronos() function in the R package ‘ape’ (λ = 1 [140];) for all phylogeny-weighted null modelling. Unless specified, all other analyses were completed in R (v. 3.5.1) using the R package ‘phyloseq’ [141].
Differential abundance testing
To identify how the bacterial microbiome differed between squirrels in forests versus the built-environment, we performed selection balance analyses after binning OTUs to genus and family [59]. Secondarily, we performed ANCOM-BC differential abundance tests and evaluated the agreement between these opposing statistical approaches [60].
Availability of data and materials
All sequence data and meta data have been archived with the NCBI SRA (under embargo; Reviewer Link: https://dataview.ncbi.nlm.nih.gov/object/-PRJNA701759?reviewer=vd6bsoqsjr0dj8urp0pqpajkf1). R script files are available at figshare.com (reviewer link: https://figshare.com/s/3a3eaeee93980f8b22bb).
Abbreviations
- ANCOM-BC:
-
Analysis of compositions of microbiomes with bias correction
- AUC-ROC:
-
Area under the curve – receiver operating characteristic
- βMNTD:
-
β-mean nearest taxon distance
- βMNTDses:
-
β-mean nearest taxon distance standardized by effect size
- DNA:
-
Deoxyribonucleic acid
- HPA:
-
Hypothalamic-pituitary-adrenal axis
- MC1R:
-
Melanocortin-1 receptor:
- MNTD:
-
Mean nearest taxon distance
- MNTDses :
-
Mean nearest taxon distance standardized by effect size
- OTU:
-
Operational taxonomic unit
- PERMANOVA:
-
Permutational multivariate analysis of variance
- PCoA:
-
principal coordinates analysis
- PCR:
-
Polymerase chain reaction
- RCbray:
-
Raup-crick bray value
- rRNA:
-
Ribosomal ribonucleic acid
References
Moloney RD, Desbonnet L, Clarke G, Dinan TG, Cryan JF. The microbiome: stress, health and disease. Mamm Genome. 2014;25(1-2):49–74. https://doi.org/10.1007/s00335-013-9488-5.
Zaneveld JR, McMinds R, Vega TR. Stress and stability: applying the Anna Karenina principle to animal microbiomes. Nat Microbiol. 2017;2:1–8.
Lynch JB, Hsiao EY. Microbiomes as sources of emergent host phenotypes. Science (80- ). 2019;365:1405–9.
Kohl KD, Carey HV. A place for host-microbe symbiosis in the comparative physiologist’s toolbox. J Exp Biol. 2016;219(22):3496–504. https://doi.org/10.1242/jeb.136325.
Gould AL, Zhang V, Lamberti L, Jones EW, Obadia B, Korasidis N, et al. Microbiome interactions shape host fitness. Proc Natl Acad Sci. 2018;115(51):E11951–60. https://doi.org/10.1073/pnas.1809349115.
Suzuki TA. Links between natural variation in the microbiome and host fitness in wild mammals. Integr Comp Biol. 2017;57:746–69.
Trevelline BK, Fontaine SS, Hartup BK, Kohl KD. Conservation biology needs a microbial renaissance: a call for the consideration of host-associated microbiota in wildlife management practices. Proc R Soc B Biol Sci. 2019;286:1–9.
Brucker RM, Bordenstein SR. The hologenomic basis of speciation: gut bacteria cause hybrid lethality in the genus Nasonia. Science (80- ). 2013;466:667–9.
Foster KR, Schluter J, Coyte KZ, Rakoff-Nahoum S. The evolution of the host microbiome as an ecosystem on a leash. Nature. 2017;548(7665):43–51. https://doi.org/10.1038/nature23292.
Rosenberg E, Zilber-Rosenberg I. The hologenome concept of evolution after 10 years. Microbiome. 2018;6:1–14.
Alberdi A, Aizpurua O, Bohmann K, Zepeda-Mendoza ML, Gilbert MTP. Do vertebrate gut metagenomes confer rapid ecological adaptation? Trends Ecol Evol. 2016;31(9):689–99. https://doi.org/10.1016/j.tree.2016.06.008.
Zilber-Rosenberg I, Rosenberg E. Role of microorganisms in the evolution of animals and plants: the hologenome theory of evolution. FEMS Microbiol Rev. 2008;32(5):723–35. https://doi.org/10.1111/j.1574-6976.2008.00123.x.
Davidson GL, Raulo A, Knowles SCL. Identifying microbiome-mediated behaviour in wild vertebrates. Trends Ecol Evol. 2020;35(11):972–80. https://doi.org/10.1016/j.tree.2020.06.014.
Thaiss CA, Zmora N, Levy M, Elinav E. The microbiome and innate immunity. Nature. 2016;535(7610):65–74. https://doi.org/10.1038/nature18847.
Nieuwdorp M, Gilijamse PW, Pai N, Kaplan LM. Role of the microbiome in energy regulation and metabolism. Gastroenterology. 2014;146(6):1525–33. https://doi.org/10.1053/j.gastro.2014.02.008.
Oliphant K, Allen-Vercoe E. Macronutrient metabolism by the human gut microbiome: major fermentation by-products and their impact on host health. Microbiome Microbiome. 2019;7:1–15.
Kohl KD, Denise DM. The woodrat gut microbiota as an experimental system for understanding microbial metabolism of dietary toxins. Front Microbiol. 2016;7:1–9.
Kohl KD, Weiss RB, Cox J, Dale C, Denise DM. Gut microbes of mammalian herbivores facilitate intake of plant toxins. Ecol Lett. 2014;17(10):1238–46. https://doi.org/10.1111/ele.12329.
Johnson MTJ, Munshi-South J. Evolution of life in urban environments. Science (80- ). 2017;358:1–11.
Rivkin LR, Santangelo JS, Alberti M, Aronson MFJ, de Keyzer CW, Diamond SE, et al. A roadmap for urban evolutionary ecology. Evol Appl. 2018;12:384–98.
Chen G, Li X, Liu X, Chen Y, Liang X, Leng J, et al. Global projections of future urban land expansion under shared socioeconomic pathways. Nat Commun. 2020;11:1–12.
Alberti M, Palkovacs EP, Roches S, Meester L, Brans KI, Govaert L, et al. The complexity of urban eco-evolutionary dynamics. Bioscience. 2020;70:1–22.
Alberti M, Correa C, Marzluff JM, Hendry AP, Palkovacs EP, Gotanda KM, et al. Global urban signatures of phenotypic change in animal and plant populations. Proc Natl Acad Sci. 2017;114(34):8951–6. https://doi.org/10.1073/pnas.1606034114.
Lambert MR, Brans KI, Roches S Des, Donihue CM, Diamond SE. Ecology & evolution adaptive evolution in cities: Progress and misconceptions. Trends Ecol Evol; 2020;36:1–19.
Zepeda Mendoza ML, Xiong Z, Escalera-Zamudio M, Runge AK, Thézé J, Streicker D, et al. Hologenomic adaptations underlying the evolution of sanguivory in the common vampire bat. Nat Ecol Evol. 2018;2(4):659–68. https://doi.org/10.1038/s41559-018-0476-8.
Nyholm L, Koziol A, Marcos S, Botnen AB, Aizpurua O. Gopalakrishnan S, et al. Holo-Omics: Integrated Host-Microbiota Multi-omics for Basic and Applied Biological Research iScience. 2020;23:1–16.
Phillips JN, Berlow M, Derryberry EP. The effects of landscape urbanization on the gut microbiome: an exploration into the gut of urban and rural white-crowned sparrows. Front Ecol Evol. 2018;6:1–10.
Berlow M, Phillips JN, Derryberry EP. Effects of urbanization and landscape on gut microbiomes in white-crowned sparrows. Microb Ecol. 2020;81:253–66.
Teyssier A, Rouffaer LO, Saleh Hudin N, Strubbe D, Matthysen E, Lens L, et al. Inside the guts of the city: urban-induced alterations of the gut microbiota in a wild passerine. Sci Total Environ. 2018;612:1276–86. https://doi.org/10.1016/j.scitotenv.2017.09.035.
Teyssier A, Matthysen E, Hudin NS, de Neve L, White J, Lens L. Diet contributes to urban-induced alterations in gut microbiota: experimental evidence from a wild passerine. Proc R Soc B Biol Sci. 2020;287:1–9.
Fuirst M, Veit RR, Hahn M, Dheilly N, Thorne LH. Effects of urbanization on the foraging ecology and microbiota of the generalist seabird Larus argentatus. PLoS One. 2018;13:1–22.
Gadau A, Crawford MS, Mayek R, Giraudeau M, McGraw KJ, Whisner CM, et al. A comparison of the nutritional physiology and gut microbiome of urban and rural house sparrows (Passer domesticus). Comp Biochem Physiol Part B Elsevier. 2019;237:1–7.
Murray MH, Lankau EW, Kidd AD, Welch CN, Ellison T, Adams HC, et al. Gut microbiome shifts with urbanization and potentially facilitates a zoonotic pathogen in a wading bird. PLoS One. 2020;15:1–16.
Laforest-Lapointe I, Messier C, Kembel SW, Brodie EL, Berkeley L. Tree leaf bacterial community structure and diversity differ along a gradient of urban intensity. mSystems. 2017;2:1–16.
Stothart MR, Palme R, Newman AEM. It’s what’s on the inside that counts: stress physiology and the bacterial microbiome of a wild urban mammal. Proc R Soc B. 2019;286:1–9.
Littleford-Colquhoun BL, Weyrich LS, Kent N, Frere CH. City life alters the gut microbiome and stable isotope profiling of the eastern water dragon (Intellagama lesueurii). Mol Ecol. 2019;28(20):4592–607. https://doi.org/10.1111/mec.15240.
Knutie SA, Chaves JA, Gotanda KM. Human activity can influence the gut microbiota of Darwin’s finches in the Galapagos Islands. Mol Ecol. 2019;28(9):2441–50. https://doi.org/10.1111/mec.15088.
Lowry H, Lill A, Wong BBM. Behavioural responses of wildlife to urban environments. Biol Rev. 2013;88(3):537–49. https://doi.org/10.1111/brv.12012.
Stothart MR, Greuel RJ, Gavriliuc S, Henry A, Wilson AJ, McLoughlin PD, et al. Bacterial dispersal and drift drive microbiome diversity patterns within a population of feral hindgut fermenters. Mol Ecol. 2021;30(2):555–71. https://doi.org/10.1111/mec.15747.
Moeller AH, Suzuki TA, Lin D, Lacey EA, Wasser SK, Nachman MW. Dispersal limitation promotes the diversification of the mammalian gut microbiota. Proc Natl Acad Sci. 2017;114(52):13768–73. https://doi.org/10.1073/pnas.1700122114.
Miller ET, Svanbäck R, Bohannan BJM. Microbiomes as metacommunities: understanding host-associated microbes through metacommunity ecology. Trends Ecol Evol. 2018;33(12):926–35. https://doi.org/10.1016/j.tree.2018.09.002.
Burns AR, Miller E, Agarwal M, Rolig AS, Milligan-myhre K, Seredick S. Interhost dispersal alters microbiome assembly and can overwhelm host innate immunity in an experimental zebrafish model. Proc Natl Acad Sci. 2017;114(42):11181–6. https://doi.org/10.1073/pnas.1702511114.
Stothart MR, Bobbie CB, Schulte-hostedde AI, Boonstra R, Palme R, Mykytczuk NCS, et al. Stress and the microbiome: linking glucocorticoids to bacterial community dynamics in wild red squirrels. Biol Lett. 2016;12:14–7.
Estellé J. Benefits from the joint analysis of host genomes and metagenomes: select the holobiont. J Anim Breed Genet. 2019;136(2):75–6. https://doi.org/10.1111/jbg.12383.
Innes S, Lavigne DM. Comparative energetics of coat colour polymorphs in the eastern grey squirrel, Sciurus carolinensis. Can J Zool. 1979;57(3):585–92. https://doi.org/10.1139/z79-068.
Ducharme MB, Larochelle J, Richard D. Thermogenic capacity in gray and black morphs the gray squirrel, Sciurus carolinensis. Physiol Zool. 1989;62(6):1273–92. https://doi.org/10.1086/physzool.62.6.30156213.
McRobie H, Thomas A, Kelly J. The genetic basis of melanism in the gray squirrel (Sciurus carolinensis). J Hered. 2009;100(6):709–14. https://doi.org/10.1093/jhered/esp059.
Gustafson EJ, VanDruff LW. Behavior of black and gray morphs of Sciurus carolinensis in an urban environment. Am Midl Nat. 1990;123(1):186–92. https://doi.org/10.2307/2425772.
Gibbs JP, Buff MF, Cosentino BJ. Understanding Urban Ecology: An Interdisciplinary Systems Approach. In: The Biological System—Urban Wildlife, Adaptation, and Evolution: Urbanization as a Driver of Contemporary Evolution in Gray Squirrels (Sciurus carolinensis). Editors: Myrna H. P. Hall & Stephen B. Balogh. Edition: 1. Cham: Springer; 2019.
Ducrest A-L, Keller L, Roulin A. Pleiotropy in the melanocortin system, coloration and behavioural syndromes. Trends Ecol Evol. 2008;23(9):502–10. https://doi.org/10.1016/j.tree.2008.06.001.
Cullender TC, Chassaing B, Janzon A, Kumar K, Muller CE, Werner JJ, Angenent LT, Bell ME, Hay AG, Peterson DA, Walter J, Vijay-Kumar M, Gewirtz AT, Ley RE Innate and adaptive immunity interact to quench microbiome flagellar motility in the gut. Cell Host Microbe. 2013;14:571–581. [cited 2018 Sep 18] Available from: http://dx.doi.org/https://doi.org/10.1016/j.chom.2013.10.009, 5
Raulo A, Allen B, Troitsky T, Husby A, Firth JA, Coulson T, et al. Social networks strongly predict the gut microbiota of wild mice. ISME J. 2021.
Stegen JC, Lin X, Konopka AE, Fredrickson JK. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 2012;6(9):1653–64. https://doi.org/10.1038/ismej.2012.22.
Stegen JC, Lin X, Fredrickson JK, Chen X, Kennedy DW, Murray CJ, et al. Quantifying community assembly processes and identifying features that impose them. ISME J. 2013;7(11):2069–79. https://doi.org/10.1038/ismej.2013.93.
Webb CO, Ackerly DD, McPeek MA, Donoghue MJ. Phylogenies and community ecology. Annu Rev Ecol Syst. 2002;33(1):475–505. https://doi.org/10.1146/annurev.ecolsys.33.010802.150448.
Ren T, Boutin S, Humphries MM, Dantzer B, Gorrell JC, Coltman DW, et al. Seasonal, spatial, and maternal effects on gut microbiome in wild red squirrels. Microbiome. 2017;5:1–14.
Dill-McFarland KA, Neil KL, Zeng A, Sprenger RJ, Kurtz CC, Suen G, et al. Hibernation alters the diversity and composition of mucosa-associated bacteria while enhancing antimicrobial defence in the gut of 13-lined ground squirrels. Mol Ecol (10.1111); 2014;23:4658–4669. [cited 2018 May 26] Available from: http://doi.wiley.com/https://doi.org/10.1111/mec.12884
Chase JM, Kraft NJB, Smith KG, Vellend M, Inouye BD. Using null models to disentangle variation in community dissimilarity from variation in α-diversity. Ecosphere. 2011;2:1–11.
Rivera-Pinto J, Egozcue JJ, Pawlowsky-Glahn V, Paredes R, Noguera-Julian M, Calle ML, et al. Balances: a new perspective for microbiome analysis. mSystems. 2018;3:1–12.
Lin H, Das PS. Analysis of compositions of microbiomes with bias correction. Nat Commun. 2020;11:1–11.
Tyakht AV, Kostryukova ES, Popenko AS, Belenikin MS, Pavlenko AV, Larin AK, et al. Human gut microbiota community structures in urban and rural populations in Russia. Nat Commun. 2013;4:1–9.
Li H, Li T, Li X, Wang G, Lin Q, Qu J. Gut microbiota in Tibetan herdsmen reflects the degree of urbanization. Front Microbiol. 2018;9:1–14.
Bosmans L, Pozo MI, Verreth C, Crauwels S, Wilberts L, Sobhy IS, et al. Habitat-specific variation in gut microbial communities and pathogen prevalence in bumblebee queens (Bombus terrestris). PLoS One. 2018;13:1–19.
Bartlewicz J, Lievens B, Honnay O, Jacquemyn H. Microbial diversity in the floral nectar of Linaria vulgaris along an urbanization gradient. BMC Ecol. 2016;16:1–11.
Grond K, Bell KC, Demboski JR, Santos M, Sullivan JM, Hird SM. No evidence for phylosymbiosis in western chipmunk species. FEMS Microbiol Ecol. 2019;96:1–10.
Goertz S, de Menezes AB, Birtles RJ, Fenn J, Lowe AE, MacColl ADC, et al. Geographical location influences the composition of the gut microbiota in wild house mice (Mus musculus domesticus) at a fine spatial scale. PLoS One. 2019;14:1–16.
Schmidt C, Treberg JR, Kinnunen RP, Garroway CJ. Serum biochemistry suggests grey squirrels (Sciurus carolinensis) have poorer physiological condition in urban settings. bioRxiv. 2019:5–10.
Schulte-Hostedde AI, Mazal Z, Jardine CM, Gagnon J. Enhanced access to anthropogenic food waste is related to hyperglycemia in raccoons (Procyon lotor). Conserv Physiol. 2018;6:1–6.
Lyons J, Mastromonaco G, Edwards DB, Schulte-Hostedde AI. Fat and happy in the city: eastern chipmunks in urban environments. Behav Ecol. 2017;28(6):1464–71. https://doi.org/10.1093/beheco/arx109.
Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3(4):213–23. https://doi.org/10.1016/j.chom.2008.02.015.
Deehan EC, Walter J. The fiber gap and the disappearing gut microbiome: implications for human nutrition. Trends Endocrinol Metab. 2016;27(5):239–42. https://doi.org/10.1016/j.tem.2016.03.001.
Umu ÖCO, Frank JA, Fangel JU, Oostindjer M, da Silva CS, Bolhuis EJ, et al. Resistant starch diet induces change in the swine microbiome and a predominance of beneficial bacterial populations. Microbiome. 2015;3:1–15.
Morotomi M, Nagai F, Watanabe Y. Parasutterella secunda sp. nov., isolated from human faeces and proposal of Sutterellaceae fam. Nov. in the order Burkholderiales. Int J Syst Evol Microbiol. 2011;61(3):637–43. https://doi.org/10.1099/ijs.0.023556-0.
Ju T, Kong JY, Stothard P, Willing BP. Defining the role of Parasutterella, a previously uncharacterized member of the core gut microbiota. ISME J. 2019;13(6):1520–34. https://doi.org/10.1038/s41396-019-0364-5.
Biddle A, Stewart L, Blanchard J, Leschine S. Untangling the genetic basis of fibrolytic specialization by Lachnospiraceae and Ruminococcaceae in diverse gut communities. Diversity. 2013;5(3):627–40. https://doi.org/10.3390/d5030627.
Heinken A, Ravcheev DA, Baldini F, Heirendt L, Fleming RMT, Thiele I. Systematic assessment of secondary bile acid metabolism in gut microbes reveals distinct metabolic capabilities in inflammatory bowel disease. Microbiome. 2019;7:1–18.
Yutin N, Galperin MY. A genomic update on clostridial phylogeny: gram-negative spore formers and other misplaced clostridia. Environ Microbiol. 2013;15(10):2631–41. https://doi.org/10.1111/1462-2920.12173.
Devendran S, Shrestha R, Alves JMP, Wolf PG, Ly L, Hernandez AG, et al. Clostridium scindens ATCC 35704: Integration of nutritional requirements, the complete genome sequence, and global transcriptional responses to bile acids. 2019;1–22.
Studer N, Desharnais L, Beutler M, Brugiroux S, Terrazos MA, Menin L, et al. Functional intestinal bile acid 7α-dehydroxylation by Clostridium scindens associated with protection from Clostridium difficile infection in a gnotobiotic mouse model. Front Cell Infect Microbiol. 2016;6:1–15.
Islam KBMS, Fukiya S, Hagio M, Fujii N, Ishizuka S, Ooka T, et al. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology. 2011;141(5):1773–81. https://doi.org/10.1053/j.gastro.2011.07.046.
Golubeva AV, Joyce SA, Moloney G, Burokas A, Sherwin E, Arboleya S, et al. Microbiota-related changes in bile acid & tryptophan metabolism are associated with gastrointestinal dysfunction in a mouse model of autism. EBioMedicine. 2017;24:166–78. https://doi.org/10.1016/j.ebiom.2017.09.020.
Edenharder R, Pfützner A, Hammann R. Characterization of NAD-dependent 3α- and 3β-hydroxysteroid dehydrogenase and of NADP-dependent 7β-hydroxysteroid dehydrogenase from Peptostreptococcus productus. Biochim Biophys Acta. 1989;1004(2):230–8. https://doi.org/10.1016/0005-2760(89)90272-5.
Bailén M, Bressa C, Martínez-lópez S, González-soltero R. Microbiota features associated with a high-fat / low-fiber diet in healthy adults. Front Nutr. 2020;7:1–13.
Hirano S, Masuda N, Oda H, Mukai H. Transformation of bile acids by Eubacterium lentum. Appl Environ Microbiol. 1981;42(3):394–9. https://doi.org/10.1128/aem.42.3.394-399.1981.
Wen K, Tao L, Tao Z, Meng Y, Zhou S, Chen J, et al. Fecal and serum metabolomic signatures and microbial community profiling of postmenopausal osteoporosis mice model. Front Cell Infect Microbiol. 2020;10:1–11.
Harris SC, Devendran S, Méndez-García C, Mythen SM, Wright CL, Fields CJ, et al. Bile acid oxidation by Eggerthella lenta strains C592 and DSM 2243 T. Gut Microbes. 2018;9(6):523–39. https://doi.org/10.1080/19490976.2018.1458180.
Granado-Serrano AB, Martín-Garí M, Sánchez V, Riart Solans M, Berdún R, Ludwig IA, et al. Faecal bacterial and short-chain fatty acids signature in hypercholesterolemia. Sci Rep. 2019;9:1–13.
Zhang Y, Yu X, Yu E, Wang N, Cai Q, Shuai Q, et al. Changes in gut microbiota and plasma inflammatory factors across the stages of colorectal tumorigenesis: a case-control study. BMC Microbiol. 2018;18:1–10.
Kasai C, Sugimoto K, Moritani I, Tanaka J, Oya Y, Inoue H, et al. Comparison of the gut microbiota composition between obese and non-obese individuals in a Japanese population, as analyzed by terminal restriction fragment length polymorphism and next-generation sequencing. BMC Gastroenterol. 2015;15:1–10.
Ozato N, Saito S, Yamaguchi T, Katashima M, Tokuda I, Sawada K, et al. Blautia genus associated with visceral fat accumulation in adults 20–76 years of age. Biofilms Microbiomes. 2019;5:1–9.
Kashtanova D, Tkacheva O, Doudinskaya E, Strazhesko I, Kotovskaya Y, Popenko A, et al. Gut microbiota in patients with different metabolic statuses: Moscow study. Microorganisms. 2018;6(4):98. https://doi.org/10.3390/microorganisms6040098.
Wu N, Yang X, Zhang R, Li J, Xiao X, Hu Y, et al. Dysbiosis signature of fecal microbiota in colorectal cancer patients. Microb Ecol. 2013;66(2):462–70. https://doi.org/10.1007/s00248-013-0245-9.
Woerther PL, Antoun S, Chachaty E, Merad M. Eggerthella lenta bacteremia in solid tumor cancer patients: pathogen or witness of frailty? Anaerobe. 2017;47:70–2. https://doi.org/10.1016/j.anaerobe.2017.04.010.
Mahler DL, King AA, Freckleton RP, Arnegard ME, Peichel CL, Schluter D, et al. Predicting and manipulating cardiac inactivation by the human gut bacterium Eggerthella lenta. Anaerobe. 2013;341:295–9.
Song H, Wang W, Shen B, Jia H, Hou Z, Chen P, et al. Pretreatment with probiotic Bifico ameliorates colitis-associated cancer in mice: transcriptome and gut flora profiling. Cancer Sci. 2018;109(3):666–77. https://doi.org/10.1111/cas.13497.
Yazici C, Wolf PG, Kim H, Cross T-WL, Vermillion K, Carroll T, et al. Race-dependent association of sulfidogenic bacteria with colorectal cancer. Gut. 2017;66:1–9.
Thomas AM, Jesus EC, Lopes A, Aguiar S, Begnami MD, Rocha RM, et al. Tissue-associated bacterial alterations in rectal carcinoma patients revealed by 16S rRNA community profiling. Front Cell Infect Microbiol. 2016;6:1–13.
Almasi B, Roulin A, Korner-Nievergelt F, Jenni-Eiermann S, Jenni L. Coloration signals the ability to cope with elevated stress hormones: effects of corticosterone on growth of barn owls are associated with melanism. J Evol Biol. 2012;25(6):1189–99. https://doi.org/10.1111/j.1420-9101.2012.02508.x.
Bennett A, Hayssen V. Measuring cortisol in hair and saliva from dogs: coat color and pigment differences. Domest Anim Endocrinol. 2010;39(3):171–80. https://doi.org/10.1016/j.domaniend.2010.04.003.
Gangoso L, Grande JM, Ducrest AL, Figuerola J, Bortolotti GR, Andrés JA, et al. MC1R-dependent, melanin-based colour polymorphism is associated with cell-mediated response in the Eleonora’s falcon. J Evol Biol. 2011;24(9):2055–63. https://doi.org/10.1111/j.1420-9101.2011.02336.x.
Jacquin L, Récapet C, Prévot-Julliard AC, Leboucher G, Lenouvel P, Erin N, et al. A potential role for parasites in the maintenance of color polymorphism in urban birds. Oecologia. 2013;173(3):1089–99. https://doi.org/10.1007/s00442-013-2663-2.
Corbel H, Legros A, Haussy C, Jacquin L, Gasparini J, Karimi B, et al. Stress response varies with plumage colour and local habitat in feral pigeons. J Ornithol. 2016;157(3):825–37. https://doi.org/10.1007/s10336-016-1331-9.
Noguera JC, Aira M, Pérez-Losada M, Domínguez J, Velando A. Glucocorticoids modulate gastrointestinal microbiome in a wild bird. R Soc Open Sci. 2018;5:1–8.
McRobie HR, King LM, Fanutti C, Symmons MF, Coussons PJ. Agouti signalling protein is an inverse agonist to the wildtype and agonist to the melanic variant of the melanocortin-1 receptor in the grey squirrel (Sciurus carolinensis). FEBS Lett. 2014;588(14):2335–43. https://doi.org/10.1016/j.febslet.2014.05.032.
Pasitschniak-Arts M, Bendell JF. Behavioural differences between locally recruiting and dispersing gray squirrels, Sciurus carolinensis. Can J Zool. 1990;68(5):935–41. https://doi.org/10.1139/z90-134.
Bohls P, Koehnle TJ. Responses of eastern gray squirrels (Sciurus carolinensis) to predator calls and their modulation by coat color. Am Midl Nat. 2017;178(2):226–36. https://doi.org/10.1674/0003-0031-178.2.226.
McRobie HR, Moncrief ND, Mundy NI. Multiple origins of melanism in two species of north American tree squirrel (Sciurus). BMC Evol Biol. 2019;19:1–14.
Tung J, Barreiro LB, Burns MB, Grenier J-C, Lynch J, Grieneisen LE, et al. Social networks predict gut microbiome composition in wild baboons. Elife. 2015;4:1–18.
Antwis RE, Lea JMD, Unwin B, Shultz S. Gut microbiome composition is associated with spatial structuring and social interactions in semi-feral Welsh Mountain ponies. Microbiome. 2018;6:1–11.
Sarkar A, Harty S, Johnson KVA, Moeller AH, Archie EA, Schell LD, et al. Microbial transmission in animal social networks and the social microbiome. Nat Ecol Evol. 2020;4(8):1020–35. https://doi.org/10.1038/s41559-020-1220-8.
Fountain-Jones NM, Clark NJ, Kinsley AC, Carstensen M, Forester J, Johnson TJ, et al. Microbial associations and spatial proximity predict north American moose (Alces alces) gastrointestinal community composition. J Anim Ecol. 2020;89(3):817–28. https://doi.org/10.1111/1365-2656.13154.
Kohl KD, Varner J, Wilkening JL, Dearing MD. Gut microbial communities of American pikas (Ochotona princeps): evidence for phylosymbiosis and adaptations to novel diets. J Anim Ecol. 2018;87(2):323–30. https://doi.org/10.1111/1365-2656.12692.
Knowles SCL, Eccles RM, Baltrūnaitė L. Species identity dominates over environment in shaping the microbiota of small mammals. Ecol Lett. 2019;22:1–12.
Engel LD, Carlen EJ, Losos JB, Winchell KM. Eastern gray squirrels (Sciurus carolinensis) differ in abundance and response to hmans across urban habitats of St. Louis. Urban Nat. 2020;33:1–6.
Becker DJ, Streicker DG, Altizer S. Linking anthropogenic resources to wildlife-pathogen dynamics: a review and meta-analysis. Ecol Lett. 2015;18(5):483–95. https://doi.org/10.1111/ele.12428.
Start D, Barbour MA, Bonner C. Urbanization reshapes a food web. J Anim Ecol. 2020;89(3):808–16. https://doi.org/10.1111/1365-2656.13136.
Alberti M. Eco-evolutionary dynamics in an urbanizing planet. Trends Ecol Evol. 2015;30(2):114–26. https://doi.org/10.1016/j.tree.2014.11.007.
Des Roches S, Brans KI, Lambert MR, Rivkin LR, Savage AM, Schell CJ, et al. Socio-eco-evolutionary dynamics in cities. Evol Appl. 2021;14(1):248–67. https://doi.org/10.1111/eva.13065.
Miles LS, Dyer RJ, Verrelli BC. Urban hubs of connectivity: contrasting patterns of gene flow within and among cities in the western black widow spider. Proc R Soc B Biol Sci. 2018;285:1–9.
Carlen E, Munshi-South J. Widespread genetic connectivity of feral pigeons across the northeastern megacity. Evol Appl. 2020;14:150–62.
Zhu L, Clayton JB, Van Haute MJ, Yang Q, Hassenstab HR, Mustoe AC, et al. Sex bias in gut microbiome transmission in newly paired marmosets (Callithrix jacchus). mSystems. 2020;5:1–12.
Koprowski JL. Natal philopatry, communal nesting, and kinship in fox squirrels and gray squirrels. J Mammal. 1996;77(4):1006–16. https://doi.org/10.2307/1382781.
Amato KR, Van Belle S, Di Fiore A, Estrada A, Stumpf R, White B, et al. Patterns in gut microbiota similarity associated with degree of sociality among sex classes of a neotropical primate. Microb Ecol. 2017;74(1):250–8. https://doi.org/10.1007/s00248-017-0938-6.
Gu S, Chen D, Zhang J-N, Lv X, Wang K, Duan L-P, et al. Bacterial community mapping of the mouse gastrointestinal tract. PLoS One. 2013;8:1–9.
Walters W, Hyde ER, Berg-lyons D, Ackermann G, Humphrey G, Parada A, et al. Improved bacterial 16S rRNA gene (V4 and V4–5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems. 2015;1:1–10.
Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013;79(17):5112–20. https://doi.org/10.1128/AEM.01043-13.
Yilmaz P, Parfrey LW, Yarza P, Gerken J, Pruesse E, Quast C, et al. The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014;42:643–8.
Westcott SL, Schloss PD. OptiClust, an improved method for assigning amplicon-based sequence data to operational taxonomic units. mSphere, 2017;2:1–11.
Gloor GB, Macklaim JM, Pawlowsky-Glahn V, Egozcue JJ. Microbiome datasets are compositional: and this is not optional. Front Microbiol. 2017;8:1–6.
Sheneman L, Evans J, Foster JA. Clearcut: a fast implementation of relaxed neighbor joining. Bioinformatics. 2006;22(22):2823–4. https://doi.org/10.1093/bioinformatics/btl478.
Kuznetsova A, Brockhoff PB. Christensen RHB lmerTest package: tests in linear mixed effects models. J Stat Softw. 2017;82:1–26.
Wang J, Shen J, Wu Y, Tu C, Soininen J, Stegen JC, et al. Phylogenetic beta diversity in bacterial assemblages across ecosystems: deterministic versus stochastic processes. ISME J. 2013;7(7):1310–21. https://doi.org/10.1038/ismej.2013.30.
Martiny JBH, Jones SE, Lennon JT, Martiny AC. Microbiomes in light of traits: a phylogenetic perspective. Science (80- ). 2015;350:1–8.
Martiny AC, Treseder K, Pusch G. Phylogenetic conservatism of functional traits in microorganisms. ISME J. 2013;7(4):830–8. https://doi.org/10.1038/ismej.2012.160.
Keck F, Rimet F, Bouchez A, Franc A. Phylosignal: an R package to measure, test, and explore the phylogenetic signal. Ecol Evol. 2016;6(9):2774–80. https://doi.org/10.1002/ece3.2051.
Kembel SW, Cowan PD, Helmus MR, Cornwell WK, Morlon H, Ackerly DD, et al. Picante: R tools for integrating phylogenies and ecology. Bioinformatics. 2010;26(11):1463–4. https://doi.org/10.1093/bioinformatics/btq166.
Russel J. MicEco: various functions for microbial community data. 2019.
Anderson MJ, Ellingsen KE, McArdle BH. Multivariate dispersion as a measure of beta diversity. Ecol Lett. 2006;9(6):683–93. https://doi.org/10.1111/j.1461-0248.2006.00926.x.
Oksanen J, Kindt R, Legendre P, O’Hara B, Simpson GL, Solymos PM, et al. vegan: Community Ecology Package [Internet]. R Packag. 2008. Available from: https://cran.r-project.org/package=vegan
Paradis E, Schliep K. Ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. 2019;35(3):526–8. https://doi.org/10.1093/bioinformatics/bty633.
McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:1–11.
Acknowledgements
We thank the rare Charitable Research Reserve, the Essex Region Conservation Authority, the Grand River Conservation Authority, the City of Guelph, the University of Guelph, the University of Waterloo, and the University of Windsor for logistical support and allowing access to their respective properties. Thanks also to Michelle Hotchkiss for her aid in data collection.
Land acknowledgement
Work at our northern sites was conducted on the traditional territories of the Neutral, Haudenosaunee, and the Anishinaabe peoples. Our southern most sites at Wawiiatanong are on the traditional territory of the Three Fires Confederacy of First Nations, which includes the Ojibwa, the Odawa, and the Potawatomie.
Funding
Research was funded through Natural Sciences and Engineering Research Council of Canada (NSERC) and University of Guelph funds to AEM and a Society of Mammalogists grant-in-aid of research to MRS. MRS was funded by an Ontario Graduate Scholarship and NSERC CGS-M while completing this research.
Author information
Authors and Affiliations
Contributions
AEM and MRS designed the study. MRS collected samples, completed lab work, and performed analyses. AEM and MRS wrote the manuscript. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Animal utilization protocol was approved by the University of Guelph Animal Care Committee (AUP no. 3506). Trapping authorization was granted by the Ontario Ministry of Natural Resources (WSCA no. 1087323).
Consent for publication
Not Applicable.
Competing interests
Not Applicable
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplementary tables and figures referenced in the main-text of Shades of grey: host phenotype dependent effect of urbanization on the bacterial microbiome of a wild mammal.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stothart, M.R., Newman, A.E.M. Shades of grey: host phenotype dependent effect of urbanization on the bacterial microbiome of a wild mammal. anim microbiome 3, 46 (2021). https://doi.org/10.1186/s42523-021-00105-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42523-021-00105-4