
Abstract
Various environmental stresses reduce crop growth and productivity. Plants respond to stresses through changes in gene expression, protein abundance, and metabolite accumulation. Among functional genomics tools, ‘omics’ techniques such as transcriptomics, proteomics, and metabolomics are extensively used to reveal these responses. Altering plant phenotype by changing transcripts, proteins, and metabolites is one of the main challenges for improving crop production. The application of ‘omics’ techniques may facilitate the development of stress-tolerant crops. This review introduces ‘omics’ techniques and their use to identify how soybean responds to flooding.
Similar content being viewed by others

Review
Background
Climate changes, which naturally occurred or resulted from human activities, are a major concern in the globe (Hashiguchi et al. 2010). Abiotic stresses are regarded as a key limiting factor that disturb plant vegetation (Bray et al. 2000) and impair growth and yield of crops around the world (Mittler and Blumwald 2010). Plants have processes of response and adaptation to abiotic stresses at molecular, cellular, physiological, and biochemical levels (Yamaguchi-Shinozaki and Shinozaki 2006). For molecular level, plants respond to stresses through changes in gene expression, protein abundance, and metabolite accumulation. Among functional genomics tools, transcriptomics, proteomics, and metabolomics are extensively used to reveal these responses. These approaches can reveal how crops respond to environmental stresses, particularly flooding, which causes significant reductions in the growth and yield of many crops, notably soybean (Komatsu et al. 2012a). Altering soybean phenotype by changing transcripts, proteins, and metabolites is one of the main challenges for improving soybean production (Komatsu et al. 2013a). This knowledge may facilitate the development of flood-tolerant crops.
Komatsu et al. (2013a) mapped 92 metabolites, 75 proteins, and 921 mRNAs of soybean whose levels differed between control and flooded conditions in young seedlings. The increased mRNA and protein levels of glycolysis-related enzymes indicated the activation of glycolysis and fermentation cascades in the early responses of soybean to flooding stress (Komatsu et al. 2009a). The accumulation of proteins and metabolites of the tricarboxylic acid (TCA) cycle but the downregulation of mRNA expression suggested that flooding stress blocks the TCA cycle (Komatsu et al. 2011a). And the accumulation of protein destination or storage proteins while mRNA levels did not change indicated that the proteins built up as a result of growth delay (Komatsu et al. 2013a). These mapping results indicate disrupted flows, accumulated metabolites, and differential transcription in the metabolic network caused by flooding stress (Komatsu et al. 2013a). They also show how a proteomic approach gave a broader perspective than other ‘omics’ approaches. The use of these techniques will have applications in both breeding and agronomy. This review introduces the techniques of proteomics, transcriptomics, and metabolomics and shows their use to identify how soybean responds to flooding.
Proteomic techniques for identifying flooding-response mechanisms in soybean
The genome of several plant species has been sequenced, and proteins can be deduced from these sequences. Proteomic techniques combined with mass spectrometry (MS) can identify a number of environmental stress-responsive proteins; two-dimensional (2D) gel-based proteomic analysis provides a visual representation of the proteins, and SDS gel-based proteomics and gel-free proteomics are able to detect the largest number of proteins. The sample type and the protein targets should determine which techniques are used (Komatsu et al. 2014a). Three major proteomics technologies are described below.
Proteomic technologies
2D gel-based proteomics: Proteins are separated in the first dimension either on an immobilized pH gradient strip or in an isoelectric focusing tube, and then in the second dimension by SDS-polyacrylamide gel electrophoresis (Komatsu 2007, 2009; Komatsu et al. 2012a). After staining, protein spots are excised from gel, reduced with dithiothreitol, alkylated with iodoacetamide, and digested with trypsin. The peptide mixtures are then analyzed by a form of MS, such as nano-liquid chromatography (LC) tandem MS. Although 2D gel-based methods give a visual representation of the proteins including profiles of intact proteins, they are not suitable for the detection and identification of low-abundance proteins or those with extreme molecular weights, isoelectric points, or hydrophobicity.
SDS gel-based proteomics: Proteins are separated directly by SDS-polyacrylamide gel electrophoresis. After staining, protein bands are excised from the gel (Komatsu et al. 2012b), reduced with dithiothreitol, alkylated with iodoacetamide, and digested with trypsin. The proteins are then analyzed by nano-LC MS/MS. This method can identify more than seven times as many proteins as 2D gel-based proteomic techniques (Komatsu et al. 2014a).
Gel-free proteomics: Both label-free and labeling methods are available. The label-free method is explained here. Protein samples are purified by chloroform-methanol extraction and reduced with dithiothreitol, alkylated with iodoacetamide, and digested with trypsin and lysyl endopeptidase. They are then analyzed by nano-LC MS/MS (Komatsu et al. 2012b). Differentially regulated proteins are identified from the spectra obtained by searching against a peptide database with MASCOT Daemon client software (Matrix Science, London, UK). Positive matches are searched against protein databases such as NCBI (www.ncbi.nlm.nih.gov) by BLASTP for identification and annotation of homologous proteins. Because this protocol is simple and can identify a lot of proteins, it is now widely used.
Applications of proteomic techniques
Under flooding, soybean seedlings show differential regulation of proteins involved in signal transduction and hormonal signaling, transcriptional control, glucose degradation/sucrose accumulation, alcohol fermentation, gamma-aminobutyric acid (GABA) shunts, suppression of active oxygen scavenging, mitochondrial impairment, ubiquitin/proteasome-mediated proteolysis, and cell wall loosening (Nanjo et al. 2011a; Komatsu et al. 2012a, 2013a; Komatsu and Hossain 2013; Hossain et al. 2013).
To reveal how soybean responds to flooding and to analyze subcellular events and posttranslational modifications, researchers have used 2D gel-based and gel-free/label-free proteomic techniques. Under flooding, changes occurred in posttranslational modifications such as glycosylation (Mustafa and Komatsu 2014), ubiquitination (Yanagawa and Komatsu 2012), and phosphorylation (Yin et al. 2014; Nanjo et al. 2010, 2012), a common signaling event that occurs in response to stress. A gel-based proteomic technique using ProQ Diamond (Thermo Fisher Scientific, Waltham, MA, USA) staining revealed that the level of eukaryotic translation initiation factor 5A2 increased under flooding (Nanjo et al. 2010). On the other hand, using a gel-free proteomic technique, Nanjo et al. (2012) revealed that proteins in the endoplasmic reticulum were phosphorylated, suggesting that flooding has a drastic effect on the function of the endoplasmic reticulum. Furthermore, Yin et al. (2014) reported that the eukaryotic translation initiation factor 4G was dephosphorylated, suggesting that the ethylene signaling pathway plays an important role via protein dephosphorylation in the initial stages of flooding stress in soybean root tip. Phosphoproteomic approaches allow us to study phosphorylation of specific regulatory proteins and to identify new components of signaling cascades.
Soybean proteins affected by flooding stress have been identified in the cell wall (Komatsu and Yanagawa 2013; Komatsu et al. 2010), endoplasmic reticulum (Komatsu et al. 2012b), mitochondria (Komatsu et al. 2011a; Komatsu et al. 2011b), plasma membrane (Komatsu et al. 2009b), and nucleus (Komatsu et al. 2013b, 2014b; Oh et al. 2014) (Figure 1). Improved protocols for the purification of subcellular proteins allow in-depth proteomic studies to generate detailed information about the intrinsic mechanisms underlying stress responses. Subcellular proteomics supported the idea that the early response of soybean to flooding is an important adaptation that ensures survival in the face of direct damage to cells by flooding. Oh et al. (2014) identified the acceleration of protein poly-ADP-ribosylation and the suppression of RNA metabolism in root tip of soybean under flooding. It is not known whether nuclear function such as transcription factors is altered. However, Komatsu et al. (2013b, 2014b) suggested that the accumulation of RACK1 in the soybean nucleus has an important role under flooding, and several nuclear-related proteins and their mRNAs were decreased in the root tip of soybean under flooding.

Subcellular responses of soybean to flooding. Subcellular proteomic techniques identify proteins with increased (red) or decreased (blue) levels in each subcellular location.
Transcriptomic techniques for identifying flooding-response mechanisms in soybean
Transcriptome analysis is one of the important omics analyses to understand plant response to environmental changes. Past remarkable advances in transcriptomics technology has contributed greatly to clarify plant biology. Here, current applicable three of transcriptomics technologies, RNA sequencing (RNA-Seq), microarray, and high-coverage gene expression profiling (HiCEP) are listed and described. Three transcriptomic technologies are described below.
Transcriptomic technologies
RNA-Seq: This technology, based on high-throughput Next Generation Sequencing (Illumina, San Diego, CA, USA), relies on high-speed sequencing of short cDNA fragments (typically 30 to 400 bp) reverse-transcribed from mRNAs. The number of reads of cDNA fragments aligned to the reference sequence indicates the abundance of the mRNA. Numerous reports of RNA-Seq technology in plants are available. RNA-Seq technology has also been applied to non-model or unsequenced organisms (Ekblom and Galindo 2011): several computer programs enable de novo assembly of the reads obtained without a reference genome sequence (Robertson et al. 2010; Grabherr et al. 2011; Zhao et al. 2011). RNA-Seq technology can also identify novel transcript (exon) sequences and greater dynamic ranges than microarray technology (below). Although management of the huge data sets generated poses challenges, this technology is becoming a mainstream of transcriptome analysis.
Microarray: Microarray technology is based on hybridization between the target DNA and probe DNA designed with known sequences. Able to cover tens of thousands of genes at a time, it has contributed greatly to research. Despite its limitations in the variety of target transcripts and in the dynamic range of quantification compared with RNA-Seq technology (Sultan et al. 2008; Wang et al. 2009), it is well established and is still a major platform for transcriptome analysis of sequenced organisms. The simplicity of data obtained allows easy processing without special data management required in RNA-Seq technology.
HiCEP: high-coverage gene expression profiling, based on the amplified-fragment-length polymorphism technique, can detect changes in transcript expression with high coverage (Fukumura et al. 2003). Amplified DNA fragments derived from mRNA are analyzed by capillary electrophoresis, and their abundances are compared as electropherogram peaks. The relevant peaks are then fractionated and sequenced. This technology can also be applied to non-model or unsequenced organisms, and has some advantages, such as high reproducibility and high quantitative performance, compared with microarray technology. Nevertheless, it is used less frequently than the other technologies.
Applications of transcriptomic techniques
HiCEP (Komatsu et al. 2009a) and microarray (Nanjo et al. 2011a; Tamang et al. 2014) techniques have been used to uncover transcriptomic responses of soybean to flooding. HiCEP analysis of root of flooded soybean seedlings (Komatsu et al. 2009a) showed that 5,831 of 29,388 detected transcripts were differentially expressed during 2 days’ flooding. Sequence identification of 108 transcripts with a tenfold change in expression revealed that flooding induced genes involved in fermentation, ethylene biosynthesis, cell wall loosening, and pathogen defense.
Microarray analysis of root of flooded soybeans using a custom-designed Agilent array (Agilent Technologies Inc., Santa Clara, CA, USA) containing 23 867 60-mer oligonucleotide probes corresponding to soybean transcripts (Nanjo et al. 2011b) showed that 6,537 to 6,893 transcripts were differentially expressed under 6 to 12 h of flooding. Genes related to photosynthesis, glycolysis, synthesis of serine, glycine and cysteine, regulation of transcription, ubiquitin-mediated protein degradation, and cell death were upregulated. Genes related to cell wall synthesis, secondary metabolism, metabolite transport, cell organization, DNA synthesis, regulation of chromatin structure, and degradation of aspartate family amino acids were downregulated. Gene set enrichment analysis using gene ontology showed the upregulation of genes related to transcription (GO:0006350) and to the regulation of gene expression (GO:0010468), macromolecule metabolic processes (GO:0050255), and metabolic processes (GO:0019222), and the downregulation of genes related to the cell cycle (GO:0007049) and to cellular amino acid and derivative (GO:0006519), secondary (GO:0019748), and carbohydrate (GO:0005975) metabolic processes.
Another microarray experiment using an Affymetrix array (Affymetrix, Santa Clara, CA, USA) containing 34 781 25-mer oligonucleotide probes corresponding to soybean transcripts (Tamang et al. 2014) showed that 5,124 transcripts commonly changed by both 1 and 5 days’ flooding in cotyledons and hypocotyls (aerial tissues) of soybean seedlings:genes were associated with anaerobic respiration, stress responses, disease resistance, protein degradation, and signaling. One day’s flooding additionally induced a limited number of genes associated with starch degradation, glycolysis, and fermentation. Five days’ flooding downregulated genes involved in carbohydrate breakdown, lipid synthesis/degradation, cell wall synthesis/degradation, protein synthesis, amino acid synthesis, photosynthesis, and secondary metabolism. Only partial overlaps with root genes (Nanjo et al. 2011b) were found.
These transcriptomic studies show overall that under flooding, a number of transcripts are affected in both roots and shoots of soybean seedlings. The upregulation of genes related to anaerobic respiration, cell wall loosening, protein degradation, disease resistance, transcription, and signaling was common to both the root (Komatsu et al. 2009a; Nanjo et al. 2011b) and aerial tissues (Tamang et al. 2014) (Figure 2). Likewise, the downregulation of genes related to cell wall synthesis, secondary metabolism, and protein synthesis was common. These transcriptional responses probably underlie processes of acclimatization to flooding. The regulation of these commonly identified genes was consistently found also in Arabidopsis (Klok et al. 2002), cotton (Christianson et al. 2010), and poplar (Kreuzwieser et al. 2009) in response to flooding or hypoxia. Thus, these studies have helped to uncover the flooding-response mechanisms of soybean seedlings. Further in-depth transcriptomic studies using various materials will help to clarify how the response is regulated.

Metabolomic techniques for identifying flooding-response mechanisms in soybean
Metabolomic analysis has emerged as a promising tool to identify metabolic networks in living cells (Fiehn et al. 2001). Nevertheless, a simultaneous detection of a great number of intracellular metabolites is a bottleneck of such studies in terms of sensitivity and selectivity of metabolites (Takahashi et al. 2005). Plant metabolomics allows a good understanding of biochemical pathways and molecular mechanisms because plants contain many metabolites.
Metabolomic technologies
The high-throughput technology has been developed to detect a number of intracellular metabolites. Platforms used routinely in plant physiology include gas chromatography MS, Fourier transform ion cyclotron resonance MS, LC-MS, capillary electrophoresis MS, and nuclear magnetic resonance (Putri et al. 2013). Metabolomic studies in plants aim to identify and quantify the complete range of primary and secondary metabolites involved in biological processes (Deshmukh et al. 2014). Several metabolomic studies based on new chromatographic and MS techniques have been performed.
Applications of metabolomic techniques
Metabolomic techniques in combination with proteomic data provide a strong tool for analyzing biological events (Komatsu et al. 2011a, 2014c). Metabolomic and proteomic techniques were used to examine whether flooding stress alters mitochondrial function in soybeans: it increased the abundance of proteins and metabolites of the TCA cycle and GABA shunts, but decreased that of inner membrane carrier proteins and proteins related to complexes III, IV, and V of the electron transport chain (Komatsu et al. 2011a). In addition, the amounts of NADH and NAD+ increased in root, but that of ATP decreased. These results suggest that flooding directly impairs the electron transport chain in soybeans, although mitochondrial NADH production increases through the TCA cycle (Komatsu et al. 2011a).
Flooding increased the abundance of methionine synthase, heat shock cognate protein, urease, and phosphoenolpyruvate carboxylase (Komatsu et al. 2014c). Capillary electrophoresis MS identified 73 flooding-responsive metabolites. Flooding increased the levels of GABA, glycine, NADH2, and phosphoenolpyruvate. Taken together, these results suggest that flooding activates the synthesis of phosphoenolpyruvate by way of oxaloacetate produced in the TCA cycle and also modulates the urea cycle in soybean root tips (Komatsu et al. 2014c).
Integration of ‘omics’ data
Schemes have been developed to integrate ‘omics’ data. One such scheme is the Soybean Proteome Database, which contains data integrated from multiple analyses (Sakata et al. 2009; Ohyanagi et al. 2012). The database provides an ‘omics’ table indicating the relationships among 89 metabolites, 51 proteins, and 106 mRNAs of soybean that vary over time under flooding stress in seedlings (Sakata et al. 2009). The database has been used in many studies, such as comparative analyses of the proteomes of leaves and flowers at various stages of development (Ahsan and Komatsu 2009).
Another scheme is based on metabolic networks, to which multiple ‘omics’ data are anchored (Komatsu et al. 2013a). The anchored data include mRNAs or proteins by reference to information on biochemical reactions held in databases such as the Kyoto Encyclopedia of Genes and Genomes (Kanehisa and Goto 2000). The relationships between mRNAs and proteins are determined by sequence similarity searches.
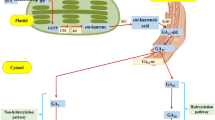
To test this scheme, we collected temporal profiles of metabolites from the glycolytic system and the TCA cycle in roots and hypocotyl tissues of 2- to 6-day-old soybean seedlings grown under control or flooded conditions (Nakamura et al. 2012). Over half of the metabolites showed differential abundance between conditions. Gel-based and gel-free proteomic approaches were used to gather evidence for the protein framework of the organs affected, and differentially abundant proteins were mapped on the metabolic network (Figure 3). The results suggested that a loop consisting of 2-oxoglutarate, glutamate, GABA, succinic semialdehyde, succinate, and succinyl-CoA, in and around the GABA shunt, was related to the possibly autocatalyzed the amplification of metabolite fluctuations under flooding. Our proteomic analyses suggested two enzymes in or near the GABA shunt that induce the amplification: succinic semialdehyde dehydrogenase (EC 1.2.1.16), which was upregulated, and malate dehydrogenase (EC 1.1.1.37), which was downregulated under flooding. These results will be useful in identifying how soybean responds to flooding.

Metabolome network around the TCA cycle. Arrows indicate metabolites in roots that showed significant changes in amount during flooding for 2 to 6 days: fat up arrow means ≥3×; thin up arrow means <3×; thin down arrow means <1×. Affected enzymes are indicated by their EC numbers: red, upregulated; blue, downregulated. (A) Control and (B) flooded. In each panel, the asterisked numbers (*1 or *2) mean the metabolic pathways are connected at the points indicated by the same numbers in the model.
Conclusion
Alcohol dehydrogenase mRNA and its protein increase temporarily in the root tips of soybean seedlings transferred to flooded anaerobic conditions (Komatsu et al. 2011b). Tougou et al. (2012) examined the response in transgenic soybean lines in which the soybean alcohol dehydrogenase (GmAdh2) gene was introduced under the control of a constitutive promoter. Protein analysis and enzyme assay confirmed the expression and activity of GmAdh2; this gene induced changes in glycolysis and alcohol fermentation and, thus, improved the germination of transgenic soybeans under flooding stress. This example indicates that the genes or proteins identified by ‘omics’ techniques are useful for gene discovery and molecular breeding.
Different ‘omics’ techniques have been used to understand how soybean responds to flooding stress. The comprehensive nature of multi-‘omics’ research provides an entirely new avenue, and research programs should adapt accordingly (Deshmukh et al. 2014). The integration of transcriptomics, proteomics, and metabolomics has identified important metabolic networks that are activated in response to flooding and has facilitated the identification of many important genes involved in stress-induced metabolic changes (Komatsu et al. 2013a). The integration of ‘omics’ data and mathematical models might improve our understanding of plant metabolism.
Abbreviations
- GABA:
-
gamma-aminobutyric acid
- HiCEP:
-
high-coverage gene expression profiling
- LC:
-
liquid chromatography
- MS:
-
mass spectrometry
- RNA-Seq:
-
RNA sequencing
- TCA cycle:
-
tricarboxylic acid cycle
- 2D-gel:
-
two-dimensional gel
References
Ahsan N, Komatsu S (2009) Comparative analyses of the proteomes of leaves and flowers at various stages of development reveal organ-specific functional differentiation of proteins in soybean. Proteomics 9:4889–4907
Bray EA, Bailey-Serres J, Weretilnyk E (2000) Responses to abiotic stresses. In: Buchannan B, Gruissem W, Jones R (eds) Biochemistry and molecular biology of plants. American Society of Plant Biologists, Rockville, MD, USA, pp 1158–1249
Christianson JA, Llewellyn DJ, Dennis ES, Wilson IW (2010) Global gene expression responses to waterlogging in roots and leaves of cotton (Gossypium hirsutum L.). Plant Cell Physiol 51:21–37
Deshmukh R, Sonah H, Patil G, Chen W, Prince S, Mutava R, Vuong T, Valliyodan B, Nguyen HT (2014) Integrating omic approaches for abiotic stress tolerance in soybean. Front Plant Sci 5:244
Ekblom R, Galindo J (2011) Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity 107:1–15
Fiehn O, Kloska S, Altmann T (2001) Integrated studies on plant biology using multiparallel techniques. Curr Opin Biotechnol 12:82–86
Fukumura R, Takahashi H, Saito T, Tsutsumi Y, Fujimori A, Sato S, Tatsumi K, Araki R, Abe M (2003) A sensitive transcriptome analysis method that can detect unknown transcripts. Nucleic Acids Res 31:e94
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotech 29:644–652
Hashiguchi A, Ahsan N, Komatsu S (2010) Proteomics application of crops in the context of climatic changes. Food Res Intl 43:1803–1813
Hossain Z, Khatoon A, Komatsu S (2013) Soybean proteomics for unraveling abiotic stress response mechanism. J Proteome Res 12:4670–4684
Kanehisa M, Goto S (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30
Klok EJ, Wilson IW, Wilson D, Chapman SC, Ewing RM, Somerville SC, Peacock WJ, Dolferus R, Dennis ES (2002) Expression profile analysis of the low-oxygen response in Arabidopsis root cultures. Plant Cell 14:2481–2494
Komatsu S (2007) Edman sequencing of proteins from 2D gels. Methods Mol Biol 355:211–217
Komatsu S (2009) Western blotting/Edman sequencing using PVDF membrane. Methods Mol Biol 536:163–171
Komatsu S, Hossain Z (2013) Organ-specific proteome analysis for identification of abiotic stress response mechanism in crop. Front Plant Sci 4:71
Komatsu S, Yanagawa Y (2013) Cell wall proteomics of crops. Front Plant Sci 4:17
Komatsu S, Wada T, Abaléa Y, Nouri MZ, Nanjo Y, Nakayama N, Shimamura S, Yamamoto R, Nakamura T, Furukawa K (2009a) Analysis of plasma membrane proteome in soybean and application to flooding stress response. J Proteome Res 8:4487–4499
Komatsu S, Yamamoto R, Nanjo Y, Mikami Y, Yunokawa H, Sakata K (2009b) A comprehensive analysis of the soybean genes and proteins expressed under flooding stress using transcriptome and proteome techniques. J Proteome Res 8:4766–4778
Komatsu S, Kobayashi Y, Nishizawa K, Nanjo Y, Furukawa K (2010) Comparative proteomics analysis of differentially expressed proteins in soybean cell wall during flooding stress. Amino Acids 39:1435–1449
Komatsu S, Deschamps T, Hiraga S, Kato M, Chiba M, Hashiguchi A, Tougou M, Shimamura S, Yasue H (2011a) Characterization of a novel flooding stress-responsive alcohol dehydrogenase expressed in soybean roots. Plant Mol Biol 77:309–322
Komatsu S, Yamamoto A, Nakamura T, Nouri MZ, Nanjo Y, Nishizawa K, Furukawa K (2011b) Comprehensive analysis of mitochondria in roots and hypocotyls of soybean under flooding stress using proteomics and metabolomics techniques. J Proteome Res 10:3993–4004
Komatsu S, Hiraga S, Yanagawa Y (2012a) Proteomics techniques for the development of flood tolerant crops. J Proteome Res 11:68–78
Komatsu S, Kuji R, Nanjo Y, Hiraga S, Furukawa K (2012b) Comprehensive analysis of endoplasmic reticulum-enriched fraction in root tips of soybean under flooding stress using proteomics techniques. J Proteomics 77:531–560
Komatsu S, Han C, Nanjo Y, Altaf-Un-Nahar M, Wang K, He D, Yang P (2013a) Label-free quantitative proteomic analysis of abscisic acid effect in early-stage soybean under flooding. J Proteome Res 12:4769–4784
Komatsu S, Shirasaka N, Sakata K (2013b) ‘Omics’ techniques for identifying flooding-response mechanisms in soybean. J Proteomics 93:169–178
Komatsu S, Hiraga S, Nouri MZ (2014a) Analysis of flooding-responsive proteins localized in the nucleus of soybean root tips. Mol Biol Rep 41:1127–1139
Komatsu S, Nakamura T, Sugimoto Y, Sakamoto K (2014b) Proteomic and metabolomic analyses of soybean root tips under flooding stress. Protein Pept Lett 21:865–884
Komatsu S, Oh MW, Jang HY, Kwon SJ, Kim HR, Ko JH, Woo SH, Nanjo Y (2014c) Proteomic analyses of soybean root tips during germination. Protein Pept Lett 21:1308–1319
Kreuzwieser J, Katharine JH, Howell A, Carroll A, Rennenberg H, Millar AH, Whelan J (2009) Differential response of gray poplar leaves and roots underpins stress adaptation during hypoxia. Plant Physiol 149:461–473
Mittler R, Blumwald E (2010) Genetic engineering for modern agriculture: challenges and perspectives. Annu Rev Plant Biol 61:443–462
Mustafa G, Komatsu S (2014) Quantitative proteomics reveals the effect of protein glycosylation in soybean root under flooding stress. Front Plant Sci 5:627
Nakamura T, Yamamoto R, Hiraga S, Nakayama N, Okazaki K, Takahashi H, Uchimiya H, Komatsu S (2012) Evaluation of metabolite alteration under flooding stress in soybeans. Jpn Agric Res Q 46:237–248
Nanjo Y, Skultety L, Ashraf Y, Komatsu S (2010) Comparative proteomic analysis of early-stage soybean seedlings responses to flooding by using gel and gel-free techniques. J Proteome Res 9:3989–4002
Nanjo Y, Maruyama K, Yasue H, Yamaguchi-Shinozaki K, Shinozaki K, Komatsu S (2011a) Transcriptional responses to flooding stress in roots including hypocotyl of soybean seedlings. Plant Mol Biol 77:129–144
Nanjo Y, Nouri MZ, Komatsu S (2011b) Quantitative proteomic analyses of crop seedlings subjected to stress conditions; a commentary. Phytochemistry 2:1263–1272
Nanjo Y, Skultety L, Uváčková L, Klubicová K, Hajduch M, Komatsu S (2012) Mass spectrometry-based analysis of proteomic changes in the root tips of flooded soybean seedlings. J Proteome Res 11:372–385
Oh MW, Nanjo Y, Komatsu S (2014) Identification of nuclear proteins in soybean under flooding stress using proteomic technique. Protein Pept Lett 21:458–467
Ohyanagi H, Sakata K, Komatsu S (2012) Soybean Proteome Database 2012: update on the comprehensive data repository for soybean proteomics. Front Plant Sci 3:110
Putri SP, Yamamoto S, Tsugawa H, Fukusaki E (2013) Current metabolomics: technological advances. J Biosci Bioeng 116:9–16
Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, Mungall K, Lee S, Okada HM, Qian JQ, Griffith M, Raymond A, Thiessen N, Cezard T, Butterfield YS, Newsome R, Chan SK, She R, Varhol R, Kamoh B, Prabhu AL, Tam A, Zhao Y, Moore RA, Hirst M, Marra MA, Jones SJ, Hoodless PA, Birol I (2010) De novo assembly and analysis of RNA-seq data. Nat Meth 7:909–912
Sakata K, Ohyanagi H, Nobori H, Nakamura T, Hashiguchi A, Nanjo Y, Mikami Y, Yunokawa H, Komatsu S (2009) Soybean proteome database: a data resource for plant differential omics. J Proteome Res 8:3539–3548
Sultan M, Schulz MH, Richard H (2008) A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 321:956–960
Takahashi H, Hotta Y, Hayashi M, Kawai-Yamada M, Komatsu S, Uchimiya H (2005) High throughput metabolome and proteome analysis of transgenic rice plants (Oryza sativa L.). Plant Biotech 22:47–50
Tamang BG, Magliozzi JO, Maroof MAS, Fukao T (2014) Physiological and transcriptomic characterization of submergence and reoxygenation responses in soybean seedlings. Plant Cell Environ 37:2350–2365
Tougou M, Hashiguchi A, Yukawa K, Nanjo Y, Hiraga S, Nakamura T, Nishizawa K, Komatsu S (2012) Responses to flooding stress in soybean seedlings with the alcohol dehydrogenase transgene. Plant Biotechnol 29:301–305
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Gen 10:57–63
Yamaguchi-Shinozaki K, Shinozaki K (2006) Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu Rev Plant Biol 57:781–803
Yanagawa Y, Komatsu S (2012) Ubiquitin/proteasome-mediated proteolysis is involved in the response to flooding stress in soybean roots, independent of oxygen limitation. Plant Sci 185–186:250–258
Yin X, Sakata K, Komatsu S (2014) Phosphoproteomics reveals the effect of ethylene in soybean root under flooding stress. J Proteome Res 13:5618–5634
Zhao QY, Wang Y, Kong YM, Luo D, Li X, Hao P (2011) Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC Bioinformatics 12:1–12
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SK, KS, and TN have prepared, read, and approved the manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Komatsu, S., Sakata, K. & Nanjo, Y. ‘Omics’ techniques and their use to identify how soybean responds to flooding. J Anal Sci Technol 6, 9 (2015). https://doi.org/10.1186/s40543-015-0052-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40543-015-0052-7