
Abstract
Background
Helicobacter pylori is an important pathogenic factor in gastric carcinogenesis. Angiogenesis (i.e., the growth of new blood vessels) is closely associated with the incidence and development of gastric cancer. Our previous study found that COX-2 stimulates gastric cancer cells to induce expression of the angiogenic growth factor VEGF through an unknown mechanism. Therefore, the aim of this study was to clarify the role of angiogenesis in H. pylori-induced gastric cancer development.
Methods
To clarify the relationship between H. pylori infection and angiogenesis, we first investigated H. pylori colonization, COX-2, VEGF, beta-catenin expression, and microvessel density (MVD) in gastric cancer tissues from 106 patients. In addition, COX-2, phospho-beta-catenin, and beta-catenin expression were measured by western blotting, and VEGF expression was measured by ELISA in H. pylori-infected SGC7901 and MKN45 human gastric cancer cells.
Results
H. pylori colonization occurred in 36.8 % of gastric carcinoma samples. Furthermore, COX-2, beta-catenin, and VEGF expression, and MVD were significantly higher in H. pylori-positive gastric cancer tissues than in H. pylori-negative gastric cancer tissues (P < 0.01). H. pylori infection was not related to sex or age in gastric cancer patients, but correlated with the depth of tumor invasion, lymph node metastasis, and tumor–node–metastasis stage (P < 0.05) and correlated with the COX-2 expression and beta-catenin expression(P < 0.01). Further cell experiments confirmed that H. pylori infection upregulated VEGF in vitro. Further analysis revealed that H. pylori-induced VEGF expression was mediated by COX-2 via activation of the Wnt/beta-catenin pathway.
Conclusions
The COX-2/Wnt/beta-catenin/VEGF pathway plays an important role in H. pylori-associated gastric cancer development. The COX-2/Wnt/beta-catenin pathway is therefore a novel therapeutic target for H. pylori-associated gastric cancers.
Similar content being viewed by others

Background
Helicobacter pylori is a Gram-negative, spiral bacillus that infects approximately half the world’s population and induces chronic inflammation of the gastric mucosa, contributing to the development of peptic ulcer and gastric malignancies [1, 2]. H. pylori has been classified as a class I carcinogen by the International Agency for Research on Cancer (IARC) and World Health Organization (WHO) [3]. However, the pathogenesis of H. pylori infection–induced gastric cancer has not been fully elucidated.
Angiogenesis is already present in early gastric cancer, and its development requires a unique tumor phenotype and necessary ingredients. As the cancer progresses toward more advanced stages, angiogenesis becomes more pronounced. Angiogenesis and the occurrence and development of gastric cancer are closely related [4]. Angiogenesis is a key step in tumor growth and metastasis [5]. Neovascularization not only provides nutrients and oxygen to the tumor cells, and carries away metabolic waste, but it also stimulates tumor growth through autocrine or paracrine modes of action. It is a complex process of angiogenesis, which is co-regulated by angiogenic and anti-angiogenic factors. Gastric cancer cells can produce a variety of proangiogenic growth factors [6], and vascular endothelial growth factor (VEGF) is the strongest and the most specific angiogenic growth factor. VEGF plays a major role in the multistep process of angiogenesis stimulation and is closely related to the development of gastric cancer [7]. Moreover, VEGF plays a pivotal role in tumor-associated microvascular angiogenesis [8] and has been demonstrated to be overexpressed in human gastric carcinomas [9–11]. Although there have been numerous reports on H. pylori infection influencing angiogenesis in gastric cancer, the exact mechanism remains unclear.
COX is a key rate-limiting enzyme in the conversion of arachidonic acid to prostanoids and thromboxanes; it exists in two forms, cyclooxygenase 1 (COX-1) and COX-2 [12, 13]. COX-1 is responsible for maintaining normal physiological function; it is expressed constitutively in most tissues. In contrast, COX-2 is an early response gene induced by growth factors, proinflammatory cytokines, tumor promoters, and bacterial toxins [14–16]. We earlier demonstrated that H. pylori can upregulate COX-2 via the p38 mitogen-activated protein kinase (MAPK)/activating transcription factor-2 (ATF-2) signaling pathway in MKN45 gastric cancer cells [17]. Caputo et al. [18] reported that H. pylori induced VEGF upregulation in MKN28 gastric cancer cells, which might be mediated by COX-2. Moreover, research shows that that H. pylori infection influences angiogenesis in gastric cancer patients [19]. Considering these results, it is reasonable to believe that COX-2 might play a role in VEGF upregulation in H. pylori-infected gastric cancers.
The Wnt/beta-catenin pathway is commonly activated during carcinogenesis [20]. In the classical Wnt signaling pathway, Wnt binding to its Fz receptor inactivates the beta-catenin destructive complex comprising adenomatous polyposis coli (APC), axin, and glycogen synthase kinase-3 beta (GSK3beta). Beta-catenin then disassociates from the complex, translocates into the nucleus, and binds to members of the lymphoid-enhancing factor/T-cell factors (Tcf/Lef) family that activate target gene transcription when the Wnt pathway is activated [21]. Normally, beta-catenin phosphorylation maintains the complex in a stable state, and unphosphorylated beta-catenin enters into the nucleus when Wnt pathway is activated. The Wnt/beta-catenin pathway is important for angiogenesis, and beta-catenin is associated with COX-2 overexpression [22] and angiogenesis [23]. However, whether the Wnt/beta-catenin pathway plays a role in H. pylori-induced angiogenesis is unclear.
In the present study, we aimed to investigate whether the Wnt/beta-catenin pathway is involved in H. pylori-induced upregulation of angiogenesis in gastric cancer.
Methods
H. pylori culture
The H. pylori cagA- and vacA-positive standard strain NCTC11637 was obtained from the Institute of Digestive Diseases, Renji Hospital, Shanghai Jiao Tong University, Shanghai, China. H. pylori was cultured on Columbia agar (Oxoid, Basingstoke Hampshire, UK) plates containing 5 % sheep blood and incubated at 37 °C under microaerophilic conditions for 48–72 h. Colonies were identified as H. pylori by Gram staining, morphology, and positive oxidase, catalase, and urease activities. Bacteria were suspended in phosphate-buffered saline (PBS) and the density was estimated by spectrophotometry (OD600 nm) and microscopic observation.
Immunohistochemical staining of COX-2, beta-catenin, VEGF, and CD34 in human gastric carcinoma tissues
A total of 106 different formalin-fixed, paraffin-embedded gastric cancer tissue samples and adjacent normal tissues were obtained from Shuguang Hospital, Shanghai University of Traditional Chinese Medicine. The use of all human tissue samples was approved by the Institutional Review Board of Shuguang Hospital, which is affiliated with Shanghai University of Traditional Chinese Medicine. Informed consent was obtained from every patient for the use of all human tissues used in this study. First, tissue samples were stained with Giemsa to determine the presence of H. pylori infection. Next, using standard methods, COX-2, beta-catenin, VEGF, and CD34 were detected immunohistochemically. Briefly, tissues were embedded in paraffin and 4-μm sections were cut, deparaffinized in xylene, and dehydrated through a graded alcohol series. Tissue sections were subjected to peroxidase clearance, antigen retrieval, and blocking of non-specific binding sites. Sections were first incubated with primary antibody (rabbit polyclonal antibodies against CD34, COX-2, beta-catenin, and VEGF (Abcam, Cambridge, MA, USA), followed by EnVision secondary antibody (Dako, Glostrup, Denmark). Sections were counterstained with hematoxylin. PBS served as a negative control for primary antibody. Staining intensity was assessed in each specimen on a scale of 0–3: 0, no staining; 1, weak staining; 2, moderate staining; and 3, strong staining.
Immunohistochemical analysis of the MVD
According to Weidner [24], areas of highest neovascularization were found by scanning tumor sections at low-power (×40) magnification. After the area of highest neovascularization was identified, individual microvessel counts were made in a single high-power (×200) magnification field. Three different visual fields were selected for microvessel counting, and the mean value was recorded. Brown-staining endothelial cells or endothelial cell clusters were considered as single, countable microvessels.
Cell culture and reagents
SGC7901 and MKN45 gastric cancer cells were obtained from the Institute of Digestive Diseases, Renji Hospital of Shanghai Jiao Tong University, Shanghai, China, and cultured in RPMI 1640(Gibco, Thermo Fisher Scientific Inc, Waltham, MA, USA) containing 10 % (v/v) fetal bovine serum (Gibco, Thermo Fisher Scientific Inc, Waltham, MA, USA) and 1 % penicillin and streptomycin (North China Pharmaceutical Company, Shijiazhuang, China). Cells were plated in 6-well plates and grown to confluency. FH535, a beta-catenin-specific inhibitor, was obtained from Cell Signaling (Beverly, MA, USA). All cells were grown in a humidified incubator containing 5 % CO2 at 37 °C.
Real-time fluorogenic quantitative polymerase chain reaction
RNA isolation
Total cellular RNA was prepared using RNAisol reagent (TaKaRa Biotechnology, Dalian, China) according to the manufacturer’s instructions. RNAisol (1 ml) was added to each sample and incubated for 5 min at room temperature. Next, 200 μl chloroform was added and samples were shaken for 15 s and incubated at room temperature for 2-3 min and then centrifuged at 12,000 g for 15 min at 4 °C after formation of a biphasic solution. For RNA precipitation, the aqueous phase (top) was transferred to a new tube and 500 μl isopropanol was added. Samples were incubated at room temperature for 5-10 min and then centrifuged at 12,000 g for 15 min at 4 °C, after which a pellet was visible. After supernatant removal, 1000 μl of 75 % ethanol was added to wash the RNA pellet; this was vortexed and centrifuged at 8000 g for 5 min at 4 °C. After the ethanol was carefully removed by pipetting, the RNA pellet was air-dried for 5-10 min and then dissolved in diethylpyrocarbonate-treated water with vortexing. RNA quality was verified by agarose gel electrophoresis and visualization of 28S and 18S ribosomal RNA. RNA was quantified by spectrophotometry (OD260/280 nm). RNA was then immediately frozen at −70 °C.
cDNA synthesis and real-time quantitative analysis
Reverse transcription was conducted using a PrimeScript RT-PCR Kit (TaKaRa Biotechnology, Dalian, China). Total RNA (1 μg) was used as a template for cDNA synthesis. Briefly, reverse transcription was carried out in a 20-μl solution including 4 μl 5× buffer, 1 μl oligo dT primer, 1 μl random 6-mers, 1 μl PrimeScript RT Enzyme Mix, and RNAse-free deionized H2O. Reverse transcription incubation conditions were 37 °C for 15 min and 85 °C for 5 s The resultant cDNA was stored at −20 °C until it was used for real-time quantitative polymerase chain reaction (PCR). Real-time PCR reactions were carried out using the ABI7300 Fast Real-Time PCR System (PE Biosystems, Foster City, CA, USA) using a PrimeScript RT-PCR Kit according to the manufacturer’s instructions. Primers and probes for human GAPDH, VEGF, and COX2 were designed and synthesized by Shanghai Shanjing Biotechnology (Shanghai, China) with FAM (6-carboxy-fluo-rescein-phosphoramidite)-labeled 5′ ends and TAMPA (carboxy-tetramethyl-rhodamine)-labeled 3′ ends. Primer and probe sequences were: human GAPDH-forward, 5′-CCACTCCTCCACCTTTGAC-3′; human GAPDH-reverse, 5′-ACCCTGTTGCTGTAGCCA-3′; GAPDH probe, 5′-TTGCCCTCAACGACCACTTTGTC-3′; human VEGF-forward, 5′-GGCCTCCGAAACCATGAACT-3′, human VEGF-reverse, 5′-ACCCTGTTGCTGTAGCCA-3′; and VEGF probe, 5′-TGTCTT GGGTGCATTGGAGC-3′. Briefly, each PCR was performed in a 20-μl reaction volume comprising 10 μl Premix EX Taq, 0.4 μl Rox reference dye, 0.4 μl each primer, 0.8 μl TaqMan probe, 6 μl deionized H2O and 2 μl cDNA. PCR cycling conditions were 95 °C for 10 s, followed by 40 cycles of 95 °C for 5 s (denaturation) and 60 °C for 31 s (annealing/extension). Each reaction was performed in triplicate, and data were analyzed by the 2−∆∆Ct method for comparing relative expression levels. GAPDH mRNA was used to normalize RNA levels from the various samples and mRNA expression was expressed as relative to the basal level without H. pylori stimulation.
Western blot analysis
Following treatment, cells were washed twice with ice-cold PBS and then protease inhibitors (Roch, Basel, Switzerland) were added. Cells were then scraped off the dish, and then cytoplasmic and nuclear fractions were prepared using a protein extraction kit (Fermentas, Waltham, MA, USA). Cell lysis buffer, nuclei washing buffer, and other reagent buffers were added to separate cytosolic proteins and nuclear proteins. The protein concentration in extracts was determined by bicinchoninic acid protein assay using a commercial kit (BCA Protein Assay Reagent; Merck, Whitehouse Station, NJ, USA). Protein samples were separated by 10 % SDS-PAGE and transferred to PVDF membrane. The membrane was incubated in blocking buffer (10 mmol/l Tris, pH 7.5, 100 mmol/l NaCl, 0.1 % Tween 20), containing 5 % nonfat powdered milk for 1 h. The membrane was then incubated with anti-phospho-beta-catenin or anti- beta-catenin polyclonal antibody (1:500; Cell Signaling Technology, USA). Following overnight incubation at 4 °C, blots were washed three times in TBS-Tween (0.05 %) solution and incubated with goat anti-rabbit antibodies conjugated to horseradish peroxidase (HRP) for 1 h at room temperature before visualizing using the Pierce ECL kit (Thermo Fisher Scientific Inc, Waltham, MA, USA). Results were analyzed by Image J software (NIH Image).
Enzyme-linked immunosorbent assay
Cell culture supernatant samples were collected and clarified at 3000 g for 5 min. ELISA was performed according to the manufacturer’s protocol. Briefly, microtiter plates were incubated with 100 μl samples at 37 °C for 120 min. After five washes in 10 mM PBS, plates were incubated with 100 μl anti-VEGF primary antibody labeled with biotin (from the ELISA kit) at 37 °C for 60 min. After five rinses with 10 mM PBS, 100 μl avidin-biotin-peroxidase complex was added to wells and incubated at 37 °C for 30 min. After extensive rinsing, 100 μl/well TMB Microwell Substrate and was added and plates were incubated in the dark at 37 °C for 15 min. The reaction was then stopped with 100 μl TMB stop solution and OD450 nm values were obtained within 30 min using a microplate reader. Finally, protein concentrations were determined from OD values using a calibration curve.
Statistical analysis
Statistical analyses were performed using the Statistical Package for the Social Sciences (SPSS version 19.0). Statistical significance was determined by t tests and one-way ANOVA followed by Fisher’s least significant difference test and differences in rates were determined by the chi-squared test. Data are presented as means ± SE and a P value of <0.05 was considered statistically significant.
Results
H. Pylori infection correlates with COX-2, VEGF, and beta-catenin upregulation and angiogenesis in gastric cancer
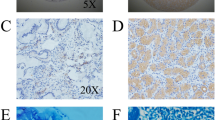
To investigate a correlation between H. pylori and gastric cancer, we first observed H. pylori colonization in 106 gastric cancer tissues. The results showed that H. pylori colonization is present in 36.8 % (39/106) of gastric carcinoma samples. Immunohistochemical analysis of gastric carcinomas and matched normal mucosa showed that the mean staining intensity of COX-2 in H. pylori-positive gastric carcinoma was 2.26 ± 0.17, significantly higher than in H. pylori-negative gastric cancer tissues (0.63 ± 0.16, P < 0.01; Fig. 1a, b). Similarly, the mean VEGF staining intensity in H. pylori-positive gastric carcinoma was significantly higher than in H. pylori-negative tissues (2.65 ± 0.11 vs 0.85 ± 0.06, P < 0.01; Fig. 1c, d). H. pylori-positive gastric cancers had a significantly higher staining intensity for beta-catenin in the gastric mucosa compared with H. pylori-negative gastric cancers (1.95 ± 0.09 vs 0.45 ± 0.075, P < 0.01; Fig. 1e, f). Blood vessel counts in H. pylori-positive and H. pylori-negative gastric cancer tissues showed that the microvessel density (MVD) was 42.9 ± 4.9 and 18.1 ± 3.5, respectively (P < 0.01; Fig. 1g, h). The presence of H. pylori infection in gastric cancer was not related to sex or age, but correlated with the depth of tumor invasion, lymph node metastasis, and tumor–node–metastasis stage(P < 0.05 Table 1) and correlated with the COX-2 expression and beta-catenin expression(P < 0.01 Table 1). These results suggest that H. pylori infection promotes COX-2, VEGF, and beta-catenin upregulation and increases MVD in gastric cancer, which might play an important role in gastric cancer development.

Immunohistochemical assessment of COX-2, beta-catenin, and VEGF expression, and MVD in human gastric cancers. a COX-2 expression in H. pylori-negative gastric cancer tissues. b. COX-2 expression in H. pylori-positive gastric cancer tissues. c VEGF expression in H. pylori-negative gastric cancer tissues. d VEGF expression in H. pylori-positive gastric cancer tissues. e Beta-catenin expression in H. pylori-negative gastric cancer tissues. f. Beta-catenin expression in H. pylori-positive gastric cancer tissues. g MVD in H. pylori-negative gastric cancer tissues. h MVD in H. pylori-positive gastric cancer tissues. Red arrows indicate positive staining. Magnification, ×200. NOTE: Specimens were obtained from 106 patients who underwent major surgical resection for gastric cancer with no preoperative chemotherapy and radiotherapy between February 2009 and December 2014 at the Department of Surgery, Shuguang Hospital (affiliated with Shanghai University of Traditional Chinese Medicine, Shanghai, PR China)
Effect of H. pylori on VEGF mRNA and protein levels in SGC7901 and MKN45 cells
To further confirm the VEGF upregulation in H. pylori-infected gastric carcinomas, we investigated the effect of H. pylori infection in gastric cancer cells on VEGF expression in vitro. After 6, 12, and 24 h incubation with H. pylori NCTC11637 strain, there was differential upregulation of VEGF mRNA in SGC7901 and MKN45 cells (Fig. 2a). To assess whether H. pylori upregulation of VEGF also occurred at the protein level, we measured VEGF at 6, 12, 24, 36, and 48 h in H. pylori-treated or untreated (control) MKN45 cells by ELISA (Fig. 2b). These data further suggest that the VEGF is upregulated by H. pylori infecting SGC7901 and MKN45 cells.

Time-dependent VEGF induction by H. pylori in SGC7901 and MKN45 cells. a VEGF mRNA expression increased in H. pylori-treated cells. Confluent SGC7901 and MKN45 cells were incubated with 100 H. pylori bacteria/cell for 0, 6, 12, 24, and 36 h and then analyzed by real-time quantitative PCR to determine VEGF mRNA expression relative to GAPDH mRNA. **P < 0.01, 12 h versus 0 h in SGC7901 and MKN45 cells. b VEGF protein content was increased in H. pylori-treated cells. SGC7901 and MKN45 cells were incubated with H. pylori for 0, 6, 12, 24, 36, and 48 h, and then analyzed by ELISA to determine VEGF protein levels. **P < 0.01, 24 h versus 0 h in SGC7901 cells; △△P < 0.01, 36 h versus 0 h in MKN45 cells
H. pylori upregulates VEGF via COX-2
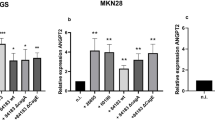
We previously reported that H. pylori upregulates COX-2 expression in vitro. To further verify whether VEGF expression is influenced by incubation with the selective COX-2 inhibitor NS398, real-time fluorogenic quantitative (RFQ) PCR analysis and western blotting were performed to measure VEGF mRNA and protein levels in SGC7901 and MKN45 cells. As shown in Fig. 3a and b, NS398 treatment of did not obviously affect basal VEGF mRNA or protein levels compared with the control group. Interestingly, higher VEGF levels were observed in SGC7901 and MKN45 cells infected with H. pylori; however, this was downregulated by COX-2 inhibition.

COX-2 inhibition attenuates H. pylori-induced VEGF upregulation in SGC7901 and MKN45 cells. a The COX-2 inhibitor NS398 attenuated H. pylori-dependent VEGF mRNA induction. Confluent SGC7901 and MKN45 cells were pretreated with 50 μM NS398 for 2 h prior to culture with or without H. pylori for 12 h. VEGF mRNA expression relative to GAPDH mRNA was determined by quantitative RT-PCR. **P < 0.01 for H. pylori-infected vs control cells. b NS398 attenuated H. pylori induction of VEGF protein. SGC7901 and MKN45 cells were pretreated with 50 μM NS-398 for 2 h prior to culture with or without H. pylori for 24 h and 36 h. The culture supernatant was then analyzed by ELISA to determine VEGF protein content. **P < 0.01 for H. pylori-infected vs control SGC7901 and MKN45 cells. c RNAi-mediated COX2 inhibition blocked VEGF upregulation by H. pylori. Confluent SGC7901 and MKN45 cells were infected with pFU-GW-COX-2-shRNA for 72 h with or without H. pylori treatment for 48 h. VEGF protein content in culture supernatant was then measured by ELISA. **P < 0.01 for H. pylori-infected vs control cells
In our previous studies, the lenti-virus based RNAi of COX-2(pFU-GW-COX-2-shRNA) was constructed to suppress the endogenous COX-2 specially [25]. To further examine the effect of COX-2 on VEGF expression, we infected human gastric cancer SGC7901 and MKN45 cells with the lpFU-GW-COX-2-shRNA to suppress COX-2 expression. RFQ-PCR and western blotting showed that COX-2 gene silencing significantly decreased COX-2 mRNA and protein levels in SGC7901 and MKN45 cells. COX-2 gene silencing or COX-2 pathway inhibition also reduced VEGF levels (Fig. 3c). Considered together, our findings suggest that H. pylori upregulates VEGF in SGC7901 and MKN45 cells via increasing COX-2 expression.
Activation of the Wnt/beta-catenin pathway by H. pylori infection
To identify whether the Wnt/beta-catenin pathway is involved in VEGF upregulation after H. pylori infection, we initially observed the effects of H. pylori infection on beta-catenin levels in SGC7901 and MKN45 cells. Western blot analysis with phospho-specific antibodies showed a time-dependent decrease in beta-catenin phosphorylation in the cytoplasm. However, H. pylori induced cytoplasmic and nuclear beta-catenin accumulation, showing that H. pylori infection could cause nuclear translocation of beta-catenin (Fig. 4). This phenomenon demonstrated that Wnt/beta-catenin might contribute to the H. pylori-induced VEGF transcription.

H. pylori affects phospho-beta-catenin and unphosphorylated beta-catenin protein levels in SGC7901 and MKN45 cells. Confluent SGC7901 and MKN45 gastric cancer cells were incubated with H. pylori at 100 bacteria/cell for 2, 6, 12, 24, and 48 h, and then p-beta-catenin and unphosphorylated beta-catenin protein levels were determined in total cellular protein extracts by western blotting. Blots were stripped and reprobed with beta-actin or PCNA to show equal protein loading. The experiment was performed for three times with similar results. a H. pylori attenuates cytoplasmic p-beta-catenin protein levels in SGC7901 and MKN45 cells. **P < 0.01 for 6 h versus 0 h in SGC7901 cells; △△P < 0.01 for 12 h versus 0 h in MKN45 cells. b H. pylori induces cytoplasmic beta-catenin accumulation in MKN45 cells. **P < 0.01 for 12 h versus 0 h in SGC7901 and MKN45 cells. c H. pylori induces nuclear beta-catenin accumulation in SGC7901 and MKN45 cells. **P < 0.01 for 12 h versus 0 h in SGC7901 and MKN45 cells
Blocking Wnt/beta-catenin attenuates H. pylori-induced VEGF upregulation in SGC7901 and MKN45 cells
Because H. pylori clearly induced the translocation of beta-catenin, additional studies were carried out with inhibitors of beta-catenin to determine the significance of Wnt/beta-catenin signaling pathways in H. pylori-induced VEGF upregulation. We found that H. pylori-induced VEGF mRNA and protein upregulation was partially blocked by FH535 (a specific inhibitor of beta-catenin activity, 20 μM; Fig. 5). Thus, the Wnt/beta-catenin signaling pathway might be primarily responsible for H. pylori-induced VEGF upregulation in SGC7901 and MKN45 cells.

Beta-catenin inhibition attenuates H. pylori-induced VEGF upregulation in SGC7901 and MKN45 cells. Confluent SGC7901 and MKN45 cells were pretreated with 20 μM FH535 for 2 h prior to culture with or without H. pylori for 12 h. Relative cellular VEGF protein levels were then measured by ELISA. **P < 0.01 for H. pylori-infected versus control cells
COX-2 inhibition attenuates H. pylori-induced effects on beta-catenin expression in SGC7901 and MKN45 cells
To determine whether COX-2 is responsible for Wnt/beta-catenin activation, SGC7901 and MKN45 cells were transfected with pFU-GW-COX-2-shRNA or the COX-2 inhibitor NS398 (50 μM, 2 h). After COX-2 gene silencing or COX-2 pathway inhibition, cytoplasmic and nuclear beta-catenin protein levels were significantly inhibited, showing that COX-2 is partly responsible for Wnt/beta-catenin activation (Fig. 6).

COX-2 inhibition attenuates H. pylori-induced beta-catenin upregulation in SGC7901 and MKN45 cells. a, b. COX-2 inhibitor NS398 attenuated H. pylori effects on cytoplasmic and nuclear beta-catenin protein expression. Confluent SGC7901 and MKN45 cells were pretreated with 50 μM NS398 for 2 h prior to culture with or without H. pylori for 12 h. Cytoplasmic and nuclear beta-catenin protein levels were then determined by western blotting. **P < 0.01 for H. pylori-infected versus control cells. c, d COX2 RNAi blocked the H. pylori induced upregulation of cytoplasmic and nuclear beta-catenin. Confluent SGC7901 and MKN45 cells were infected with pFU-GW-COX-2-shRNA for 72 h with or without H. pylori incubation for 12 h. Cytoplasmic and nuclear beta-catenin protein expression levels were then measured by western blotting. **P < 0.01 for MKN45+ H. pylori versus MKN45 control and COX2 siRNA+MKN45+ H. pylori cells
Discussion
Malignant tumor growth and metastasis are complex processes related not only to the characteristics of the tumor itself but also to those of the tumor growth environment. Numerous studies have shown that increased MVD is an important feature of the tumor growth environment, which is a key factor in promoting tumor growth. H. pylori-induced angiogenesis in the gastric mucosa is important for the occurrence and development of gastric cancer. We first observed H. pylori colonization, COX-2, VEGF, beta-catenin levels and MVD in gastric cancer tissues. We found that approximately one third of gastric cancer tissues have H. pylori colonization, which is not a very high proportion. This suggests that many factors lead to gastric cancer development and H. pylori is only one of these. This may also explain why the gastric mucosa can no longer support H. pylori survival after gastric carcinogenesis. We also found that COX-2, VEGF, and beta-catenin expression and MVD were significantly higher in H. pylori-positive gastric cancer tissues than in H. pylori-negative gastric cancer tissues, indicating that H. pylori plays an important role in blood vessel formation in gastric cancer.
Some investigators have suggested an association between H. pylori infection and angiogenesis. Sasaki et al. previously reported tumor vascularity was greater in H. pylori-infected gastric cancer patients than in gastric cancer patients after H. pylori eradication [26]. H. pylori heat shock protein 60 (HSP60) enhances angiogenesis via CXCR2/PLCbeta2/Ca2+ signal transduction in HUVECs [27], but until now, the mechanism of H. pylori induced angiogenesis has remained poorly understood.
Tumor angiogenesis results from an imbalance between positive and negative angiogenic factors released by tumor and host cells into the neoplastic tissue microenvironment [28]. Gastric cancer cells produce many angiogenic factors, including VEGF, interleukin-8 and platelet-derived endothelial growth factor. Furthermore, H. pylori infection increases the expression of these angiogenic factors. Of these, VEGF is the key regulator of tumor-associated angiogenesis. The mechanism of H. pylori-induced VEGF upregulation is complicated and unclear. Our previous study found that the p38 MAPK-COX-2-EP2/EP4 axis regulates H. pylori-induced VEGF upregulation in gastric cells, providing a theoretical basis for investigating the pathogenesis of H. pylori-induced gastric cancer [25]. H. pylori also stimulates host VEGF gene expression via MEK/ERK-dependent activation of Sp1 and Sp3 [29]. Our findings suggest that infection with H. pylori standard strain NCTC11637 leads to a remarkable increase in VEGF expression in SGC7901 and MKN45 gastric epithelial cells via increasing the expression of COX-2, an important factor in gastric cancer development.
Many studies have shown that multiple intracellular pathways are activated by H. pylori [30–33], including the Wnt/beta-catenin pathway [34]. In the absence of Wnt signaling, beta-catenin is present in the cellular beta-catenin destruction complex. This multiprotein complex contains axin and adenomatous APC scaffolds that bind beta-catenin to facilitate its phosphorylation by casein kinase 1 (CKI) at Ser-45 and by GSK3beta at Ser-33, Ser-37, and Thr-41 [35]; it is targeted for degradation by the proteasome. Wnt pathway activation leads to depolymerization of the destruction complex, resulting in cytoplasmic beta-catenin accumulation and further transcription in the nucleus [36, 37]. The Wnt/beta-catenin pathway is involved in the development of a variety of malignant tumors, including gastric cancer [35, 38]. The H pylori cag secretion system activates beta-catenin, p120, and PPARδ, which promote gastric epithelial cell proliferation and might therefore contribute to gastric adenocarcinoma development in humans [39]. Studies have also shown that the Wnt/beta-catenin signaling plays an important role in gastric cancer angiogenesis. Beta-catenin is an important part of the angiogenesis pathway [40, 41]. However, the role of Wnt/beta-catenin in gastric cancer angiogenesis and the mechanism responsible for this remain to be elucidated.
Our results have demonstrated that H. pylori infection upregulates COX-2, which reduces cytoplasmic beta-catenin protein phosphorylation and induces cytoplasmic and nuclear beta-catenin accumulation, thus showing that H. pylori infection can induce the nuclear translocation of beta-catenin. We also confirmed that H pylori stimulates gastric epithelial cells to secrete VEGF, a proangiogenic factor, via the COX-2/Wnt/beta-catenin pathway, which may be an important drug target for preventing and treating H. pylori infection. Future studies should aim to determine which bacterial components induce angiogenesis in H. pylori-infected cells. Moreover, COX-2 and Wnt inhibitors have been used to treat tumors in clinical trials, and further studies may reveal whether these inhibitors can be used to treat gastric cancer [42].
Conclusions
This study indicates H. pylori infection correlated with the depth of tumor invasion, degree of lymph node metastasis, and TNM stage. Moreover, H. pylori infection promoted COX-2, VEGF, and beta-catenin expression and increased MVD in gastric cancer. Further studies revealed that H. pylori infection induces VEGF upregulation via the COX-2/Wnt/beta-catenin pathway.
Ethics approval and consent to participate
Human tissue is involved in the study, and the study was approved by the IRB of Shuguang Hospital, Shanghai University of TCM.
Consent for publication
Not applicable.
Availability of data and materials
We state that data will not be shared. The National Natural Science Foundation of China (81273958) has not yet been completed, so the date is not open.
Abbreviations
- ANOVA:
-
analysis of variance
- APC:
-
adenomatous polyposis coli
- ATF-2:
-
activating transcription factor-2
- ATF-2:
-
adenomatous polyposis coli
- BCA:
-
bicinchoninic acid
- CA:
-
State of California
- COX-2:
-
cyclooxygenase 2
- DMEM:
-
Dulbecco’s modified eagle medium
- ELISA:
-
enzyme linked immunosorbent assay
- FAM:
-
6-carboxy-fluo-rescein-phosphoramidite
- GSK3beta:
-
glycogen synthase kinase 3 beta
- H. pylori :
-
Helicobacter pylori
- HSP60:
-
heat shock protein 60
- IARC:
-
International Agency for Research on Cancer
- LEF1/TCF:
-
lymphoid-enhancing factor/T-cell factors
- MAPK:
-
mitogen-activated protein kinases
- MVD:
-
microvessel density
- NIH:
-
National Institutes of Health
- OD:
-
optical density
- PBS:
-
phosphate-buffered saline
- PE Biosystems:
-
PerkinElmer Biosystems
- PVDF:
-
polyvinylidene fluoride
- RFQ-PCR:
-
real-time fluorogenic quantitative polymerase chain reaction
- RNA:
-
ribonucleic acid
- RPMI:
-
Roswell Park Memorial Institute
- RT-PCR:
-
reverse transcription polymerase chain reaction
- SE:
-
standard error
- TAMPA:
-
carboxy-tetramethyl-rhodamine
- TBM:
-
3,3′,5,5′-Tetramethylbenzidine
- TBS:
-
tris-buffered saline
- USA:
-
United States of America
- VEGF:
-
vascular endothelial growth factor
- WHO:
-
World Health Organization
References
Crowe SE. Helicobacter infection, chronic inflammation, and the development of malignancy. Curr Opin Gastroenterol. 2005;21(1):32–8.
Pandey R, Misra V, Misra SP, Dwivedi M, Kumar A, Tiwari BK. Helicobacter pylori and gastric cancer. Asian Pac J Cancer Prev. 2010;11(3):583–8.
Lee A, O’Rourke J, De Ungria MC, Robertson B, Daskalopoulos G, Dixon MF. A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterology. 1997;112(4):1386–97.
He LF, Wang TT, Gao QY, Zhao GF, Huang YH, Yu LK, et al. Stanniocalcin-1 promotes tumor angiogenesis through up-regulation of VEGF in gastric cancer cells. J Biomed Sci. 2011;18:39.
Dai J, Peng L, Fan K, Wang H, Wei R, Ji G, et al. Osteopontin induces angiogenesis through activation of PI3K/AKT and ERK1/2 in endothelial cells. Oncogene. 2009;28(38):3412–22.
Kitadai Y. Cancer-stromal cell interaction and tumor angiogenesis in gastric cancer. Cancer Microenviron. 2010;3(1):109–16.
Wu CY, Wang CJ, Tseng CC, Chen HP, Wu MS, Lin JT, et al. Helicobacter pylori promote gastric cancer cells invasion through a NF-kappaB and COX-2-mediated pathway. World J Gastroenterol. 2005;11(21):3197–203.
Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. 2014;515(7525):130–3.
Park do J, Thomas NJ, Yoon C, Yoon SS. Vascular endothelial growth factor a inhibition in gastric cancer. Gastric Cancer. 2015;18(1):33–42.
Sawada T, Yashiro M, Sentani K, Oue N, Yasui W, Miyazaki K, et al. New molecular staging with G-factor supplements TNM classification in gastric cancer: a multicenter collaborative research by the Japan Society for Gastroenterological Carcinogenesis G-Project committee. Gastric Cancer. 2015;18(1):119–28.
Wang X, Chen X, Fang J, Yang C. Overexpression of both VEGF-A and VEGF-C in gastric cancer correlates with prognosis, and silencing of both is effective to inhibit cancer growth. Int J Clin Exp Pathol. 2013;6(4):586–97.
de Vries EF. Imaging of cyclooxygenase-2 (COX-2) expression: potential use in diagnosis and drug evaluation. Curr Pharm Des. 2006;12(30):3847–56. PubMed:17073683.
Arber N. Cyclooxygenase-2 inhibitors in colorectal cancer prevention: point. Cancer Epidemiol Biomarkers Prev. 2008;17(8):1852–7.
Prescott SM, Fitzpatrick FA. Cyclooxygenase-2 and carcinogenesis. Biochim Biophys Acta. 2000;1470(2):M69–78.
Funk CD, Funk LB, Kennedy ME, Pong AS, Fitzgerald GA. Human platelet/erythroleukemia cell prostaglandin G/H synthase: cDNA cloning, expression, and gene chromosomal assignment. FASEB J. 1991;5(9):2304–12.
Gately S. The contributions of cyclooxygenase-2 to tumor angiogenesis. Cancer Metastasis Rev. 2000;19(1-2):19–27.
Li Q, Liu N, Shen B, Zhou L, Wang Y, Wang Y, et al. Helicobacter pylori enhances cyclooxygenase 2 expression via p38MAPK/ATF-2 signaling pathway in MKN45 cells. Cancer Lett. 2009;278(1):97–103.
Caputo R, Tuccillo C, Manzo BA, Zarrilli R, Tortora G, Blanco Cdel V, et al. Helicobacter pylori VacA toxin up-regulates vascular endothelial growth factor expression in MKN 28 gastric cells through an epidermal growth factor receptor-, cyclooxygenase-2-dependent mechanism. Clin Cancer Res. 2003;9(6):2015–21.
Yamac D, Ayyildiz T, Coşkun U, Akyürek N, Dursun A, Seckin S, et al. Cyclooxygenase-2 expression and its association with angiogenesis, Helicobacter pylori, and clinicopathologic characteristics of gastric carcinoma. Pathol Res Pract. 2008;204(8):527–36.
Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8(5):387–98.
Hseu YC, Tsou HT, Kumar KJ, Lin KY, Chang HW, Yang HL. The antitumor activity of Antrodia camphorata in melanoma cells: modulation of Wnt/beta-Catenin signaling pathways. Evid Based Complement Alternat Med. 2012;2012:197309.
Kawasaki T, Nosho K, Ohnishi M, Suemoto Y, Kirkner GJ, Dehari R, et al. Correlation of beta-catenin localization with cyclooxygenase-2 expression and CpG island methylator phenotype (CIMP) in colorectal cancer. Neoplasia. 2007;9(7):569–77.
Matono H, Tamiya S, Yokoyama R, Saito T, Iwamoto Y, Tsuneyoshi M, Oda Y. Abnormalities of the Wnt/beta-catenin signalling pathway induce tumour progression in sporadic desmoid tumours: correlation between beta-catenin widespread nuclear expression and VEGF overexpression. Histopathology. 2011;59(3):368–75.
Weidner N. Current pathologic methods for measuring intratumoral microvessel density within breast carcinoma and other solid tumors. Breast Cancer Res Treat. 1995;36(2):169–80.
Liu N, Wu Q, Wang Y, Sui H, Liu X, Zhou N, et al. Helicobacter pylori promotes VEGF expression via the p38 MAPK-mediated COX-2-PGE2 pathway in MKN45 cells. Mol Med Rep. 2014;4:2123–9.
Sasaki A, Kitadai Y, Ito M, Sumii M, Tanaka S, Yoshihara M, et al. Helicobacter pylori infection influences tumor growth of human gastric carcinomas. Scand J Gastroenterol. 2003;38(2):153–8.
Lin CS, He PJ, Hsu WT, Wu MS, Wu CJ, Shen HW, et al. Helicobacter pylori-derived Heat shock protein 60 enhances angiogenesis via a CXCR2-mediated signaling pathway. Biochem Biophys Res Commun. 2010;397(2):283–9.
Kitadai Y. Angiogenesis and lymphangiogenesis of gastric cancer. J Oncol. 2010;2010:468725.
Strowski MZ, Cramer T, Schäfer G, Jüttner S, Walduck A, Schipani E, et al. Helicobacter pylori stimulates host vascular endothelial growth factor-A (vegf-A) gene expression via MEK/ERK-dependent activation of Sp1 and Sp3. FASEB J. 2004;18(1):218–20.
Allen LA, Allgood JA, Han X, Wittine LM. Phosphoinositide3-kinase regulates actin polymerization during delayed phagocytosis of Helicobacter pylori. J Leukoc Biol. 2005;78(1):220–30.
Krueger S, Hundertmark T, Kalinski T, Peitz U, Wex T, Malfertheiner P, et al. Helicobacter pylori encoding the pathogenicity island activates matrix metalloproteinase 1 in gastric epithelial cells via JNK and ERK. J Biol Chem. 2006;281(5):2868–75.
Kaebisch R, Mejías-Luque R, Prinz C, Gerhard M. Helicobacter pylori cytotoxin-associated gene A impairs human dendritic cell maturation and function through IL-10-mediated activation of STAT3. J Immunol. 2014;192(1):316–23.
Buommino E, Donnarumma G, Manente L, De Filippis A, Silvestri F, Iaquinto S, et al. The Helicobacter pylori protein HspB interferes with Nrf2/Keap1 pathway altering the antioxidant response of Ags cells. Helicobacter. 2012;17(6):417–25.
Neal JT, Peterson TS, Kent ML, Guillemin K. H. pylori virulence factor CagA increases intestinal cell proliferation by Wnt pathway activation in a transgenic zebrafish model. Dis Model Mech. 2013;6(3):802–10.
Sokolova O, Bozko PM, Naumann M. Helicobacter pylori suppresses glycogen synthase kinase 3beta to promote beta-catenin activity. J Biol Chem. 2008;283(43):29367–74.
Lim K, Han C, Xu L, Isse K, Demetris AJ, Wu T. Cyclooxygenase-2-derived prostaglandin E2 activates beta-catenin in human cholangiocarcinoma cells: evidence for inhibition of these signaling pathways by omega 3 polyunsaturated fatty acids. Cancer Res. 2008;68(2):553–60.
Franco AT, Israel DA, Washington MK, Krishna U, Fox JG, Rogers AB, et al. Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci U S A. 2005;102(30):10646–51.
Gnad T, Feoktistova M, Leverkus M, Lendeckel U, Naumann M. Helicobacter pylori-induced activation of beta-catenin involves low density lipoprotein receptor-related protein 6 and Dishevelled. Mol Cancer. 2010;9:31.
Nagy TA, Wroblewski LE, Wang D, Piazuelo MB, Delgado A, Romero-Gallo J, et al. beta-Catenin and p120 mediate PPARδ-dependent proliferation induced by Helicobacter pylori in human and rodent epithelia. Gastroenterology. 2011;141(2):553–64.
Wang Z, Humphries B, Xiao H, Jiang Y, Yang C. Epithelial to mesenchymal transition in arsenic-transformed cells promotes angiogenesis through activating beta-catenin-vascular endothelial growth factor pathway. Toxicol Appl Pharmacol. 2013;271(1):20–9.
Fei HR, Cui LY, Zhang ZR, Zhao Y, Wang FZ. Caudatin inhibits carcinomic human alveolar basal epithelial cell growth and angiogenesis through modulating GSK3beta/beta-catenin pathway. J Cell Biochem. 2012;113(11):3403–10.
Ng K, Meyerhardt JA, Chan AT, Sato K, Chan JA, Niedzwiecki D, Saltz LB, Mayer RJ, Benson AB 3rd, Schaefer PL, Whittom R, Hantel A, Goldberg RM, Venook AP, Ogino S, Giovannucci EL, Fuchs CS. Aspirin and COX-2 inhibitor use in patients with stage III colon cancer. J Natl Cancer Inst. 2014;107(1):345.
Acknowledgments
Ashley Craig is thanked for English editing.
Funding
This study was supported by grants from the National Natural Science Foundation of China (81273958, 81072955 & 81202663); the Natural Science Foundation of Shanghai, China (12ZR1449300), the Shanghai Municipal Education Commission (12ZZ118, 09YZ132), and the Shanghai Health and Family Planning Commission (XBR2011061, 2010019, 20134309, 13YZ045).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
QL and NL designed the manuscript. QL designed the in vivo experiments. NZ and NL performed the in vivo experiments with the help of XL and HJ. QW and NC performed the in vitro experiments. NL wrote the paper. All authors reviewed and approved the manuscript.
Authors’ information
All authors are oncologists in China. QL is is professor in the Department of Medical Oncology, Shuguang Hospital, Shanghai University of Traditional Chinese Medicine.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liu, N., Zhou, N., Chai, N. et al. Helicobacter pylori promotes angiogenesis depending on Wnt/beta-catenin-mediated vascular endothelial growth factor via the cyclooxygenase-2 pathway in gastric cancer. BMC Cancer 16, 321 (2016). https://doi.org/10.1186/s12885-016-2351-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-016-2351-9