
Abstract
Lung cancer is a heterogeneous and complex disease. Genomic and transcriptomic profiling of lung cancer not only further our knowledge about cancer initiation and progression, but could also provide guidance on treatment decisions. The fact that targeted treatment is most successful in a subset of tumors indicates the need for better classification of clinically related molecular tumor phenotypes based on better understanding of the mutations in relevant genes, especially in those oncogenic driver mutations. EGFR gene mutations, KRAS gene mutations, EML4-ALK rearrangements and altered MET signaling are widely recognized alterations that play important roles in both the biological mechanisms and the clinical sensitivity to treatment in lung cancer. In this article, we reviewed the discovery of the clinical values of these oncogenic driver mutations and the clinical studies revealing the prognostic and predictive values of these biomarkers for clinical sensitivity and resistance to anti-EGFR therapy or other targeted therapies. These form the basis of personalized treatment in lung cancer based on biomarker profiles of individual tumor, leading to therapeutic advancement and betterment.
Similar content being viewed by others


Review
Lung cancer is the leading cause of cancer-related death worldwide. In the past, therapeutic decisions have been based on histological classifications, which distinguish small cell lung cancers (SCLC) and non-small cell lung cancer (NSCLC). The latter comprises three major subtypes: squamous cell carcinoma, large cell carcinoma, and adenocarcinoma [1]. Although the histological features have been proven to play an important role in the selection of chemotherapy [1], the overall survival remains very poor as a result of presentation of disease at advanced stage.
A diversity of genomic and epigenetic abnormalities has been reported in NSCLC. Oncogenic driver mutations refer to mutations that are responsible for both the initiation and maintenance of the cancer. These mutations are often found in genes that encode for signaling proteins that are critical for maintaining normal cellular proliferation and survival. The presence of mutations on these genes will confer growth advantage on cancer cells, favoring their being selected during tumor progression [2]. NSCLC, especially lung adenocarcinomas, can be further sub-classified by their genetic mutation profiles, making personalized treatment strategies based on the identification of oncogenic driver mutations feasible.
Epidermal Growth Factor Receptor (EGFR) gene mutations were the first targets for targeted treatment in NSCLC. Clinical efficacy and outcomes of EGFR-tyrosine kinase inhibitors (EGFR-TKI) have been reviewed thoroughly [3–7]. Deletions in exon 19 and the missense mutation L858R or L861Q in exon 21 exhibit an association with favorable response to reversible EGFR-TKIs whereas the secondary mutation T790M in exon 20 and insertions in exon 20 will confer resistance to gefitinib and erlotinib, while the nature and clinical sensitivity of other less common mutations like those mutations in exon 18 in the tyrosine kinase (TK) domain of EGFR are not well defined [8].
Apart from EGFR targeted therapy, more molecular targeted agents have developed to improve therapeutic outcomes, for example, bevacizumab which is an angiogenesis inhibitor targeting vascular endothelial growth factor (VEGF) [9], pemetrexed inhibiting thymidylate synthase and other folate dependent enzymes [10], as well as a mammalian target of rapamycin (mTOR) inhibitor everolimas [11]. Meanwhile, other oncogenic mutations that can define clinically relevant molecular subsets of NSCLC have been identified. The presence of individual driver gene is usually found to be mutually exclusive to each other. In this article, we review the four commonly known oncogenic driver mutations in lung cancer – EGFR mutations at exons 18 – 21, KRAS gene mutation at codons 12 and 13, EML4-ALK fusion genes and deregulation of MET signaling.
EGFR mutations
The epidermal growth factor receptor family of tyrosine kinases consists of four forms: EGFR (ErbB1, HER1), ErbB2 (HER2), ErbB3 (HER3) and ErbB4 (HER4) [6]. The binding of specific ligands leads to homo- and hetero-dimerization, with subsequent autophosphorylation of the intracellular receptor TK domain. The activated TK activity subsequently recruits appropriate downstream components of the TK signaling pathway which are involved in multiple cellular processes, including cell proliferation, cell survival, cell motility and cell invasion [12].
EGFR mutations have been extensively studies in NSCLC with about 27% overall incidence (according to COSMIC database, http://www.sanger.ac.uk). EGFR-mutant NSCLC, which often exhibits adenocarcinoma histology, has been found to be associated with a better prognosis compared to EGFR wild-typed NSCLC in most populations [13–18], except for a study in Chinese patients [19]. The activating EGFR mutations have been identified in exons 18 to 21 of the TK domain, about 90% of which are deletions in exon 19 (Figure 1) and the point mutation L858R in exon 21 (Figure 2) [6]. The mutant EGFR shows a preferential binding of gefitinib or erlotinib to ATP, thus correlating with higher sensitivity to these two anti-EGFR TKIs [20, 21]. Several trials have revealed the clinical role of activating EGFR mutations in EGFR-TKI therapy. A randomized prospective Phase III study (NEJ002) with 230 Japanese advanced, untreated and EGFR-mutant NSCLC patients sustained improved progression-free survival in the first-line gefitinib versus standard chemotherapy [22]. According to a study in Spain, erlotinib also showed similar effectiveness in EGFR-mutant patients [23].

Analysis of EGFR exon 19 mutation by direct sequencing. (A) Wild-type EGFR exon 19; (B) An example of inframe deletion in EGFR exon 19 (2481_2495del). The arrows indicate the span of deletion in each amplified sequence of exon 19.

EGFR exon 21 mutation analysis via direct sequencing. (A) Wild-type EGFR exon 21; (B) One missense mutation (L858R) in EGFR exon 21. The arrow indicates the position of the mutant nucleotide. Only forward sequences are shown.
Some EGFR mutations, although they occur in exons 18 to 21, are related to primary resistance to EGFR-TKIs, for example, small insertions or duplications in exon 20. Additionally, the presence of other types of mutations can induce acquired resistance to EGFR inhibitors, such as T790M (exon 20), L747S (exon 19), D761Y (exon 19) and T854A (exon 21) [24]. Other genetic alterations contribute to primary or acquired resistance to EGFR-TKIs, including KARS mutations, PIK3CA mutations, loss of PTEN function, MET amplification and altered EGFR-related signaling [25]. Therefore, strategies to overcome resistance to EGFR-TKIs have been under investigation, including development of second- and third-generation of EGFR-TKIs, combinational therapy targeting compensatory pathways, alternative TKI dosing to delay drug resistance, or continuation therapy with EGFR-TKIs [13, 25].
Apart from erlotinib and gefitinib, second-generation irreversible EGFR-TKIs have been under extensive investigation, for example, afatinib and dacomitinib. Afatinib (BIBW 2992; Boehringer Ingelheim; Ingelheim, Germany) is a highly selective and irreversible inhibitor of both EGFR and HER2 [26]. It has been found to be effective in NSCLC with EGFR mutations, especially with de novo T790M mutations [27]. Several phase III trials of afatinib are undergoing, including LUX-Lung 3), in which afatinib is compared to cisplatin/pemetrexed in the first-line treatment of advanced NSCLC patients with activating EGFR mutations, as well as LUX-Lung 6 comparing afatinib with cisplatin/gemcitabine in the same population as LUX-Lung 3 with more recruitment in China, India and South Korea. Dacomitinib (PF-00299804, Pfizer; New London, CT, USA) is an irreversible pan-HER inhibitor, which showed remarkable activity to gefitinib-resistant EGFR T790M mutations or HER2 mutations [28]. A phase I/II study of this inhibitor in Asian populations who were refractory to chemotherapy and erlotinib or gefitinib, 15% response rate and 32% 6-month overall survival achieved [29]. Therefore, a randomized phase III trial (JBR-26) comparing dacomitinib to placebo in the third-line treatment in patients have failed chemotherapy and EGFR TKIs is ongoing.
KRAS mutations
A milestone study in 1984 identified a KRAS mutation in a squamous cell lung carcinoma with the absence of this point mutation in the corresponding normal bronchial or parenchymal tissue [30]. Since then KRAS mutations have been found frequently in NSCLC, and according to clinical trials, the incidence of KRAS mutations in NSCLC ranges from 8% to 24% [31]. Most activating KRAS mutations in NSCLC are located in codons 12 or 13, and are also reported in lung adenocarcinomas [32]. The study in which KRAS mutations were analyzed in nearly 500 lung adenocarcinomas showed that KRAS mutations were found in 15%, 22% and 25% of tumors from never smokers, former smokers and current smokers respectively [33]. Whereas EGFR mutations tend to occur more frequently in never-smokers with lung cancer, the presence of KRAS mutations cannot be easily predicted based on smoking status alone [34]. Another novel finding of this study was that KRAS transition mutations were more common in never smokers, whereas former/current smokers were more likely to harbor KRAS transversion mutations. This implies that transversions may be smoking related.
The mutated KRAS proteins exhibit impaired GTPase activity, resulting in constitutive activation of RAS signaling. Since KRAS is downstream of EGFR, constitutive activation of KRAS renders resistance to anti-EGFR therapy. Many studies have observed lower efficacy of EGFR-TKI therapy in KRAS mutated NSCLC patients [35, 36]. The presence of EGFR mutations and KRAS mutations are mutually exclusive in the same tumor. However, there was a report on a case series of three patients with both activating EGFR mutations and KRAS mutations demonstrated that all EGFR+/KRAS+ patients showed a positive response to gefitinib or erlotinib [37]. Several large clinical observational studies failed to identify KRAS mutation to be a significant response predictor to EGFR-TKIs [38–40]. As a result, KRAS positivity cannot be established as a criterion to exclude NSCLC patients from EGFR-TKI therapy.
The development of therapeutic agents targeting mutated KRAS signaling is under intensive investigation. An obstacle is that mutant KRAS proteins entail loss-of-function. It is more difficult to inhibit loss-of-function proteins than gain-of-function ones like mutant EGFR. Current approaches try either to inhibit protein synthesis of mutated KRAS or to impede downstream effectors of mutant KRAS.
EML4-ALK rearrangement
The anaplastic lymphoma kinase (ALK) protein is a receptor tyrosine kinase in the insulin receptor superfamily. In 2007, Soda et al. identified a small inversion within the short arm of chromosome 2, resulting in the fusion of the N-terminal of the echinoderm microtubule-associated protein-like 4 (EML4) gene with the ALK gene [41]. Up to now, at least 11 different EML4-ALK variants have been reported, all of which involve the coiled-coil domain of EML4 and the intracellular tyrosine kinase domain of ALK [42]. Furthermore, all EML4-ALK variants exhibit dimerization and constitutive activation of the fusion proteins [42, 43].
There is no “gold standard” method to screen the ALK gene rearrangements. Three detection technologies have been evaluated, including fluorescence in situ hybridization (FISH), reverse transcriptase PCR, and immunohistochemistry (IHC). The frequency of EML4-ALK fusions in unselected NSCLC patients, according to previous studies mostly with East Asians, ranges from 1.6% to 11.6% [42, 44–46]. Although other histological sub-types rarely contain EML4-ALK rearrangements, lung adenocarcinoma has been reported to be the major type showing EML4-ALK translocations. The majority of Caucasian lung adenocarcinoma harboring EML4-ALK show the signet-ring cell histology, whereas the acinar pattern is pre-dominant in ALK-positive Asian adenocarcinomas [47]. ALK-positivity tends to be found in younger NSCLC patients [44]. Similar to EGFR mutations, EML4-ALK fusions are associated with never or light smokers [48]. EML4-ALK translocations and EGFR mutations are mutually exclusive except for rare cases [45].
In Shaw et al. study, wild-type NSCLC patients and ALK-positive NSCLC patients displayed a similar response to chemotherapy and no significant differences in overall survival, however, EML4-ALK translocations have been found to be associated with resistance to EGFR-TKIs [44]. Similarly, EML4-ALK fusion gene was not a significant prognostic factor based on the analysis of 720 resected lung adenocarcinomas [49]. Consistent with these findings, EML4-ALK status did not affect the sensitivity of advanced NSCLC patients to platinum-based combination chemotherapy in terms of response rate and progression-free survival although overall survival of EML4-ALK positive patients tended to be shorter than that of the EGFR mutated cohort nut resembled that in the wild-type cohort [50]. In contrast, according to a study including EGFR wild-type and advanced-stage lung adenocarcinoma patients who received either monotherapy or platinum-doublet chemotherapy, ALK positivity conferred superior overall survival [51]. Compared with aforementioned studies, this study enrolled more EML4-ALK translocated patients (39 out of 116, 34%), which may partially explain better outcome. Additionally, in Chinese patients with advanced NSCLC, response rate to EGFR-TKI was similar between ALK rearranged and EGFR mutated patients although median progression-free survival was significantly shorter in those with EML4-ALK gene [52]. The apparent discrepancy between findings of this study and that of the study by Shaw et al. may be explained by predominantly Asian population in this study and the limited sample sizes in both studies. The clinicopathological features of EML4-ALK rearrangement in NSCLC patients needs further investigation.
In order to treat ALK-positive patients, selective tyrosine kinase inhibitors of ALK are currently under clinical trials, including an ALK/MET inhibitor PF-02341066 (crizotinib) [53]. Unfortunately, acquired resistance will emerge in EML4-ALK-positive tumors. Several secondary mutations in the ALK gene, such as L1196M and C1156Y, have been revealed to contribute to resistance to crizotinib [54, 55]. In order to overcome resistance, new molecules continue to be developed. CH5424802, a selective, potent, and orally available ALK inhibitor, have exhibited remarkable activity against C1156Y- and L1196M-resistant EML4-ALK mutants [56]. Clinical trials have been conducted to confirm safety and efficacy of this agent in ALK- positive NSCLC [46].
MET signaling
The MET gene, which is located on chromosome 7, encodes a receptor tyrosine kinase composed of an extracellular α-chain and a transmembrane β-chain. The natural ligand for MET receptor is hepatocyte growth factor (HGF), also called scatter factor (SF). Since MET interacts with numerous downstream effectors, the activation of MET signaling affects many pathways and regulates various cellular processes, including cell proliferation, cell motility, cell scattering, cell invasion, metastasis, angiogenesis and epithelial to mesenchymal transition (EMT) [57–59]. Therefore, it is not surprising that deregulation of this signaling pathway can be considered as a driving force in tumor initiation and tumor maintenance.
In transformed cells, MET can be altered via overexpression, genomic amplification, mutations, or alternative splicing [57, 60, 61]. These alterations result in aberrant MET activation which can be mediated through HGF-dependent or HGF-independent mechanisms.
MET overexpression has been observed in NSCLC. In a study using 32 lung cancer tissues, all tumor samples expressed MET with no significant MET staining in corresponding normal lung tissues, and 61% (14 of 23) of NSCLC showed strong MET expression examined by IHC. Furthermore, an increase in MET activity identified by higher levels of phosphorylated MET (p-MET) at sites Y1003 and Y1230/1234/1235 came along with MET overexpression in this study. It was also mentionable that the activated p-MET was preferentially expressed in tumor cells located at the invasive front of NSCLC tissues [62]. In another study with 183 lung adenocarcinomas, MET amplification was observed in 8 (4%) patients with wild-typed EGFR and wild-typed KRAS, indicating that the presence of MET gene amplification might be mutually exclusive with EGFR and KRAS mutations. Phosphorylation of MET at sites Y1234/1235 has been found to be associated with poor survival in patients who have complete resection of lung tumors [63].
MET gene copy number variations have also been reported in NSCLC. MET status was analyzed with FISH in 447 NSCLC patients and high MET gene copy number (≥5 copies/cell) was observed in 48 cases (11.1%), and patients with high MET gene copy number (MET-positive) exhibited shorter survival than MET-negative patients [64].
Mutations provide another mechanism for MET dysregulation. Mutated MET allows the kinase to overcome inhibitory mechanisms, thus becoming constructively activated or hyperresponsive to stimuli. Mutations could also prolong the duration of stimulating signals by increasing the level of activation or by preventing the degradation of the kinase. One study examined individual exons of semaphorin, juxtamembrane, and tyrosine kinase domains of MET in 141 East Asians, 76 Caucasians and 66 African Americans [65]. The results showed that MET mutations varied with ethnicity. N375S, occurred within the semaphorin domain, was the most frequent non-synonymous mutation, and the frequency of this mutation was higher in East Asians compared to Caucasians. In both East Asians and Caucasians, the frequency of N375S was higher in squamous cell carcinoma than in other non-small cell lung cancer. Among East Asians, the frequency of N375S in males was much higher compared with females [65]. Since there is a relationship between ethnic differences and MET mutations, greater knowledge of this correlation can help us understand incidence, prognosis and treatment of lung cancer.
Several agents targeting MET signaling are under investigation. Among them, a non-ATP competitive MET inhibitor, tivatinib (ARQ197), has been used in combination with erlotinib (EGFR-TKI) as second-line treatment for previously-treated non-small cell lung cancer [66]. The dual MET-EGFR combinatorial inhibition is well-tolerated in advanced stage lung cancer patients. Although no significant change in progression-free survival (PFS) or overall survival (OS) has been reported in the intent-to-treat population, improvement in PFS and/or OS can be seen when including key prognostic factors and/or biomarkers, for example, presence of KRAS mutations. More MET-targeted agents are under preclinical and clinical studies, making MET the next major biomarkers in lung cancer.
Other oncogenic driver mutations
The identification of oncogenic driver mutations reveals the complexity and heterogeneity of NSCLC. A collaborative study investigated 623 candidate cancer genes in 188 lung adenocarcinomas. 26 genes were discovered to be somatically mutated at high frequencies and thus may be related to tumorigenesis, including ERBB3, ERBB4, VEGFR, multiple ephrin receptors genes and NTRK genes [67].
Significance of oncogenic driver mutations in lung cancer severity and therapy
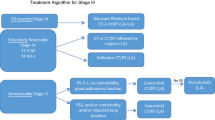
Table 1 lists these four molecular targets with their respective detection methods and inhibitors. The presence of oncogenic driver mutations leads to a phenomenon called ‘oncogene addiction’ wherein tumor cells tend to be dependent on the specific mutant oncogene for their own survival and growth. Blocking the relevant oncogenic pathway by specific inhibitors may induce ‘oncogenic shock’ which ultimately results in cancer cell apoptosis [68]. This hypothesis suggests that the promising future of lung cancer treatment is indeed personalized therapy with drugs targeting specific driver oncogenes that “drive” tumorigenesis. The selection of proper therapeutic approach should be based on both histological features and the tumor mutation profiles of individual patient (Figure 3). This combination can contribute to better prediction of the malignant behavior and to improved clinical management. EGFR mutations could be present in early stage NSCLC [69, 70], suggesting that it may be possible to detect of lung cancer at more early stages via the molecular testing of mentioned driver mutations in susceptible individuals.

A suggested schema for guidance of clinical testing for oncogenic driver mutations which aid in personalized treatment in lung cancer.
Conclusion
As we further our understanding into the biological mechanisms underlying these oncogenic driver mutations, the clinical relevance of these driver mutations will allow for further advancement into targeted therapeutics in lung cancer.
References
Bronte G, Rizzo S, La Paglia L, Adamo V, Siragusa S, Ficorella C, Santini D, Bazan V, Colucci G, Gebbia N: Driver mutations and differential sensitivity to targeted therapies: a new approach to the treatment of lung adenocarcinoma. Canc Treat Rev 2010,36(Suppl 3):S21-S29.
Stratton MR, Campbell PJ, Futreal PA: The cancer genome. Nature 2009,458(7239):719–724. 10.1038/nature07943
Linardou H, Dahabreh IJ, Bafaloukos D, Kosmidis P, Murray S: Somatic EGFR mutations and efficacy of tyrosine kinase inhibitors in NSCLC. Nat Rev Clin Oncol 2009,6(6):352–366. 10.1038/nrclinonc.2009.62
Riely GJ, Politi KA, Miller VA, Pao W: Update on epidermal growth factor receptor mutations in Non-small cell lung cancer. Clin Cancer Res 2006,12(24):7232–7241. 10.1158/1078-0432.CCR-06-0658
Rosell R, Morán T, Carcereny E, Quiroga V, Molina MÁ, Costa C, Benlloch S, Tarón M: Non-small-cell lung cancer harbouring mutations in the EGFR kinase domain. Clin Transl Oncol 2010,12(2):75–80. 10.1007/S12094-010-0473-0
Sharma SV, Bell DW, Settleman J, Haber DA: Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007,7(3):169–181.
Stella GM, Luisetti M, Inghilleri S, Cemmi F, Scabini R, Zorzetto M, Pozzi E: Targeting EGFR in non-small-cell lung cancer: lessons, experiences, strategies. Respir Med 2012,106(2):173–183. 10.1016/j.rmed.2011.10.015
Zhou Q, Zhang XC, Chen ZH, Yin XL, Yang JJ, Xu CR, Yan HH, Chen HJ, Su J, Zhong WZ: Relative abundance of EGFR mutations predicts benefit from gefitinib treatment for advanced non-small-cell lung cancer. J Clin Oncol 2011,29(24):3316–3321. 10.1200/JCO.2010.33.3757
Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, Lilenbaum R, Johnson DH: Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med 2006,355(24):2542–2550. 10.1056/NEJMoa061884
Cullen MH, Zatloukal P, Sorenson S, Novello S, Fischer JR, Anil AJ, Zereu M, Peterson P, Visseren-Grul C, Iscoe N: Pemetrexed for the treatment of advanced non-small cell lung cancer (NSCLC): results from a randomized phase III dose finding trial in patients who progressed following platinum-containing chemotherapy. J Thorac Oncol 2007,2(8):S316-S317.
Ramalingam SS, Harvey RD, Saba N, Owonikoko TK, Kauh J, Shin DM, Sun SY, Strychor S, Tighiouart M, Egorin MJ: Phase 1 and pharmacokinetic study of everolimus, a mammalian target of rapamycin inhibitor, in combination with docetaxel for recurrent/refractory nonsmall cell lung cancer. Cancer 2010,116(16):3903–3909. 10.1002/cncr.25264
Scaltriti M, Baselga J: The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res 2006,12(18):5268–5272. 10.1158/1078-0432.CCR-05-1554
Pao W, Chmielecki J: Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer 2010,10(11):760–774. 10.1038/nrc2947
Marks JL, Broderick S, Zhou Q, Chitale D, Li AR, Zakowski MF, Kris MG, Rusch VW, Azzoli CG, Seshan VE: Prognostic and therapeutic implications of EGFR and KRAS mutations in resected lung adenocarcinoma. J Thorac Oncol 2008,3(2):111–116. 10.1097/JTO.0b013e318160c607
Eberhard DA, Johnson BE, Amler LC, Goddard AD, Heldens SL, Herbst RS, Ince WL, Janne PA, Januario T, Johnson DH: Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol 2005,23(25):5900–5909. 10.1200/JCO.2005.02.857
Kosaka T, Yatabe Y, Onozato R, Kuwano H, Mitsudomi T: Prognostic implication of EGFR, KRAS, and TP53 gene mutations in a large cohort of Japanese patients with surgically treated lung adenocarcinoma. J Thorac Oncol 2009,4(1):22–29. 10.1097/JTO.0b013e3181914111
Liu HP, Isaac Wu HD, Chang JW, Wu YC, Yang HY, Chen YT, Hsieh WY, Chen YR, Huang SF: Prognostic implications of epidermal growth factor receptor and KRAS gene mutations and epidermal growth factor receptor gene copy numbers in patients with surgically resectable non-small cell lung cancer in Taiwan. J Thorac Oncol 2010,5(8):1175–1184. 10.1097/JTO.0b013e3181e2f4d6
Tsao MS, Sakurada A, Ding K, Aviel-Ronen S, Ludkovski O, Liu N, Le Maitre A, Gandara D, Johnson DH, Rigas JR: Prognostic and predictive value of epidermal growth factor receptor tyrosine kinase domain mutation status and gene copy number for adjuvant chemotherapy in non-small cell lung cancer. J Thorac Oncol 2011,6(1):139–147. 10.1097/JTO.0b013e3181fd83a4
Gao J, Chen JQ, Zhang L, Liang ZY: Relationship between EGFR and KRAS mutations and prognosis in Chinese patients with non-small cell lung cancer: a mutation analysis with real-time polymerase chain reaction using scorpion amplification refractory mutation system. Zhonghua Bing Li Xue Za Zhi 2012,41(10):652–656.
Carey KD, Garton AJ, Romero MS, Kahler J, Thomson S, Ross S, Park F, Haley JD, Gibson N, Sliwkowski MX: Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res 2006,66(16):8163–8171. 10.1158/0008-5472.CAN-06-0453
Mulloy R, Ferrand A, Kim Y, Sordella R, Bell DW, Haber DA, Anderson KS, Settleman J: Epidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinib. Cancer Res 2007,67(5):2325–2330. 10.1158/0008-5472.CAN-06-4293
Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I: Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 2010,362(25):2380–2388. 10.1056/NEJMoa0909530
Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, Majem M, Lopez-Vivanco G, Isla D, Provencio M: Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009,361(10):958–967. 10.1056/NEJMoa0904554
Avizienyte E, Ward RA, Garner AP: Comparison of the EGFR resistance mutation profiles generated by EGFR-targeted tyrosine kinase inhibitors and the impact of drug combinations. Biochem J 2008,415(2):197–206. 10.1042/BJ20080728
Lin L, Bivona TG: Mechanisms of resistance to epidermal growth factor receptor inhibitors and novel therapeutic strategies to overcome resistance in NSCLC patients. Chemother Res Pract 2012, 2012: 817297.
Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A, Himmelsbach F: BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008,27(34):4702–4711. 10.1038/onc.2008.109
Vadakara J, Borghaei H: Personalized medicine and treatment approaches in non-small-cell lung carcinoma. Pharmgenomics Pers Med 2012, 5: 113–123.
Engelman JA, Zejnullahu K, Gale CM, Lifshits E, Gonzales AJ, Shimamura T, Zhao F, Vincent PW, Naumov GN, Bradner JE: PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res 2007,67(24):11924–11932. 10.1158/0008-5472.CAN-07-1885
Ou SH: Second-generation irreversible epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs): a better mousetrap? a review of the clinical evidence. Crit Rev Oncol Hematol 2012,83(3):407–421. 10.1016/j.critrevonc.2011.11.010
Santos E, Martin-Zanca D, Reddy E, Pierotti M, Della Porta G, Barbacid M: Malignant activation of a K-RAS oncogene in lung carcinoma but not in a normal tissue of the same patient. Science 1984, 223: 661–664. 10.1126/science.6695174
Langer CJ: Roles of EGFR and KRAS mutations in the treatment of patients with Non-small-cell lung cancer. P T 2011,36(5):263–279.
Riely GJ, Marks J, Pao W: KRAS mutations in Non-small cell lung cancer. Proc Am Thorac Soc 2009,6(2):201–205. 10.1513/pats.200809-107LC
Riely GJ, Kris MG, Rosenbaum D, Marks J, Li A, Chitale DA, Nafa K, Riedel ER, Hsu M, Pao W: Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin Cancer Res 2008,14(18):5731–5734. 10.1158/1078-0432.CCR-08-0646
Thunnissen FB, Prinsen C, Hol B, Van der Drift M, Vesin A, Brambilla C, Montuenga L, Field JK: Smoking history and lung carcinoma: KRAS mutation is an early hit in lung adenocarcinoma development. Lung Cancer 2012,75(2):156–160. 10.1016/j.lungcan.2011.07.013
Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG, Varmus HE: KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2005,2(1):e17. 10.1371/journal.pmed.0020017
Garassino MC, Borgonovo K, Rossi A, Mancuso A, Martelli O, Tinazzi A, Di Cosimo S, La Verde N, Sburlati P, Bianchi C: Biological and clinical features in predicting efficacy of epidermal growth factor receptor tyrosine kinase inhibitors: a systematic review and meta-analysis. Anticancer Res 2009,29(7):2691–2701.
Benesova L, Minarik M, Jancarikova D, Belsanova B, Pesek M: Multiplicity of EGFR and KRAS mutations in non-small cell lung cancer (NSCLC) patients treated with tyrosine kinase inhibitors. Anticancer Res 2010,30(5):1667–1671.
Douillard JY, Shepherd FA, Hirsh V, Mok T, Socinski MA, Gervais R, Liao ML, Bischoff H, Reck M, Sellers MV: Molecular predictors of outcome with gefitinib and docetaxel in previously treated Non-small-cell lung cancer: data from the randomized phase III INTEREST trial. J Clin Oncol 2009,28(5):744–752.
Zhu CQ, da Cunha Santos G, Ding K, Sakurada A, Cutz JC, Liu N, Zhang T, Marrano P, Whitehead M, Squire JA: Role of KRAS and EGFR as biomarkers of response to erlotinib in national cancer institute of canada clinical trials group study BR.21. J Clin Oncol 2008,26(26):4268–4275. 10.1200/JCO.2007.14.8924
Brugger W, Triller N, Blasinska-Morawiec M, Curescu S, Sakalauskas R, Manikhas GM, Mazieres J, Whittom R, Ward C, Mayne K: Prospective molecular marker analyses of EGFR and KRAS from a randomized, placebo-controlled study of erlotinib maintenance therapy in advanced Non-small-cell lung cancer. J Clin Oncol 2011,29(31):4113–4120. 10.1200/JCO.2010.31.8162
Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara SI, Watanabe H, Kurashina K, Hatanaka H: Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007,448(7153):561–566. 10.1038/nature05945
Choi YL, Takeuchi K, Soda M, Inamura K, Togashi Y, Hatano S, Enomoto M, Hamada T, Haruta H, Watanabe H: Identification of novel isoforms of the EML4-ALK transforming gene in Non-small cell lung cancer. Cancer Res 2008,68(13):4971–4976. 10.1158/0008-5472.CAN-07-6158
Horn L, Pao W: EML4-ALK: honing in on a New target in Non-small-cell lung cancer. J Clin Oncol 2009,27(26):4232–4235. 10.1200/JCO.2009.23.6661
Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, Solomon B, Stubbs H, Admane S, McDermott U: Clinical features and outcome of patients with Non-small-cell lung cancer Who harbor EML4-ALK. J Clin Oncol 2009,27(26):4247–4253. 10.1200/JCO.2009.22.6993
Zhang X, Zhang S, Yang X, Yang J, Zhou Q, Yin L, An S, Lin J, Chen S, Xie Z: Fusion of EML4 and ALK is associated with development of lung adenocarcinomas lacking EGFR and KRAS mutations and is correlated with ALK expression. Mol Cancer 2010,9(1):188. 10.1186/1476-4598-9-188
Casaluce F, Sgambato A, Maione P, Rossi A, Ferrara C, Napolitano A, Palazzolo G, Ciardiello F, Gridelli C: ALK inhibitors: a new targeted therapy in the treatment of advanced NSCLC. Target Oncol 2013. Jan [Epub ahead of print]
Crystal AS, Shaw AT: New targets in advanced NSCLC: EML4-ALK. Clin Adv Hematol Oncol 2011,9(3):207–214.
Sasaki T, Rodig SJ, Chirieac LR, Jänne PA: The biology and treatment of EML4-ALK non-small cell lung cancer. Eur J Cancer 2010,46(10):1773–1780. 10.1016/j.ejca.2010.04.002
Fukui T, Yatabe Y, Kobayashi Y, Tomizawa K, Ito S, Hatooka S, Matsuo K, Mitsudomi T: Clinicoradiologic characteristics of patients with lung adenocarcinoma harboring EML4-ALK fusion oncogene. Lung Cancer 2012,77(2):319–325. 10.1016/j.lungcan.2012.03.013
Takeda M, Okamoto I, Sakai K, Kawakami H, Nishio K, Nakagawa K: Clinical outcome for EML4-ALK-positive patients with advanced non-small-cell lung cancer treated with first-line platinum-based chemotherapy. Ann Oncol 2012,23(11):2931–2936. 10.1093/annonc/mds124
Wu SG, Kuo YW, Chang YL, Shih JY, Chen YH, Tsai MF, Yu CJ, Yang CH, Yang PC: EML4-ALK translocation predicts better outcome in lung adenocarcinoma patients with wild-type EGFR. J Thorac Oncol 2012,7(1):98–104. 10.1097/JTO.0b013e3182370e30
Wang Z, Zhang X, Bai H, Zhao J, Zhuo M, An T, Duan J, Yang L, Wu M, Wang S: EML4-ALK rearrangement and its clinical significance in Chinese patients with advanced non-small cell lung cancer. Oncology 2012,83(5):248–256. 10.1159/000341381
Ou SH, Bazhenova L, Camidge DR, Solomon BJ, Herman J, Kain T, Bang YJ, Kwak EL, Shaw AT, Salgia R: Rapid and dramatic radiographic and clinical response to an ALK inhibitor (crizotinib, PF02341066) in an ALK translocation-positive patient with non-small cell lung cancer. J Thorac Oncol 2010,5(12):2044–2046. 10.1097/JTO.0b013e318200f9ff
Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, Yatabe Y, Takeuchi K, Hamada T, Haruta H: EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. New Engl J Med 2010,363(18):1734–1739. 10.1056/NEJMoa1007478
Heuckmann JM, Holzel M, Sos ML, Heynck S, Balke-Want H, Koker M, Peifer M, Weiss J, Lovly CM, Grutter C: ALK mutations conferring differential resistance to structurally diverse ALK inhibitors. Clin Cancer Res 2011,17(23):7394–7401. 10.1158/1078-0432.CCR-11-1648
Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, Fukami TA, Oikawa N, Tsukuda T, Ishii N, Aoki Y: CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011,19(5):679–690. 10.1016/j.ccr.2011.04.004
Feng Y, Thiagarajan PS, Ma PC: MET signaling: novel targeted inhibition and its clinical development in lung cancer. J Thorac Oncol 2012,7(2):459–467. 10.1097/JTO.0b013e3182417e44
Guo A, Villen J, Kornhauser J, Lee KA, Stokes MP, Rikova K, Possemato A, Nardone J, Innocenti G, Wetzel R: Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci 2008,105(2):692–697. 10.1073/pnas.0707270105
Organ SL, Tsao MS: An overview of the c-MET signaling pathway. Ther Adv Med Oncol 2011,3(1 Suppl):S7-S19. 10.1177/1758834011422556
Lawrence RE, Salgia R: MET molecular mechanisms and therapies in lung cancer. Cell Adh Migr 2009,4(1):146–152.
Sattler M, Reddy MM, Hasina R, Gangadhar T, Salgia R: The role of the c-Met pathway in lung cancer and the potential for targeted therapy. Ther Adv Med Oncol 2011,3(4):171–184. 10.1177/1758834011408636
Ma PC: Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in Non-small cell lung cancer. Cancer Res 2005,65(4):1479–1488. 10.1158/0008-5472.CAN-04-2650
Onitsuka T, Uramoto H, Ono K, Takenoyama M, Hanagiri T, Oyama T, Izumi H, Kohno K, Yasumoto K: Comprehensive molecular analyses of lung adenocarcinoma with regard to the epidermal growth factor receptor, K-ras, MET, and hepatocyte growth factor status. J Thorac Oncol 2010,5(5):591–596.
Cappuzzo F, Marchetti A, Skokan M, Rossi E, Gajapathy S, Felicioni L, del Grammastro M, Sciarrotta MG, Buttitta F, Incarbone M: Increased MET gene copy number negatively affects survival of surgically resected Non-small-cell lung cancer patients. J Clin Oncol 2009,27(10):1667–1674. 10.1200/JCO.2008.19.1635
Krishnaswamy S, Kanteti R, Duke-Cohan JS, Loganathan S, Liu W, Ma PC, Sattler M, Singleton PA, Ramnath N, Innocenti F: Ethnic differences and functional analysis of MET mutations in lung cancer. Clin Cancer Res 2009,15(18):5714–5723. 10.1158/1078-0432.CCR-09-0070
Sequist LV, von Pawel J, Garmey EG, Akerley WL, Brugger W, Ferrari D, Chen Y, Costa DB, Gerber DE, Orlov S: Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J Clin Oncol 2011,29(24):3307–3315. 10.1200/JCO.2010.34.0570
Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB: Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008,455(7216):1069–1075. 10.1038/nature07423
Sharma SV, Settleman J: Oncogenic shock: turning an activated kinase against the tumor cell. Cell Cycle 2006,5(24):2878–2880. 10.4161/cc.5.24.3598
Ji H, Li D, Chen L, Shimamura T, Kobayashi S, McNamara K, Mahmood U, Mitchell A, Sun Y, Al-Hashem R: The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell 2006,9(6):485–495. 10.1016/j.ccr.2006.04.022
Tang X, Shigematsu H, Bekele BN, Roth JA, Minna JD, Hong WK, Gazdar AF, Wistuba II: EGFR tyrosine kinase domain mutations are detected in histologically normal respiratory epithelium in lung cancer patients. Cancer Res 2005,65(17):7568–7572.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors’ contributions
Susan Y Luo (SYL) carried out the molecular tests and drafted the manuscript. David CL Lam (DCL) planned, wrote and reviewed the manuscript. All authors read and approved the final manuscript.
Declaration of competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Luo, S.Y., Lam, D.C. Oncogenic driver mutations in lung cancer. Transl Respir Med 1, 6 (2013). https://doi.org/10.1186/2213-0802-1-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2213-0802-1-6