
Abstract
Current databases of Phyllostachys pubescens full-length cDNAs (FL-cDNAs) provide a rich source of sequences for the development of potential FL-cDNA simple sequence repeat (SSR) markers. We screened 10,608 P. pubescens cDNAs, discovering 1614 SSRs in 1382 SSR-containing FL-cDNAs. The SSRs were more abundant within transposable elements (TEs) than expressed sequence tags (ESTs) and genome survey sequences (GSSs), and specific dinucleotide repeats tended to associate with particular TE families: (TA)n with En/Spm and (CT)n with Mutator. A selected panel of 100 FL-cDNAs containing type I SSRs yielded 68 functional SSR markers with an average polymorphism information content (PIC) value of 0.12, among which 22 loci contained polymorphisms. These markers became less transferrable (83.1% → 69.9% → 49.3%) but more polymorphic (79.4% → 92.3% → 92.8%) with increasing phylogenetic distance (intra-genus → intra-subtribe → intra-family). Transferability and polymorphism also depended on the location of the marker, with those located in the coding region being more transferrable (69.1%) and less polymorphic (89.4%) than those in the 5′-UTR (63.4% transferable, 90.7% polymorphic) and the 3′-UTR (61.8% transferable, 91.4% polymorphic). As proof of principle, we were able to use our FL-cDNA SSR markers to identify the parental stocks in interspecific hybrids of bamboo within and beyond P. pubescens, and estimate the outcrossing rate for P. pubescens. Our research should facilitate molecular breeding in bamboo species where original genetic markers are scarce.
Similar content being viewed by others


Background
Bambusoideae is a subfamily of the grass family Poaceae and is further divided into nine subtribes comprising more than 80 bamboo genera and about 1400 species worldwide. Fifty genera and more than 500 species are found in China, among which Phyllostachys pubescens (synonym: P. edulis) is commercially the most important species providing the third largest source of timber and the most predominant source of bamboo shoots. P. pubescens plantations cover an area of 3 million ha (approximately 2% of the total forest area), which has doubled over the last 30 years and taken on a more important ecological role (Fu 2001). Compounds extracted from P. pubescens have recently shown potential for the treatment of obesity and other diseases (Higa et al. 2012). However, various problems associated with P. pubescens plantations including its simultaneous flowering intervals of more than 60 years and recovers from a limited number of clones (Janzen 1976; Watanabe et al. 1982). Additionally, the little knowledge of its basic biology, genetics and breeding system bring about the practical difficulties associated with the identification and characterization of superior genotypes.
Molecular markers developing from microsatellites, also known as simple sequence repeats (SSRs) with characterization of high genome coverage, random dispersion, co-dominant inheritance, reproducibility and amenability to automation in high throughout genotyping, have gained considerable spotlight recently. By now, microsatellite markers have been developed for several other bamboo species, e.g. six loci for Bambusa arundinacea (Nayak and Rout 2005), eight loci for Sasa senanensis (Miyazaki et al. 2009) and eight loci for S. cernua (Kitamura and Kawahara 2009). We identified 19 GenBank microsatellite markers in P. pubescens and related species (Tang et al. 2010), and 15 expressed sequence tag (EST) SSR markers for Bambusa species (Dong et al. 2011). Recently, the Bamboo Full-Length cDNA Project (Peng et al. 2010) has generated a vast amount of publicly-available P. pubescens cDNA sequence data that can be used for gene discovery, comparative genomics/transcriptomics and marker development. Microsatellites derived from cDNAs or ESTs are highly transferable to closely related species (Zhang et al. 2005) facilitating the development of gene-based maps that may increase the efficiency of marker-assisted selection through the use of candidate genes (Rossi et al. 2003; Lu et al. 2006).
Here, we report the use of P. pubescens full-length cDNA (FL-cDNA) sequences to 1) analyze the association between SSRs and transposable elements (TEs) in the transcriptome; 2) develop and validate FL-cDNA SSR markers and determine their transferability to other bamboo species; and 3) apply the polymorphic SSR markers to estimate outcrossing rates in P. pubescens and identify bamboo interspecies hybrids.
Results and discussion
Association between SSRs and TEs in the P. pubescens transcriptome
We analyzed 10,608 P. pubescens FL-cDNA sequences available in NCBI GenBank, representing ~7171 kb of DNA. EST-trimmer was used to remove poly(A/T) runs, and the remaining sequence data were screened using MISA, identifying 2330 SSRs in 2014 cDNAs, the remaining cDNAs lacking SSRs. The sequences were clustered with CAP3, reducing the collection to 1614 non-redundant SSRs in 1382 cDNA contigs (Additional file 1: Figure S1). Peng et al. (2010) described the distribution of SSRs in the P. pubescens transcriptome in detail. Therefore, we selectively analyzed the non-redundant cDNA sequences and contigs with RepeatMasker to determine the association between SSRs and TEs because previous reports have shown that many SSRs are located in TEs (Richard et al. 2008), e.g. 50% of SSRs in the human genome (Scherer 2008), and that SSRs are closely associated with TEs in rice (Akagi et al. 2001; Temnykh et al. 2001) and barley (Wei et al. 2002). The results revealed 95 TEs, representing 13.52 kb (0.27%) of the total cDNA sequence data. Further analysis showed that 29 TEs contained a total of 39 SSRs, accounting for 822 bp (6.41%) of the total TE DNA sequences. In comparison, the non-redundant EST sequence data (7089 cDNAs refined from the original 10,608 sequences) contained 1614 SSRs, accounting for 2.60% of the total cDNA sequences in length. Therefore, SSRs were approximately 2.5-, 65.4-fold more abundant in TEs compared to cDNAs (Table 1) and whole genome (0.098% based on the analysis of genome survey sequences (GSSs; Tang et al. 2010)). It is possible that SSRs within TEs are also involved in the regulation of gene expression (Tomilin 2008).
Some studies have also suggested associations between specific SSR motifs and particular TE families, e.g. (TA)n is often found in the 5′-UTR of Micron element transposase genes in rice (Akagi et al. 2001; Temnykh et al. 2001). We also investigated the distribution of SSRs among DNA transposons, and found they were most likely to occur in En/Spm elements (33.87% of the total En/Spm DNA sequence). Six SSRs were found in five En/Spm elements, with one element containing two SSRs (Table 2). Mutator elements were the next most likely to contain SSRs (14.19% of the total Mutator DNA sequence). Thirteen SSRs were found in 12 Mutator elements, again with one element containing two SSRs (Table 2). The situation was very different among retrotransposons, with only 0.30% of the total Ty1-copia DNA sequence and 0% of Ty3-gypsy DNA sequence made up of SSRs. More detailed investigation of specific repeat motifs showed that four of the six SSRs found in En/Spm elements were TA/AT repeats, and 10 of the 13 SSRs found in Mutator elements were CT/AG repeats. All 13 of the Mutator SSRs and six of the En/Spm SSRs were located in the 5′-UTR. It has been reported that TE molecular markers (mPing) showed significantly higher levels of polymorphism than all other molecular markers in closely-related rice cultivars (Monden et al. 2009). Considering that it is difficult to detect genetic variation in P. pubescens using ordinary markers (Lin et al. 2009; Tang et al. 2010), SSRs in TEs therefore appear to be promising markers for bamboo species.
Development and polymorphism assessment of FL-cDNA SSR markers for P. pubescens
Original collection of 10,680 P. pubescens FL-cDNA sequences produced 1382 cDNA contigs containing SSRs. Sequences containing mononucleotide repeat motifs were excluded, leaving 1051 cDNA sequences containing SSRs with 2–6 nt repeats motifs (Additional file 1: Figure S1). Following the procedure already adopted for rice (Temnykh et al. 2001). We were able to design primer pairs for 583 (55%) of these cDNAs, the remainder offering either insufficient flanking DNA (over half of the SSRs were found in the 5′ or 3′ UTRs) or flanking DNA that was unsuitable for primer design. Only 325 (24.1%) of the SSRs were type I repeats (>20 bp), which offer greater potential for marker development. The 100 most promising sequences were selected for PCR validation, including dinucleotide repeats with ≥12 repeat units, trinucleotide repeats with ≥8 repeat units, tetranucleotide repeats with ≥6 repeat units, pentanucleotide and hexanucleotide repeats with ≥5 repeat units and some compound SSRs with >24 repeats (Table 3). We found that 32 of the selected cDNAs were unsuitable because the PCR failed to generate a product (four cDNAs) or generated products lacking SSRs (28 cDNAs), but the remaining 68 sequences allowed the development of FL-cDNA SSR markers (Table 3). These contained 18 compound SSRs, 19 dinucleotide repeats, 18 trinucleotide repeats, four tetranucleotide repeats, three pentanucleotide repeats and six hexanucleotide repeats. Interestingly, although 45 of the cDNAs (66.2%) generated the anticipated PCR product, 16 (23.5%) generated products with more repeats than expected, five (7.4%) generated products with fewer repeats than expected, and two (PBM050 and PBM055) generated products with different repeats and flanking sequences than those anticipated. The unanticipated amplification resulted in three SSR markers (PBM036, PBM055 and PBM 077) containing type II repeats (12–19 bp in length) and one marker (PBM079) shorter than 12 bp. In total, 67 sequences were deposited in GenBank (accession nos GU644371–GU644438). Based on BLASTX analysis, putative functions were assigned to most (66.2%) of the cDNA sequences with significant similarity to known proteins, whereas 27.9% matched unknown/hypothetical proteins and 5.9% were novel sequences (Table 3).
One hundred and seven primer pairs finally yielded 68 FL-cDNA SSR markers for P. pubescens, which is towards the lower end of the 60–90% success rate previously reported in sugarcane (Cordeiro et al. 2001), barley (Thiel et al. 2003), wheat (Yu et al. 2004) and peanut (Liang et al. 2009). Squirrell et al. (2003) defined the successive loss of sequenced fragments and designed primers, until arriving at a final collection of “working SSRs” producing discrete bands of the expected size, as the “attrition rate”. Kofler et al. (2008) reported a high attrition rate when developing SSR markers from enriched libraries, BAC-end sequences and ESTs in rye, possibly reflecting the large number of TEs in the rye genome. Tero et al. (2006) found that the number of SSR markers was reduced when the markers were predominantly located within TEs. Squirrell et al. (2003) suggested that SSR marker development would be challenging in polypoid species and species such as wheat and rye with large numbers of TEs. P. pubescens has 2n = 48 chromosomes and is thought to be tetraploid (Li et al. 1999). The genome is >2000 Mb, which is approximately 5.4 times larger than diploid cultivated rice (Gui et al. 2007), and it contains a large number of TEs (Zhong et al. 2010; Zhou et al. 2010a, [b], [c]). The slightly higher attrition rate we encountered therefore seems reasonable when considering the chromosomal polyploidy, size and TE content of the genome. We also encountered a higher attrition rate in B. oldhamii (Li et al. 2001), a hexaploid bamboo species with a large genome (data unpublished) in which we developed 15 EST-SSR markers from 52 promising sequences selected from 3406 non-redundant ESTs (Dong et al. 2011).
We surveyed the allelic variability of the markers by genotyping 50 open-pollinated seedlings germinated from the year 2010 seedlot (Table 3). Among the 68 FL-cDNA SSR markers, only 22 (32.4%) showed polymorphism. The polymorphism information content (PIC) values of the 68 markers ranged from 0 to 0.51 with a mean value of 0.12. For the 22 polymorphic loci, the PIC values ranged from 0.19 to 0.51 with a mean value of 0.36, and the top ten markers in terms of polymorphism were PBM075, PBM069, PBM095, PBM046, PBM066, PBM080, PBm087, PBM044, PBM084 and PBM091. SSR polymorphism in P. pubescens is much lower than observed in cereals (Thiel et al. 2003; Yu et al. 2004), coffee (Aggarwal et al. 2007) and the rubber tree (Feng et al. 2009). Bamboo P. pubescens has a long flowering interval of more than 60 years (Janzen 1976; Watanabe et al. 1982). Therefore, open pollination (DNA recombination) appears to have limited the amount of replication slippage, which diversifies SSR alleles (Richards and Sutherland 1994; Jakupiak and Wells 1999). Clonal propagation in the interim periods of flowering has reduced the SSR diversity in bamboo (Nayak and Rout 2005). In a previous study, we discovered almost no allelic variation in the panel of 11 varieties and 17 provenances of P. pubescens using 19 GSS-SSRs (Tang et al. 2010).
Interspecific transferability and polymorphism of P. pubescens FL-cDNA SSR markers
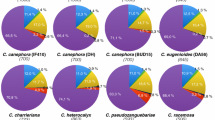
Although more than 1000 bamboo species have been described, the vast majority of publically-available sequence data are derived from P. pubescens (Tang 2009). Therefore, the development of a set of transferable P. pubescens FL-cDNA SSR markers suitable for other bamboo species would help to accelerate genetic research and comparative genomics in the Bambusoideae subfamily. Previously, we developed 19 P. pubescens GSS-SSR markers and successfully transferred them to six other Phyllostachys species with an average transferability of 75.3% and 66.7% polymorphism (Tang et al. 2010). In B. arundinacea, 100% and 83.3% transferability were achieved with 6 SSR markers in eight other Bambusa species and 10 species of other genera, respectively (Nayak and Rout 2005). In B. oldhamii, we achieved an average 59.6% transferability and 51.4% polymorphism with 15 markers in 14 bamboo species including four species within the same genus (Dong et al. 2011). We tested the transferability and polymorphism of these 68 putative FL-cDNA SSR markers across 41 diverse species in six tribes of the Bambusoideae subfamily, as defined by Das et al. (2008) and Yang et al. (2008) (Additional file 2: Table S1 and Additional file 3: Table S2). Successful amplification became less likely with increasing phylogenetic distance from P. pubescens, with an 83.1% success rate within the genus Phyllostachys, a 79.4% success rate across genera within the subtribe Shibataeeae, and a 49.3% average success rate for other subtribes, ranging from 36.8–76.5% (Table 4 and Figure 1). In contrast, the number of markers showing polymorphism increased with phylogenetic distance, with 79.4% of markers showing polymorphism within the genus Phyllostachys, 91.3% showing polymorphism within the Shibataeeae, and 92.8% showing polymorphism when comparing other subtribes. Markers in coding sequences were on average the most transferrable (69.1%) and the least polymorphic (89.4%), compared to those located in 5′-UTRs (63.4% transferrable, 90.7% polymorphic) and 3′-UTRs (61.8% transferrable, 91.4% polymorphic). These trends were exacerbated with increasing phylogenetic distance. These matches the results from a metastudy of 601 loci in 35 plant species showing an average 89.8% transferability at the subgenus level, 76.4% at the genus level and 35.2% at the family level (Rossetto 2001). Interestingly, more than 17 (25%) of the markers were transferrable to more than 85% of the tested species (Additional file 3: Table S2). This success rate suggests that FL-cDNA SSRs and their flanking regions are sufficiently conserved (Zhang et al. 2005), and it is therefore possible to transfer P. pubescens FL-cDNA SSR markers to other bamboo species for evolutionary studies and phylogenetic reconstructions (Sharma et al. 2008).

Polyacrylamide gel electrophoresis bands representing microsatellites derived from FL-cDNA sequences, tested on a panel of selected bamboo species to evaluate transferability and polymorphism in locus of PBM042 (above) and PBM064 (nether). M: size marker. Mb, Cp, etc.: Bamboo species abbreviations are listed in Supplementary Table 1.
Using polymorphic FL-cDNA SSR markers to estimate outcrossing rates and identify interspecific bamboo hybrids
Sexual propagation increases genetic diversity by creating progenies of different genotypes through recombination (i.e. outcrossing). This is advantageous for predominantly clonal plants such as most bamboo species, which rely mostly on vegetative regeneration interspersed with occasional flowering (Janzen 1976). The analysis of the reproductive system is therefore fundamental to elucidate primary genetic diversity and the structure of regenerating bamboo populations, and to adopt strategies for genetic improvement. Previous studies on the bamboo reproductive system based on field data and artificial pollination showed that self-compatibility is predominant in Sasa species (Nishiwaki and Konno 1990), and the selfing rate could approach 0.99 in Merostachys riedeliana (Guilherme and Ressel 2001). Outcrossing rate was estimated using SSR-based analysis as reported in S. cernua (Kitamura and Kawahara 2011).
Among the 22 polymorphic SSR markers described above, the ten most polymorphic (PIC ≥ 0.36) were used to detect polymorphisms in 50 open-pollinated half-sib seeds (year 2011) from three flowering sites in the Guangxi Province separated by at least 100 km. Polymorphism in the PBM044, PBM080 and PBM095 loci was identical in the seeds from all three flowering sites, whereas PBM084 and PBM091 featured additional alleles from Lipu, PBM069, PBM075, PBM087 and PBM091 featured additional alleles from Lingchuan, and PBM069, PBM075 and PBM084 featured additional alleles from Guanyang (Table 5). This indicated that flowering culms in different sites featured diverse SSR genotypes and produced genetically-diverse half-sib seed sources. Therefore, we used these eight polymorphic loci to estimate the outcrossing rates and other related genetic parameters for P. pubescens (Table 5). The overall estimates of tm and ts for three culms were 0.089 for both parameters, with no standard deviation. The estimates for individual culms showed small differences of 0.067 in Lipu and Lingchuan, and 0.133 in Guanyan, again for both parameters. Estimation of F is for the overall population was 0.195, indicating homozygote excess. We found that the outcrossing rate was 0.089, estimated from eight polymorphic multilocus datasets in P. pubescens, which is slight lower than the 0.148 reported in S. cernua using six multilocus SSR datasets (Kitamura and Kawahara 2011). This indicated that the reproductive system of P. pubescens predominantly involves self-fertilization with an adequate proportion of crossing to ensure genetic diversity as reported for S. cernua (Kitamura and Kawahara 2011).
The grow-out test for bamboo interspecific hybrids is time-consuming and laborious because it involves growing plants to maturity (which takes at least 5 years), assessing several anatomical, morphological and floral (long-term interval) characteristics that distinguish the hybrid. The polymorphic SSR markers could also help in the rapid and accurate identification of interspecies hybrids, as reported in poplar (Rajora and Rahman 2003) and wheat-barley (Malysheva et al. 2003). To obtain proof of principle that our novel SSR markers are suitable for hybrid characterization, we next selected several highly-transferable and polymorphic FL-cDNA SSR markers. PBM032, PMB049, PMB063 and PMB064, each with a number of species-restricted alleles, were used to test uncharacterized bamboo samples. Marker PMB063 identified the parental species in one hybrid as P. kwangsiensis and P. bambusoides, because all sequenced bands contained the (TCCA)n motif although with a variable number of repeats (Figure 2). Similarly, marker PMB064 identified the parental species B. pervariabilis and Dendrocalamus latiflorus which are distantly related to P. pubescens, with a variable number of repeats in the (GAGT)n motif (Figure 3). As previously shown using GSS-SSR markers, such high levels of transferability and polymorphism within the Bambusoideae subfamily should allow the use of FL-cDNA SSR markers to identify interspecific hybrids and their parents, both within the genus Phyllostachys (Tang et al. 2010) and in more distant taxa within subtribe of Shibataeeae (Lu et al. 2009). We have also developed several putative EST-SSR markers in B. oldhamii and have used these to identify some other sympodial bamboo interspecies hybrids (Wu et al. 2009; Dong et al. 2011). The SSR markers developed in the present study were used to identify not only interspecific hybrids from monopodial Phyllostachys but also intergeneric hybrids with sympodial rhizomes, which are distantly related to P. pubescens. Our data confirmed that microsatellites, especially SSR markers based on cDNAs and ESTs, are ideal for the identification of bamboo interspecies hybrids.

A, Microsatellite DNA fingerprints of P. kwangsiensis (line 1), P. bambusoides (line 3) and a presumed hybrid (line 2) at locus PBM063. B, Alignment of the nucleotide sequences of the microsatellite alleles at locus PBM014 amplified from P. kwangsiensis, P. bambusoides and two presumed hybrids. Nucleotides conserved among these sequences (relative to P. kwangsiensis) are shown by dots. The lines indicate the primer sequences used to amplify this microsatellite locus. The box highlights the microsatellite. The suffix numbers after bamboo species correspond to the DNA bands marked in part (a).

A, Microsatellite DNA fingerprints of Bambusa pervariabilis , Dendrocalamus latiflorus and their presumed hybrids at locus PBM064. B, Alignment of the nucleotide sequences of the microsatellite alleles at locus PBM064 amplified from B. pervariabilis, D. latiflorus and their presumed hybrids. Nucleotides conserved among these sequences (relative to B. pervariabilis) are shown by dots. The lines indicate the primer sequences used to amplify this microsatellite locus. The box highlights the microsatellite. The suffix numbers after the bamboo species correspond to the DNA bands marked in part (A).
Conclusions
Our data provide insight into the association between SSRs and TEs in FL-cDNAs from the P. pubescens transcriptome, allowing us to develop and evaluate 68 FL-cDNA SSR markers that can be used in P. pubescens and partially for many other bamboo species, to estimate the reproductive system of P. pubescens and identify several interspecific hybrids. These FL-cDNA SSR markers enrich the molecular marker resources currently available for bamboo. When a large set of polymorphic markers becomes available, we can use genome-wide association mapping in bamboo, in the absence of structured populations, to identify markers for traits of interest that can be used for marker-assisted selection in the Bambusoideae subfamily.
Methods
Full-length cDNA mining and SSR/TE detection
We obtained 10,608 FL-cDNA sequences from NCBI Entrez (http://www.ncbi.nlm.nih.gov/) on July 1, 2010. These cDNA sequences were assembled from five cDNA libraries constructed from breaking-out shoots, young (40-cm) shoots and young leaves from plants, and shoots and roots from germinated seeds (Peng et al. 2010). We used EST Trimmer (http://pgrc.ipk-gatersleben.de/misa/download/est_trimmer.pl) to remove poly(A/T) runs from the 5′ and 3′ ends until there were no occurrences of (T)5 or (A)5 within a 50-bp range. Redundant sequences were eliminated and overlapping sequences were spliced together using CAP3 (http://seq.cs.iastate.edu/cap3.html) (Huang and Madan 1999).
After pre-treatment, we used MISA (http://pgrc.ipk-gatersleben.de/misa/misa.html) to screen for SSRs including mononucleotide repeats ≥10 bp in length, dinucleotide to hexanucleotide repeats with ≥6 repeat units, and interrupted composite SSRs with ≤100 bp of intervening DNA. Putative annotations were assigned to non-redundant ESTs containing SSRs using BLAST against the Moso Bamboo cDNA Database (http://202.127.18.228/mbcd/) and the Gramene Ontologies Database (http://archive.gramene.org/plant_ontology/). TEs were identified using RepeatMasker and RepeatProteinMask (http://www.repeatmasker.org) based on similar elements present in the rice genome, and SSRs within TEs were screened using MISA with the same parameters as above. Additional file 1: Figure S1 provides a flow chart for the data mining and marker development process.
Plant material and DNA extraction
We used P. pubescens samples collected from the Anji Bamboo Germplasm Garden, Anji, Zhejiang Province, to identify and characterize putative FL-cDNA SSR markers. The polymorphism of these SSR markers was evaluated using 50 seedlings germinated from an open-pollinated seedlots (mixed seed sources, mainly from different flowering sites in the counties of Lipu, Lingchuan and Guanyang, Guangxi Province in the year 2010). Another 50 seedlings were germinated from open-pollinated half-sib seeds (year 2011) from three flowering culms in the same three counties (>100 km between sites) and were used to estimate the P. pubescens outcrossing rate. We obtained 41 representative bamboo species from 38 genera within six subtribes mainly found in China to test the transferability and polymorphism of the FL-cDNA SSR markers (Additional file 2: Table S1). We obtained three Phyllostachys interspecific hybrids from Jiangxi Province, China, and two intergeneric hybrids from Yoshinaka Bamboo Germplasm Garden, Fukuoka, Japan, for the hybrid identification tests. Genomic DNA was extracted from young leaves using the hexadecyltrimethylammonium bromide (CTAB) method (Doyle and Doyle 1987), with some modifications.
Amplification and sequencing of SSR loci
Primer pairs designed according to the available cDNA sequences were synthesized by Shanghai Sangon Biological Engineering Technology & Services Co., Ltd. P. pubescens DNA was amplified in 20-μl reactions comprising 50–100 ng of template DNA, 0.2 μM of each primer, 200 μM of each dNTP and 1 unit of Taq DNA polymerase with 1× PCR universal buffer (10 μM Tris–HCl, pH 8.3 at 25°C; 50 μM KCl), and 1.5 μM MgCl2 (Shanghai Sangon Biological Engineering Technology & Services Co., Ltd). The reaction was heated to 95°C for 5 min using an ABI PE9700 thermocycler, followed by 30 cycles of 1 min denaturation at 95°C, 1 min annealing at 46–59°C depending on the primer pair (Table 3), and 2 min extension at 72°C, followed by a final hold at 72°C for 5 min. Amplified microsatellite loci were tested in 41 diverse species in six tribes of the Bambusoideae subfamily (Table 4) and interspecific hybrids (Figures 2 and 3). The annealing temperature was lowered by 2–5°C according to the evolutionary distance between species based on molecular markers (Das et al. 2008) and nuclear and chloroplast sequences (Yang et al. 2008), as suggested by Rossetto (2001). PCR products were separated on 6% polyacrylamide denaturing gels, and marker bands were revealed by silver staining as described by Panaud et al. (1996). Specific bands were excised directly from the silver staining polyacrylamide gel, purified using the EZ-10 Spin Column DNA Gel Extraction Kit (Biobasic Inc.) and ligated into the pUC18 vector (TaKaRa, Japan). Three positive clones for each bamboo species were selected for sequencing using BigDye terminator V3.1 in a cycle sequencing protocol according to the manufacturer’s specifications (PE Applied Biosystems, ABI PRISM 3100-Avant Automatic DNA Sequencer). Vector sequences were removed then edited using Vector NTI software (version 10.0, Invitrogen Co., USA). Sequences were deposited in NCBI GenBank (accession nos GU644371–GU644438).
Data analysis
The polymorphism information content (PIC) (Botstein et al. 1980) of our SSR markers was determined using Powermarker v3.25 (Liu and Muse 2005). All 68 selected primer pairs were used to amplify template DNA from 41 bamboo species covering 35 genera in six subtribes (Additional file 2: Table S1) and the statistical methods of Nayak and Rout (2005) and Sharma et al. (2009) were used to calculate the cross-taxon transferability and polymorphism (Additional file 3: Table S2), in which polymorphism is calculated only from the loci that were successfully transferred across taxa (Rossetto 2001). Single locus and multilocus outcrossing rates and relative parameters were analyzed separately under the mixed mating model of Ritland & Jain (1981) and Ritland (2002), implemented using MLTR v3.4 (Ritland 1996).
References
Aggarwal RK, Hendre PS, Varshney RK, Bhat PR, Krishnakumar V, Singh L: Identification, characterization and utilization of EST-derived genic microsatellite markers for genome analyses of coffee and related species. Theor Appl Genet 2007, 114(2):359-372. 10.1007/s00122-006-0440-x
Akagi H, Yokozeki Y, Inagaki A, Mori K, Fujimura T: Micron, a microsatellite-targeting transposable element in the rice genome. Mol Genet Genomics 2001, 266: 471-480. 10.1007/s004380100563
Botstein D, White RL, Skolnick M, Davis RW: Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 1980, 32: 314-331.
Cordeiro GM, Casu R, McIntyre CL, Manners JM, Henry RJ: Microsatellite markers from sugarcane ( Saccharum spp.) ESTs cross-transferable to erianthus and sorghum. Plant Sci 2001, 160(6):1115-1123. 10.1016/S0168-9452(01)00365-X
Das M, Bhattacharya S, Singh P, Filgueiras TS, Pal A: Bamboo taxonomy and diversity in the era of molecular markers. In Advances in Botanical Research. Edited by: Jean-Claude K, Michel D. San Diego, CA 92101-4495, USA: Academic Press; 2008:225-268.
Dong WJ, Wu MD, Lin Y, Tang DQ: Evaluation of 15 caespitose bamboo EST-SSR markers for cross-species/genera transferability and ability to identify interspecies hybrids. Plant Breed 2011, 130: 596-600. 10.1111/j.1439-0523.2011.01860.x
Doyle JJ, Doyle JL: A rapid isolation procedure for small quantities of fresh leaf materials. Phytochem Bull 1987, 19: 11-15.
Feng S, Li W, Huang H, Wang J, Wu Y: Development, characterization and cross-species/genera transferability of EST-SSR markers for rubber tree ( Hevea brasiliensis ). Mol Breed 2009, 23(1):85-97. 10.1007/s11032-008-9216-0
Fu J: Chinese Moso Bamboo: Its importance. Bamboo 2001, 22(5):5-7.
Gui Y, Sheng W, Quan L, Zhou C, Long S, Zheng H, Jin L, Zhang X, Ma N, Fan L: Genome size and sequence composition of moso bamboo: A comparative study. Sci China Series C: Life Sci 2007, 50(5):700-705.
Guilherme FAG, Ressel K: Biologia floral e sistema de reprodução de Merostachys riedeliana (Poaceae: Bambusoideae). Revta brasil Bot 2001(in Spanish with English abstract), 24: 205-211. (in Spanish with English abstract) 10.1590/S0100-84042001000200011
Higa JK, Liang Z, Williams PG, Panee J: Phyllostachys edulis compounds inhibit palmitic acid-induced monocyte chemoattractant protein 1 (MCP-1) production. PLoS One 2012, 7(9):e45082. 10.1371/journal.pone.0045082
Huang X, Madan A: CAP3: A DNA sequence assembly program. Genome Res 1999, 9: 868-877. 10.1101/gr.9.9.868
Jakupiak JP, Wells RD: Genetic instabilities in (CTG.CAG) repeats occur by recombination. J Biol Chem 1999, 274: 23468-23479. 10.1074/jbc.274.33.23468
Janzen DH: Why bamboos wait so long to flower. Ann Rev Eco Syst 1976, 7: 347-391. 10.1146/annurev.es.07.110176.002023
Kitamura K, Kawahara T: Clonal identification by microsatellite loci in sporadic flowering of a dwarf bamboo species, Sasa cernua . J Plant Res 2009, 122(3):299-304. 10.1007/s10265-009-0220-1
Kitamura K, Kawahara T: Estimation of outcrossing rates at small-scale flowering sites of the dwarf bamboo species, Sasa cernua . J Plant Res 2011, 124: 683-688. 10.1007/s10265-010-0398-2
Kofler R, Bartos J, Gong L, Stift G, Sucha′nkova P, Suchankova P, Simkova H, Berenyi M, Burg K, Dolezel J, Lelley T: Development of microsatellite markers specific for the short arm of rye ( Secale cereale L.) chromosome 1. Theor Appl Genet 2008, 117(6):915-926. 10.1007/s00122-008-0831-2
Li XL, Liu S, Shong WQ, Chen RY, Wang WZ: Chromosome numbers of forty species of scattered bamboo. Acta Phytotax Sinica 1999(in Chinese with English abstract), 37(6):541-544. (in Chinese with English abstract)
Li XL, Lin RS, Fen XL, Qi ZX, Song WQ, Chen RY: Chromosome numbers of ninety-four species of caespitose bamboo. Acta Phytotax Sinica 2001(in Chinese with English abstract), 39(5):433-442. (in Chinese with English abstract)
Liang X, Chen X, Hong Y, Liu H, Zhou G, Li S, Guo B: Utility of EST-derived SSR in cultivated peanut ( Arachis hypogaea L.) and Arachis wild species. BMC Plant Biol 2009, 9: 35. 10.1186/1471-2229-9-35
Lin XC, Ruan XS, Lou YF, Guo XQ, Fang W: Genetic similarity among cultivars of Phyllostachys pubescens . Plant Syst Evol 2009, 277: 67-73. 10.1007/s00606-008-0104-1
Liu KJ, Muse SV: PowerMarker: An integrated analysis environment for genetic marker date. Bioinformatics 2005, 21(9):2128-2212. 10.1093/bioinformatics/bti282
Lu HJ, Fellers JP, Friesen TL, Meinhardt SW, Faris JD: Genomic analysis and marker development for the Tsn1 locus in wheat using bin-mapped ESTs and flanking BAC contigs. Theor Appl Genet 2006, 112(6):1132-1142. 10.1007/s00122-006-0215-4
Lu JJ, Yoshinaga K, Fang W, Tang DQ: Identification of the bamboo hybrids by SSR markers. Sci Silv Sin 2009(in Chinese with English abstract), 45(3):29-34. (in Chinese with English abstract)
Malysheva L, Sjakste T, Matzk F, Roder M, Ganal M: Molecular cytogenetic analysis of wheat-barley hybrids using genomic in situ hybridization and barley microsatellite markers. Genome 2003, 46: 314-322. 10.1139/g02-117
Miyazaki Y, Ohnishi N, Hirayama K, Nagata J: Development and characterization of polymorphic microsatellite DNA markers for Sasa senanensis (Poaceae: Bambuseae). Conserv Genet 2009, 10: 585-587. 10.1007/s10592-008-9575-4
Monden Y, Naito K, Okumoto Y, Saito H, Oki N, Tsukiyama T, Ideta O, Nakazaki T, Wessler SR, Tanisaka T: High potential of a transposon mPing as a marker system in japonica x japonica cross in rice. DNA Res 2009, 16(2):131-140. 10.1093/dnares/dsp004
Nayak S, Rout GR: Isolation and characterization of microsatellites in Bambusa arundinacea and cross species amplification in other bamboos. Plant Breed 2005, 124: 559-602.
Nishiwaki A, Konno Y: Pollination system in two dwarf bamboo species. Bamboo J 1990, 8: 17-20.
Panaud O, Chen X, McCouch SR: Development of microsatellite markers and characterization of simple sequence length polymorphism (SSLP) in rice ( Oryza sativa L.). Mol Gen Genet 1996, 252: 597-607.
Peng Z, Lu T, Li L, Liu X, Gao Z, Hu T, Yang X, Feng Q, Guan J, Weng Q, Fan D, Zhu C, Lu Y, Han B, Jiang Z: Genome wide characterization of the biggest grass, bamboo, based on 10,608 putative full-length cDNA sequences. BMC Plant Biol 2010, 10: 116. 10.1186/1471-2229-10-116
Rajora P, Rahman H: Microsatellite DNA and RAPD fingerprinting, identification and genetic relationships of hybrid poplar ( Populus x canadensis ) cultivars. Theor Appl Genet 2003, 106: 470-477.
Richard GF, Kerrest A, Dujon B: Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Micr Mol Bio Rev 2008, 72: 686-727. 10.1128/MMBR.00011-08
Richards RI, Sutherland GR: Simple repeat DNA is not replicated simply. Nature Genet 1994, 6: 114-116. 10.1038/ng0294-114
Ritland K: Multilocus mating system program – MLTR. 1996. Available free of charge at http://genetics.forestry.ubc.ca/ritland/programs.html
Ritland K: Extensions of models for the estimation of mating systems using n independent loci. Heredity 2002, 88: 221-228. 10.1038/sj.hdy.6800029
Ritland K, Jain SK: A model for the estimation of outcrossing rate and gene frequencies using n independent loci. Heredity 1981, 47: 35-52. 10.1038/hdy.1981.57
Rossetto M: Sourcing of SSR markers from related plant species. In Plant genotyping---the DNA fingerprinting of plant. Edited by: Henry RJ. Wallingford, Oxon OX10 8DE, UK: CABI Publishing; 2001:211-224.
Rossi M, Araujo PG, Paulet F, Garsmeur O, Dias VM, Chen H, Van Sluys MA, D′Hont A: Genomic distribution and characterization of EST-derived resistance gene analogs (RGAs) in sugarcane. Mol Genet Genomics 2003, 269(3):406-419. 10.1007/s00438-003-0849-8
Scherer S: A short guide to the human genome. Cold Spring Harbor University Press, Cold Spring, NY; 2008.
Sharma RK, Gupta P, Sharma V, Sood A, Mohapatra T, Ahuja PS: Evaluation of rice and sugarcane SSR markers for phylogenetic and genetic diversity analyses in bamboo. Genome 2008, 51(2):91-103. 10.1139/G07-101
Sharma V, Bhardwaj P, Kumar R, Sharma RK, Sood A, Ahuja PS: Identification and cross-species amplification of EST derived SSR markers in different bamboo. Conserv Genet 2009, 10: 721-724. 10.1007/s10592-008-9630-1
Squirrell J, Hollingsworth PM, Woodhead M, Russell J, Lowe AJ, Gibby M, Powell W: How much effort is required to isolate nuclear microsatellites from plants? Mol Ecol 2003, 12(6):1339-1348. 10.1046/j.1365-294X.2003.01825.x
Tang DQ: Genomic sequencing and its application for biological and evolutional research in bamboo. Bamboo J 2009, 26: 1-10.
Tang DQ, Lu JJ, Fang W, Zhang S, Zhou MB: Development, characterization and utilization of GenBank microsatellite markers in Phyllostachys pubescens and related species. Mol Breed 2010, 25: 299-311. 10.1007/s11032-009-9333-4
Temnykh S, DeClerck G, Lukashova A, Lipovich L, Cartinhour S, McCouch SR: Computational and experimental analysis of microsatellites in rice ( Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res 2001, 11: 1441-1452. 10.1101/gr.184001
Tero N, Neumeier H, Gudavalli R, Schlo¨tterer C: Silene tatarica microsatellites are frequently located in repetitive DNA. J Evol Biol 2006, 19(5):1612-1619. 10.1111/j.1420-9101.2006.01118.x
Thiel T, Michalek W, Varshney RK, Graner A: Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley ( Hordeum vulgare L.). Theor Appl Genet 2003, 106: 411-422.
Tomilin NV: Regulation of mammalian gene expression by retroelements and non-coding tandem repeats. Bioessays 2008, 30: 338-348. 10.1002/bies.20741
Watanabe M, Ueda K, Manabe I, Akai T: Flowering, seeding, germination and flowering periodicity of Phyllostachys pubescens . J Jpn For Soc 1982, 64: 107-111.
Wei F, Wing RA, Wise RP: Genome dynamics and evolution of the Mla (powdery mildew) resistance locus in barley. Plant Cell 2002, 14: 1903-1917. 10.1105/tpc.002238
Wu M, Dong W, Tang DQ: Identification of four caespitose hybrid bamboos by using SSR markers. Mol Plant Breed 2009(in Chinese with English abstract), 7(5):959-965. (in Chinese with English abstract)
Yang HQ, Yang JB, Gao J, Yang YM, Peng S, Li DZ: A molecular phylogenetic and fruit evolutionary analysis of the major groups of the paleotropical woody bamboos (Gramineae: Bambusoideae) based on nuclear ITS, GBSSI gene and plastid trnL -F DNA sequences. Mol Phylogenet Evol 2008, 48(3):809-824. 10.1016/j.ympev.2008.06.001
Yu JK, Dake TM, Singh S, Benscher D, Li W, Gill B, Sorrells ME: Development and mapping of EST-derived simple sequence repeat markers for hexaploid wheat. Genome 2004, 47(5):805-818. 10.1139/g04-057
Zhang LY, Bernard M, Leroy P, Feuillet C, Sourdille P: High transferability of bread wheat EST-derived SSRs to other cereals. Theor Appl Genet 2005, 111(4):677-687. 10.1007/s00122-005-2041-5
Zhong H, Zhou MB, Xu CM, Tang DQ: Diversity and evolution of Pong -like elements in Bambusoideae subfamily. Biochem Syst Ecol 2010, 38: 750-758. 10.1016/j.bse.2010.06.010
Zhou MB, Lu JJ, Zhong H, Liu XM, Tang DQ: Distribution and diversity of PIF -like transposable elements in the Bambusoideae subfamily. Plant Sci 2010, 179: 257-266. 10.1016/j.plantsci.2010.05.012
Zhou MB, Lu JJ, Zhong H, Tang KX, Tang DQ: Distribution and polymorphism of mariner -like elements in the Bambusoideae subfamily. Plant Syst Evol 2010, 289: 1-11. 10.1007/s00606-010-0323-0
Zhou MB, Zhong H, Zhang QH, Tang KX, Tang DQ: Diversity and evolution of Ty1- copia retroelements in representative tribes of Bambusoideae subfamily. Genetica 2010, 138: 861-868. 10.1007/s10709-010-9469-5
Acknowledgements
This work was financially supported by grants from the “973” Program (2012CB723008), National Natural Science Foundation of China (31170623), and Agricultural Projects of Zhejiang Province (2010C12011, 2012C12908-2).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
DQT conceived the study, participated in its design, coordination, data analysis and interpretation, and drafted, reviewed and improved the manuscript. YL and JJL carried out genotyping, sequencing and the identification of hybrids. WJD and MBZ carried out mining of EST data, unigene prediction, and analysis of repeat type and frequency of microsatellites. WF and YI helped in interpretation the data and improve the manuscript. All authors have read and approved the final manuscript.
Electronic supplementary material
40064_2014_1193_MOESM1_ESM.doc
Additional file 1: Figure S1: Scheme used for database mining and the development of SSR markers from P. pubescens FL-cDNA sequences. (DOC 31 KB)
40064_2014_1193_MOESM2_ESM.xls
Additional file 2: Table S1: Species used to test cross species/genus amplification of P. pubescens FL-cDNA SSR loci. (XLS 34 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Lin, Y., Lu, JJ., Wu, MD. et al. Identification, cross-taxon transferability and application of full-length cDNA SSR markers in Phyllostachys pubescens. SpringerPlus 3, 486 (2014). https://doi.org/10.1186/2193-1801-3-486
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2193-1801-3-486