
Abstract
Angiogenesis is essential for promoting growth and metastasis of solid tumors by ensuring blood supply to the tumor mass. Targeting angiogenesis is therefore an attractive approach to therapeutic intervention of cancer. Tumor angiogenesis is a process that is controlled by a complex network of molecular components including sensors, signaling transducers, and effectors, leading to cellular responses under hypoxic conditions. Positioned at the center of this network are the hypoxia-inducible factors (HIFs). HIF-1 is a major transcription factor that consists of two subunits, HIF-1α and HIF-1β. It mediates transcription of a spectrum of gene targets whose products are essential for mounting hypoxic responses. HIF-1α protein level is very low in the normoxic condition but is rapidly elevated under hypoxia. This dramatic change in the cellular HIF-1α level is primarily regulated through the proteosome-mediated degradation process. In the past few years, scientific progress has clearly demonstrated that HIF-1α phosphorylation is mediated by several families of protein kinases including GSK3β and ERKs both of which play crucial roles in the regulation of HIF-1α stability. Recent research progress has identified that Polo-like kinase 3 (Plk3) phosphorylates HIF-1α at two previously unidentified serine residues and that the Plk3-mediated phosphorylation of these residues results in destabilization of HIF-1α. Plk3 has also recently been found to phosphorylate and stabilize PTEN phosphatase, a known regulator of HIF-1α and tumor angiogenesis. Given the success of targeting protein kinases and tumor angiogenesis in anti-cancer therapies, Plk3 could be a potential molecular target for the development of novel and effective therapeutic agents for cancer treatment.
Similar content being viewed by others

Introduction
When tumors grow to a certain size, a severe hypoxic microenvironment develops within the tumor mass [1]. Simple diffusion of oxygen and nutrients becomes insufficient to meet the demand of fast growing tumors [1]. It is believed that tumor size generally will not exceed 1 centimeter in diameter without the support of angiogenesis [2]. Tumor angiogenesis is a process leading to formation of vasculature that supplies the growth of solid tumors [1, 2]. Logically, tumor angiogenesis tends to be more pronounced in aggressive and malignant tumors [3]. Since the initial discovery of tumor angiogenesis four decades ago [4], tumor angiogenesis has emerged as an important target for cancer therapy [1, 2, 5–7]. Many inhibitors of tumor angiogenesis have been developed and used in clinical application or is in the pipeline leading to clinics [1, 2, 6, 7]. Since highly vascular tumors tend to have much higher potential for metastasis [8], these inhibitors of angiogenesis may have the added value of blocking tumor metastasis, thus making them more effective for cancer treatment.
A primary consequence of increased tumor size is hypoxia, a result of oxygen restriction in the tumor mass. Hypoxia triggers tumor angiogenesis by activating the cellular hypoxia response pathways [1]. Located in the center of these pathways are the hypoxia inducible factors (HIFs) including the well characterized HIF-1 [1, 9–11]. HIF-1 is a transcription factor that promotes transcription of a series of genes such as vascular endothelial growth factor (VEGF) that are critical for the cellular hypoxic response [1, 5].
HIF-1α is the inducible subunit of the HIF-1 transcription factor, whose protein level can be dramatically induced upon exposure to hypoxia [1, 9–11]. The cellular level of HIF-1α protein is regulated primarily at the post-translational level [1, 9–11]. The HIF-1α gene is constitutively transcribed and translated [1, 9–11]. Under normoxic conditions, HIF-1α protein is hydroxylated by prolyl hydroxylases (PHDs), recognized and polyubiquitinated by and degraded by a proteosome-dependent mechanism [1, 9–11]. Hypoxia greatly stabilizes HIF-1α because of a reduced activity of PHDs due to low oxygen tension [1, 9–11]. In addition to hydroxylation, phosphorylation by protein kinases also regulates HIF-1α stability and/or localization [12–17]. Compared to hydroxylation, the significance of HIF-1α phosphorylation in regulating its stability is far less appreciated. Most recent studies have demonstrated that Plk3 is an endogenous kinase of HIF-1α that regulate its stability [17]. In addition, Plk3 phosphorylates and stabilizes PTEN phosphatase [18], which can in turn contribute to HIF-1α stability by suppression of the PDK/Akt signaling axis. Supporting this, PLK3 knock-out murine embryonic fibroblasts (MEFs) display significantly enhanced expression of HIF-1α and tumor angiogenesis in response to hypoxia [17, 19]. In this review, we will discuss significance of Plk3 in HIF-1α regulation, tumor angiogenesis, and its potential as a therapeutic target for cancer treatment.
Polo-like kinase 3
Polo-like kinases (Plk) are named after Drosophila Polo, the founding member of a family of evolutionarily conserved protein serine/threonine kinases. Polo is involved in the regulation of mitosis and meiosis [20, 21]. The mammalian Plk family consists of 5 members: Plk1, Plk2, Plk3, Plk4 and Plk5 [21, 22]. These proteins share significant amino acid sequence homology to Polo and to one another [21]. Structurally, all Plks are comprised of a highly conserved kinase domain at the amino-terminus and a Polo box domain (PBD) at the carboxyl-terminus [21, 23] (Figure 1). However, human Plk5 has a truncated kinase domain that lacks kinase activity [22]. PBD is believed to be important for the subcellular localization and the substrate recognition by these kinases [21, 23]. The biological functions of mammalian Plks are more diverse than those of their counterparts in lower eukaryotes [21]. Among the Polo kinase family members, Plk1 is the most extensively studied. It primarily plays a role in mitosis, thus functionally closest to that of Polo in Drosophila and Cdc5 in the budding yeast. On the other hand, the functions of other Plk members seem to be more diverse and much less understood [17, 18, 21, 24–29]. Extensive studies in the past have shown that Plk1, Plk3, and Plk4 are involved in the genesis and/or development of tumors [30–34].

Domain structures of human Plks. All Plks have a highly conserved kinase domain and a polo box domain (PBD). Plk 1-3 and 5 have two polo boxes (PB) while Plk4 has one discernable PB. Human Plk5 has a truncated kinase domain.
Murine Plk3 was initially identified as a fibroblast growth factor-inducible kinase (Fnk) in NIH 3T3 cells in a differential display study [35]. Human Plk3 was later cloned and named as Prk (p roliferation-r elated k inase) [33]. The amino acid sequence of human Plk3 shares 36% and 33% overall identity to Plk1 and Polo, respectively [33]. Plk3 has the structure that is typical of a Plk family member, with an N-terminal kinase domain and a C-terminal PBD with two Polo boxes [21] (Figure 1).
The Plk3 mRNA level was found to be inducible by mitogen stimulation with peak expression at the G1 phase, whereas its protein level seems to be fairly constant in mitogen-stimulated cells throughout the cell cycle [25, 36, 37]. However, a more recent report, shows that Plk3 protein level is also cell cycle-regulated with a pattern similar to that of its mRNA [38]. Plk3 is considered an immediate early response gene, whose expression does not depend on de novo protein synthesis [25, 33, 35]. The kinase activity of Plk3 has also been shown to oscillate during the cell cycle and it is subjected to regulation by a variety of stress conditions, including genotoxic insults, hypoxic treatment, and osmotic shock [27–29, 36, 39–41].
The function of Plk3 appears to be rather diverse. Plk3 is involved in multiple phases of the cell cycle [38, 42]. Plk3 is closely associated with spindle poles, mitotic spindles, and midbody during mitosis [43]. Plk3 overexpression induces cell cycle arrest at the M phase followed by apoptosis, which likely results from deregulated microtubule dynamics and centrosomal function [43]. Plk3 may also regulate the onset and/or progression of mitosis and meiosis [44]. Plk3 phosphorylates and regulates the subcellular localization of Cdc25C phosphatase [44, 45], a key regulator of the G2/M transition [46]. Plk3 is reported to mediate the G1/S transition and appears to be required for the S phase entry [38, 47]. In addition, Plk3 phosphorylates proteins that are important for regulating DNA replication, such as topoisomerase IIα and DNA polymerase δ (Pol δ) [42, 48, 49]. Furthermore, Plk3 regulates Golgi fragmentation during mitotic entry in mammalian cells [50–52].
Plk3 also has a role in apoptosis and stress responses. Ectopic expression of Plk3 causes cell cycle arrest followed by chromatin condensation and apoptosis in cultured cells [39, 43, 53, 54]. Plk3 is activated by cellular responses to DNA damage that produces ROS [27–29, 36, 39, 40, 55]. Genotoxic agents such as hydrogen peroxide (H2O2), ionizing radiation, methylmethane sulfonate, ultraviolet light, and adriamycin strongly activate Plk3 [27–29, 36, 39, 40, 55], which in turn phosphorylates p53 at Ser-20 [55]. Ser-20 phosphorylaiton is known to be associated with the stability of p53 [56]. Thus, Plk3 may function to reinforce DNA damage checkpoint responses or to preferentially mediate certain types of DNA damage insults. Plk3 also directly regulates Chk2 and vice versa [36, 40]. These results underscore the dynamic role of Plk3 in the DNA damage checkpoint control. Activation of Plk3 by hypoxia and osmotic stress has also been reported in certain cell type [27, 28]. Despite the diverse functions of Plk3 discovered in various cell-based in vitro assays, PLK3 null mice develop rather normally and are fertile [19]. It is likely that the lack of noticeable phenotype is a result of compensatory mechanisms at the whole animal level. Figure 2 summarizes known functions of Plk3.

Known functions of Plk3. Plk3 regulates a number of cellular activities by modulating a wide array of cellular components. Arrows indicate activation, T bars indicate inhibition.
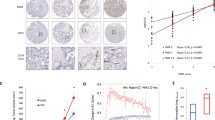
Plk3 expression is often deregulated in human cancers. Reduced mRNA levels of PLK3 in many types of cancer has been reported, including lung cancer, head and neck cancer, and colon cancer [33, 34, 57], suggesting that reduced Plk3 expression may contribute to tumor development. Consistent with these early studies, we have demonstrated that Plk3 protein levels are also reduced in several other human malignancies including those of kidney, liver, stomach, and rectum (Figure 3). Moreover, we have observed that expression of Plk3 mRNA and protein is significantly deregulated in human melanoma cell lines and tumor tissues (Figure 4). In fact, Plk3 has been generally regarded as having a tumor-suppressing function [30, 58]. This notion has been supported by a mouse genetic study showing that, compared with wild-type littermates, PLK3 null mice are prone to the development of malignancies in several organs later in the life [19]. The lower levels of PLK3 mRNA in cancer cells appear to be a result of reduced mRNA transcription [33]. Of note, mutations of PLK3 coding sequences seem to be rare, at lease in lung cancers [59]. Thus, the expression level of Plk3 may play a major role in Plk3's association with cancer development. It is also noteworthy to point out that the PLK3 gene is located at chromosome 1p34 where a high frequency of loss of heterozygosity occurs in many human cancers. This implies its close relationship between the loss of its function and tumorigenesis [34, 60, 61].

Plk3 expression in selected human tumors. Immunohistochemistry was performed on paraffin sections of indicated human tumor and corresponding normal tissue samples using the Plk3 antibody. IgGs were used as control.

Plk3 mRNA and protein expression in selected human melanoma cell lines and tissue samples. A. Real-time PCR was performed on RNA samples from normal melanocytes and selected human melanoma cell lines with DNA primers for Plk3. B. Western blot was performed on normal melanocytes and selected human melanoma cell lines with Plk3 and β-actin antibodies. C. Immunohistochemistry was performed on paraffin sections of human melanoma specimens and corresponding normal skin tissues using the Plk3 antibody.
Plk3 and tumor angiogenesis
Plk3 regulates tumor angiogenesis through HIF-1α
Neovascularization as the result of angiogenesis is essential for progression of solid tumor as the fast growing tumor mass needs to be supported by oxygen and nutrients from the blood [1, 2]. One of the key events that triggers tumor angiogenesis is cellular hypoxia, which is a consequence of blood restriction in the inner portion of a solid tumor [2, 3, 8]. Thus, cellular responses to hypoxia play a pivotal role in tumor angiogenesis [2, 3, 8].
HIF-1 is the primary player in cellular hypoxic responses and the initiating factor for tumor angiogenesis [1, 9, 10]. HIF-1 is an oxygen sensing transcription factor containing a constitutively expression β subunit (HIF-1 β) and an inducible α subunit (HIF-1α) [1, 10, 11, 62]. Upon activation by hypoxia, HIF-1 trans-activates an array of genes whose products in turn drive the cellular machinery for hypoxic responses [1, 10, 11]. Among the most important HIF-1 inducible proteins are VEGF, fibroblast growth factor (FGF), and glucose transporter [1, 2]. These proteins are known to stimulate the formation of vasculature or nutrient transport [1, 2].
The activity of HIF-1 is controlled by the availability of its α subunit (HIF-1α), which is primarily regulated at the post-translational level [10, 11]. HIF-1α is subjected to multiple types of covalent modifications including hydroxylation, phosphorylation, acetylation, and ubiquitination [10, 11]. Although most of these modifications are associated with HIF-1α stability, hydroxylation and ubiquitination are the primary mechanisms by which the HIF-1α protein level is negatively controlled within the cell [10, 11]. In normoxia, HIF-1α is hydroxylated by prolyl hydroxylases at two proline residues (Pro-402 and Pro-564 in human) and by factor inhibiting HIF-1 at the asparagine residue (Asn-803) [10, 11] (Figure 5). HIF-1α hydroxylation facilitates its association with ubiquitin E3 ligase pVHL and subsequent ubiquitination that leads to HIF-1α degradation by the proteosome [10, 11]. Under hypoxic condition, HIF-1α hydroxylation is inhibited, resulting in reduced ubiquitination with concomitant stabilization of HIF-1α protein. In turn, the elevated HIF-1α, in conjunction with HIF-1β, promotes expression of HIF-1-responsive genes [10, 11].

Domain structure of human HIF-1α, showing Plk3 phosphorylation sites. Human HIF-1α contains a basic helix-loop-helix domain (bHLH), a PAS domain, an oxygen-dependent degradation domain (ODDD), an N-terminal transactivation domain (N-TAD), and a C-terminal transactivation domain (C-TAD). The hydroxylation sites (P402, P564, and N803) are indicated. The Plk3 phosphorylation sites (S576, S657) are bolded. NES: nuclear exporting signal. NLS: nuclear localization signal.
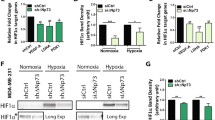
The association of Plk3 with tumor angiogenesis was discovered in a recent genetic study demonstrating that PLK3 null mice display an increased tumor incidence [19]. Importantly, tumors developed in these mice are frequently large in mass and more vasculated [19]. Further biochemical analysis revealed that Plk3 modulates HIF-1α protein levels in response to hypoxic conditions [17, 19]. Specifically, compared with wild-type mouse embryonic fibroblasts (MEFs), PLK3 null MEFs exhibit an elevated level of HIF-1α and a hyper-sensitive response to the treatment with hypoxia or with nickel ions that elicit hypoxia-like cellular responses [17, 19]. Consistently, expression of VEGF-A, a major HIF-1α response protein, is also higher in PLK3-/- MEFs than in wild-type MEFs [19]. Ex vivo analyses confirmed the role of Plk3 in regulating the stability of HIF-1α [17, 19]. Furthermore, ectopically expressed Plk3 was able to suppress the nuclear accumulation of HIF-1α in HeLa cells [19]. This inhibition of HIF-1α nuclear translocation appears to be dependent on the kinase activity of Plk3 since overexpression of only the Plk3 kinase domain is sufficient to suppress HIF-1α accumulation in the nucleus under hypoxic conditions [19]. These studies strongly suggest that Plk3 regulates tumor angiogenesis through modulating the expression level of HIF-1α.
Plk3 directly phosphorylates HIF-1α and regulates its stability
Phosphorylation of HIF-1α by several protein kinases has been reported to regulate HIF-1α stability and/or nuclear translocation [12–17, 19]. The MAP kinases, ERKs, phosphorylate HIF-1α at residues Ser-641 and Ser-643 [15] (Figure 5). This phosphorylation facilitates translocation of HIF-1α from the cytoplasm to the nucleus, thereby promoting HIF-1-mediated transcriptional activity [15, 16]. These phosphorylation sites are located far outside of the nuclear localization signal (NLS) but within the nuclear exporting signal (NES) of HIF-1α, suggesting that phosphorylation of these sites regulates nuclear export rather than import of HIF-1α [16] (Figure 5). Glycogen synthase kinase 3 β (GSK3β) phosphorylates HIF-1α at three serine residues (Ser-551, Ser-555, and Ser-589) located within its oxygen-dependent degradation domain (ODDD) [12] (Figure 5). Phosphorylation at these residues by GSK3β enhances HIF-1α degradation in a pVHL-independent manner, resulting in suppression of the HIF-1 activity [12, 13].
A recent study identifies Plk3 as a new player in the regulation of HIF-1α stability through direct phosphorylation [17]. This study was prompted by genetic evidence showing the involvement of Plk3 in tumor angiogenesis and HIF-1α regulation [19]. The most obvious possibility is that Plk3 phosphorylates HIF-1α and destabilizes HIF-1α in a manner similar to that of GSK3β [12, 13]. Kinase assays using purified recombinant HIF-1α and Plk3 reveal that Plk3, but not the kinase-dead counterpart, strongly phosphorylates HIF-1α in vitro [17]. HIF-1α phosphorylation by Plk3 results in a significant mobility shift on a denaturing gel [17]. Pull-down and Co-IP analyses confirm that Plk3 interacts with HIF-1α [17]. Two serine residues (Ser-576 and Ser-657) of HIF-1α have subsequently been identified as direct targets of Plk3 by mass spectrometry [17] (Figure 5). These two residues are evolutionarily conserved among higher animals and have not been shown to be targets of any other kinases [17]. Ser-576 is located within ODDD whereas Ser-657 is right downstream of the NES of HIF-1α (Figure 5), suggesting that Plk3 can potentially regulate both HIF-1α degradation and its nuclear localization. Further analyses reveal that recombinant HIF-1α proteins containing mutations of these residues were much more stable when they are ectopically expressed in cells [17]. Specifically, a single mutation that replaces either Ser-576 or Ser-657 with alanine greatly stabilizes the HIF-1α protein level when the mutant is expressed in HEK293 cells [17]. Replacing both residues with alanines further stabilizes HIF-1α [17], underscoring the importance of phosphorylation of these residues in controlling HIF-1α stability. It is worthwhile to note that mutation of Ser-576, which is located within the ODDD of HIF-1α, appears to be more effective in stabilizing HIF-1α than mutation of Ser-657. These results are consistent with the previous finding that phosphorylation of residues in this domain by GSK3β compromises HIF-1α stability [12]. Similar to that of GSK3β, the Plk3-dependent destabilization of HIF-1α also appears to be hydroxylation- and pVHL-independent as mutations of Plk3-phosphorylation sites can further stabilize HIF-1α with both key proline residues for hydroxylation mutated to alanines [12, 17].
Plk3 indirectly regulates HIF-1a through PTEN phosphatase
PTEN and tumor angiogenesis
The lipid phosphatase and tensin homologue (PTEN) is an important tumor suppressor that is mutated or deleted in many cancers [63, 64]. PTEN inhibits the phosphatidylinositol 3-kinases kinase (PI3K) signaling pathway by dephosphorylating the phosphoinositides on the 3' position of the inositol ring, thereby reversing the effect of PI3K leading to activation AKT [65, 66]. The close tie between PTEN and tumor angiogenesis has been well established [67–71]. PTEN is capable of suppressing tumor angiogenesis in vivo in multiple tumors [67–71]. The inhibitory effect of PTEN on tumor angiogenesis is through the inhibition of the PI3K signaling cascade leading to activation of AKT [67–71]. The PI3K pathway is known to facilitate tumor angiogenesis by increasing the HIF-1α level [5, 11, 68]. Accordingly, growth factors and cytokines, such as EGF and insulin, increase HIF-1α protein level [5], since these factors activate the PI3K pathway [66]. AKT may increase HIF-1α through its downstream targets such as mTOR or GSK3β, which regulate the protein synthesis and stability of HIF-1α, respectively [5, 11, 12]. A more recent study provided direct evidence that GSK3β directly phosphorylates HIF-1α and promotes its degradation in a hydroxylation-independent manner [12] (see previous section). In the PI3K signaling cascade, AKT directly phosphorylates GSK3β at Ser-9, thereby inhibiting its activity [72–74]. Thus, PTEN inhibits tumor angiogenesis by suppressing HIF-1 activity through the reduction of the HIF-1α protein level. This is accomplished by the inhibition of the PI3K pathway that leads to lower GSK3β activity.
Regulation of PTEN by phosphorylation
PTEN is subjected to phosphorylation by multiple kinases [18, 75–82] (Figure 6). Phosphorylation of PTEN affects PTEN activity and stability [18, 75–77, 79–85]. Most of the identified PTEN phosphorylation sites are located at the C-terminal region of the protein [18, 75–77, 79] (Figure 6). The C-terminal domain is thus considered the regulatory domain of PTEN [75, 86, 87]. Casein kinase 2 (CK2) seems to be the major kinase that phosphorylates PTEN [75–77]. The two major sites phosphorylated by CK2 are Ser-370 and Ser-385, although to a lesser extent, Thr-366, Ser-380, Ser-382, and Ser-383 [75–77]. Ser-385, in conjunction with one of the preceding serine sites (Ser-380 or Ser-382, and Ser-385) has also been proposed to be a target of liver kinase B1 [79]. GSK3β has been shown to phosphorylate PTEN at Ser-362 and Thr-366 [78] (Figure 6). Interestingly, the phosphorylation of Thr-366 is strongly enhanced by prior phosphorylation of CK2, suggesting that there is cooperative mechanism between different kinases in PTEN phosphorylation [78]. PTEN phosphorylation by Src tyrosine kinase, a microtubule-associated kinase, and RhoA associated kinase has also been reported [80–82]. A most recent study identified Plk3 as a PTEN kinase that phosphorylates Thr-366 and Ser-370 [18] (Figure 6).

Domain structure of human PTEN, showing Plk3 phosphorylation sites. Human PTEN contains an N-terminal phosphatase domain, a C-terminal C2 domain, and a PDZ binding domain. Major phosphorylation sites at the C-terminal region are indicated. Plk3 phosphorylation sites are bolded.
The effect of PTEN phosphorylation on PTEN function appears to be multifaceted. Phosphorylation of Ser-370 and Ser-385 by CK2 has been reported to suppress the PTEN activity [76, 77]. So does the tyrosine phosphorylation by Src [81]. In contrast, mutations of Ser-380, Ser-382, and Ser-383 destabilize PTEN, suggesting that phosphorylation of these sites may increase its stability [85, 88]. Phosphorylation of PTEN by CK2 at Ser-370 and Ser-385 and by Plk3 at Thr 366 and Ser-370 also stabilize PTEN [18, 77]. Interestingly, deletion of the tail-domain or mutation of Ser-380, Ser-382, and Ser-383 greatly reduces the stability PTEN but activates its phosphatase activity [84, 88]. A model has been proposed that upon phosphorylation, the C-terminal domain, stabilizes the protein due to a change in protein conformation that inhibits its phosphatase activity [84, 87, 88]. Dephoshorylation of PTEN releases the inhibitory conformation and renders PTEN more active [84, 87, 88]. However, a change in the overall PTEN level can have great biological consequences [89]. For instance, haploinsufficiency of PTEN also results in enhanced tumor development [90, 91], underscoring the importance of the PTEN protein level in controlling cell proliferation and malignant transformation. Given the fact that PTEN is phosphorylated by multiple protein kinases, it is conceivable that a correct combination of phosphorylation is needed to fine-tune the activity or level of PTEN for a given physiological state.
Plk3 phosphorylates and regulates PTEN stability
PLK3 ablation also results in significantly increased activity of the PI3K signaling pathway. Specifically, enhanced phosphorylation of Ser-473 of AKT, Ser-9 of GSK3β, and Ser-2448 of mTOR were observed in PLK3 null mouse embryonic fibroblasts (MEFs) compared to wild-type MEFs [18]. Further analyses on the upstream components of the PI3K pathway revealed that PLK3 null MEFS contained lower levels of PTEN protein [18]. These findings raised the possibility that Plk3 may directly regulate PTEN by phosphorylation. In vitro kinase assays using recombinant PTEN and Plk3 confirmed that Plk3 indeed strongly phosphorylates PTEN [18]. Mass spectrometric analysis identified Thr-366 and Ser-370 as residues that were phosphorylated by Plk3 [18]. The phosphorylation of these two sites by Plk3 was further confirmed using a phospho-specific antibody that recognized phospo-Thr-366 and phospo-Ser-370 [18].
Phosphorylation of these two sites was found to affect the stability of PTEN. PTEN constructs with Thr-366 and Ser-370 mutated to alanines were less stable when transfected into HEK 293 cells [18]. This result is therefore consistent with the reduced endogenous PTEN protein level in PLK3 null MEFs [18]. The lower PTEN level in PLK3 null MEFs corresponds to a reduced overall PTEN activity as phosphorylation of phosphoinositide-dependent protein kinase 1 (PDK-1) at Ser-241, an indirect target of PTEN [92], was enhanced in PLK3 null MEFs [18]. Thus, phosphorylation of Thr-366 and Ser-370 by Plk3 functions to stabilize PTEN, resulting in an increased overall PTEN phosphatase activity in the cell. Of note, these two sites have been reported to be the targets of CK2 [76, 77]. Thr-366 can also be phosphorylated by GSK3β [78]. Therefore, Thr-366 and Ser-370 of PTEN appear to be subjected to modification by multiple protein kinases.
PTEN stability affected by Plk3 phosphorylation appears to be proteosome-dependent since proteosome inhibitor was able to increase PTEN protein level in PLK3 null MEFs [18]. This is consistent with previous findings that PTEN is subjected to ubiquitination and degradation by the proteosome [85, 93, 94]. Notably, there is considerable phosphorylation of Ser-370 and Thr-366 of PTEN at the resting state that is readily detectable by the phospho-specific antibody [18]. This constitutive phosphorylation is partially inhibited by LiCl, an inhibitor of GSK3β and CK2 [95], supporting the previous findings that one or both of these two kinases phosphorylate these two sites [76, 77].
Given the known effect of the PI3K pathway on HIF-1α stability and the effect of Plk3 on PTEN, it is conceivable that Plk3 may affect HIF-1α stability and HIF-1 activity indirectly through regulation of the PI3K signaling pathway. Combined, these studies yield an attractive model for Plk3 regulation of HIF-1α stability/HIF-1 activity both directly by phosphorylating HIF-1α and indirectly by phosphorylating PTEN (Figure 7). This model nicely explains that the function of Plk3 has in the suppression of tumor angiogenesis, as well as tumor development in vivo.

Proposed model for Plk3 regulation of HIF-1α. Plk3 directly phosphorylates HIF-1α and facilitates its degradation. Plk3 also phosphorylates PTEN and stabilizes it, thereby increasing overall PTEN activity. PTEN inhibits the PI3K pathway and leads to activation of GSK3β, which phosphorylates and destabilize HIF-1α. Inhibition of HIF-1α by Plk3 through direct and indirect mechanisms results in reduced tumor angiogenesis.
Plk3 as a potential therapeutic target for cancer treatment
The newly discovered role of Plk3 in tumor angiogenesis highlights its potential as a therapeutic target for cancer treatment. Tumor angiogenesis has been well established as a valuable cellular process for drug intervention [2, 6, 7]. Protein kinases have been favored by the pharmaceutical industry for cancer drug development [96]. Screening small chemicals that interfere with the ATP binding capacity of kinases thereby inhibiting their kinase activities is the conventional approach for identifying drug leads [96, 97]. Among the Plk family kinases, Plk1 has been the major focus of drug development [58, 98, 99]. Several ATP-competitive and non-ATP competitive compounds have been identified as having the potential to inhibit Plk1 activity [58, 98, 99]. These compounds possess the ability, albeit highly variable, to slow cell proliferation of many cancer cell lines [58, 98, 99]. Some of these compounds have been tested for their efficacy in clinical trials for the treatment of cancers of various origins [58, 98, 99]. Many Plk1 inhibitors also block Plk3 activity to a certain extent [58]. However, in contrast to Plk1, which plays a positive role in cell proliferation and tumor development, Plk3 is regarded as having a tumor-suppressing function [21, 30]. The latest study on the effect of Plk3 on tumor angiogenesis has provided results that are consistence with this notion [18]. It is conceivable that a chemical compound that inhibits Plk3 activity specifically might have no beneficial effect to cancer treatment or management. Therefore, it is likely that the anti-tumor effect of those compounds which have an activity toward both Plk1 and Plk3 may result from Plk1 inhibition. Consequently, the discovery of Plk3 activators would be a more rational approach for anti-cancer drug development.
Targeting Plk3-mediated tumor angiogenesis may offer several advantages over Plk1 in cancer treatment. It have been shown that Plk1 knock-down via RNAi results in cell cycle arrest followed by apoptosis and that Plk1 knock-out mice is embryonically lethal [32, 100]. These results suggest that Plk1 is vital for normal cell proliferation and survival. Consequently, inhibition of Plk1 is likely to have a detrimental effect on normal cells in addition to cancer cells. In contrast, PLK3 ablation in mice does not noticeably perturb animal development except for enhanced tumor angiogenesis in mice of advanced age [19]. Therefore targeting Plk3 would likely to have less side-effect in vivo. In addition, therapeutic intervention that activates Plk3 should lead to enhanced apoptosis in targeting cells as the effect has been observed in cells that overexpress Plk3 [21].
Concluding remarks
In spite of extensive research in the past which has yielded a sizable body of knowledge about the biochemical and cellular functions of Plk3, it remains as a protein kinase we know very little about, especially with regard to its physiological and patho-physiological roles. Mouse genetic studies have yielded valuable information about Plk3's role in suppressing tumor angiogenesis through negative regulation of the PI3K signaling network and the HIF-1 activity. Additional analyses are clearly needed to fully understand the physiological role of PLK3 in animal development and stress responses. Given its documented roles in suppressing cell proliferation in vitro and tumor development in vivo, Plk3 represents an attractive target for the development of anti-cancer compounds.
References
Pugh CW, Ratcliffe PJ: Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 2003, 9: 677–684. 10.1038/nm0603-677
Folkman J: Angiogenesis. Annu Rev Med 2006, 57: 1–18. 10.1146/annurev.med.57.121304.131306
Liao D, Johnson RS: Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev 2007, 26: 281–290. 10.1007/s10555-007-9066-y
Folkman J: Tumor angiogenesis: therapeutic implications. N Engl J Med 1971, 285: 1182–1186. 10.1056/NEJM197111182852108
Semenza GL: Targeting HIF-1 for cancer therapy. Nat Rev Cancer 2003, 3: 721–732. 10.1038/nrc1187
Heath VL, Bicknell R: Anticancer strategies involving the vasculature. Nat Rev Clin Oncol 2009, 6: 395–404. 10.1038/nrclinonc.2009.52
Jain RK: Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005, 307: 58–62. 10.1126/science.1104819
Zetter BR: Angiogenesis and tumor metastasis. Annu Rev Med 1998, 49: 407–424. 10.1146/annurev.med.49.1.407
Kaelin WG Jr, Ratcliffe PJ: Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 2008, 30: 393–402. 10.1016/j.molcel.2008.04.009
Ke Q, Costa M: Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol 2006, 70: 1469–1480. 10.1124/mol.106.027029
Yee-Koh M, Spivak-Kroizman TR, Powis G: HIF-1 regulation: not so easy come, easy go. Trends Biochem Sci 2008, 33: 526–534. 10.1016/j.tibs.2008.08.002
Flugel D, Gorlach A, Michiels C, Kietzmann T: Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1alpha and mediates its destabilization in a VHL-independent manner. Mol Cell Biol 2007, 27: 3253–3265. 10.1128/MCB.00015-07
Mottet D, Dumont V, Deccache Y, Demazy C, Ninane N, Raes M, Michiels C: Regulation of hypoxia-inducible factor-1alpha protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta pathway in HepG2 cells. J Biol Chem 2003, 278: 31277–31285. 10.1074/jbc.M300763200
Richard DE, Berra E, Gothie E, Roux D, Pouyssegur J: p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J Biol Chem 1999, 274: 32631–32637. 10.1074/jbc.274.46.32631
Mylonis I, Chachami G, Samiotaki M, Panayotou G, Paraskeva E, Kalousi A, Georgatsou E, Bonanou S, Simos G: Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1alpha. J Biol Chem 2006, 281: 33095–33106. 10.1074/jbc.M605058200
Mylonis I, Chachami G, Paraskeva E, Simos G: Atypical CRM1-dependent nuclear export signal mediates regulation of hypoxia-inducible factor-1alpha by MAPK. J Biol Chem 2008, 283: 27620–27627. 10.1074/jbc.M803081200
Xu D, Yao Y, Lu L, Costa M, Dai W: Plk3 functions as an essential component of the hypoxia regulatory pathway by direct phosphorylation of HIF-1alpha. J Biol Chem 2010, 285: 38944–38950. 10.1074/jbc.M110.160325
Xu D, Yao Y, Jiang X, Lu L, Dai W: Regulation of PTEN stability and activity by Plk3. J Biol Chem 2010, 285: 39935–39942. 10.1074/jbc.M110.166462
Yang Y, Bai J, Shen R, Brown SA, Komissarova E, Huang Y, Jiang N, Alberts GF, Costa M, Lu L, Winkles JA, Dai W: Polo-like kinase 3 functions as a tumor suppressor and is a negative regulator of hypoxia-inducible factor-1 alpha under hypoxic conditions. Cancer Res 2008, 68: 4077–4085. 10.1158/0008-5472.CAN-07-6182
Fenton B, Glover DM: A conserved mitotic kinase active at late anaphase-telophase in syncytial Drosophila embryos. Nature 1993, 363: 637–640. 10.1038/363637a0
Dai W: Polo-like kinases, an introduction. Oncogene 2005, 24: 214–216. 10.1038/sj.onc.1208270
Andrysik Z, Bernstein WZ, Deng L, Myer DL, Li YQ, Tischfield JA, Stambrook PJ, Bahassi el M: The novel mouse Polo-like kinase 5 responds to DNA damage and localizes in the nucleolus. Nucleic Acids Res 2010, 38: 2931–43. 10.1093/nar/gkq011
Lowery DM, Lim D, Yaffe MB: Structure and function of Polo-like kinases. Oncogene 2005, 24: 248–259. 10.1038/sj.onc.1208280
Archambault V, Glover DM: Polo-like kinases: conservation and divergence in their functions and regulation. Nat Rev Mol Cell Biol 2009, 10: 265–275. 10.1038/nrm2653
Winkles JA, Alberts GF: Differential regulation of polo-like kinase 1, 2, 3, and 4 gene expression in mammalian cells and tissues. Oncogene 2005, 24: 260–266. 10.1038/sj.onc.1208219
Seeburg DP, Pak D, Sheng M: Polo-like kinases in the nervous system. Oncogene 2005, 24: 292–298. 10.1038/sj.onc.1208277
Wang L, Payton R, Dai W, Lu L: Hyperosmotic stress-induced ATF-2 activation through Polo-like kinase 3 in human corneal epithelial cells. J Biol Chem 2011, 286: 1951–1958. 10.1074/jbc.M110.166009
Wang L, Gao J, Dai W, Lu L: Activation of Polo-like kinase 3 by hypoxic stresses. J Biol Chem 2008, 283: 25928–25935. 10.1074/jbc.M801326200
Wang L, Dai W, Lu L: Stress-induced c-Jun activation mediated by Polo-like kinase 3 in corneal epithelial cells. J Biol Chem 2007, 282: 32121–32127. 10.1074/jbc.M702791200
Eckerdt F, Yuan J, Strebhardt K: Polo-like kinases and oncogenesis. Oncogene 2005, 24: 267–276. 10.1038/sj.onc.1208273
Takai N, Hamanaka R, Yoshimatsu J, Miyakawa I: Polo-like kinases (Plks) and cancer. Oncogene 2005, 24: 287–291. 10.1038/sj.onc.1208272
Lu LY, Wood JL, Minter-Dykhouse K, Ye L, Saunders TL, Yu X, Chen J: Polo-like kinase 1 is essential for early embryonic development and tumor suppression. Mol Cell Biol 2008, 28: 6870–6876. 10.1128/MCB.00392-08
Li B, Ouyang B, Pan H, Reissmann PT, Slamon DJ, Arceci R, Lu L, Dai W: Prk, a cytokine-inducible human protein serine/threonine kinase whose expression appears to be down-regulated in lung carcinomas. J Biol Chem 1996, 271: 19402–19408. 10.1074/jbc.271.32.19402
Dai W, Li Y, Ouyang B, Pan H, Reissmann P, Li J, Wiest J, Stambrook P, Gluckman JL, Noffsinger A, Bejarano P: PRK, a cell cycle gene localized to 8p21, is downregulated in head and neck cancer. Genes Chromosomes Cancer 2000, 27: 332–336. 10.1002/(SICI)1098-2264(200003)27:3<332::AID-GCC15>3.0.CO;2-K
Donohue PJ, Alberts GF, Guo Y, Winkles JA: Identification by targeted differential display of an immediate early gene encoding a putative serine/threonine kinase. J Biol Chem 1995, 270: 10351–10357. 10.1074/jbc.270.17.10351
Bahassi el M, Conn CW, Myer DL, Hennigan RF, McGowan CH, Sanchez Y, Stambrook PJ: Mammalian Polo-like kinase 3 (Plk3) is a multifunctional protein involved in stress response pathways. Oncogene 2002, 21: 6633–6640. 10.1038/sj.onc.1205850
Chase D, Feng Y, Hanshew B, Winkles JA, Longo DL, Ferris DK: Expression and phosphorylation of fibroblast-growth-factor-inducible kinase (Fnk) during cell-cycle progression. Biochem J 1998,333(Pt 3):655–660.
Zimmerman WC, Erikson RL: Polo-like kinase 3 is required for entry into S phase. Proc Natl Acad Sci USA 2007, 104: 1847–1852. 10.1073/pnas.0610856104
Xie S, Wu H, Wang Q, Cogswell JP, Husain I, Conn C, Stambrook P, Jhanwar-Uniyal M, Dai W: Plk3 functionally links DNA damage to cell cycle arrest and apoptosis at least in part via the p53 pathway. J Biol Chem 2001, 276: 43305–43312. 10.1074/jbc.M106050200
Xie S, Wu H, Wang Q, Kunicki J, Thomas RO, Hollingsworth RE, Cogswell J, Dai W: Genotoxic stress-induced activation of Plk3 is partly mediated by Chk2. Cell Cycle 2002, 1: 424–429. 10.4161/cc.1.6.271
Kis E, Szatmari T, Keszei M, Farkas R, Esik O, Lumniczky K, Falus A, Safrany G: Microarray analysis of radiation response genes in primary human fibroblasts. Int J Radiat Oncol Biol Phys 2006, 66: 1506–1514. 10.1016/j.ijrobp.2006.08.004
Xie S, Xie B, Lee MY, Dai W: Regulation of cell cycle checkpoints by polo-like kinases. Oncogene 2005, 24: 277–286. 10.1038/sj.onc.1208218
Wang Q, Xie S, Chen J, Fukasawa K, Naik U, Traganos F, Darzynkiewicz Z, Jhanwar-Uniyal M, Dai W: Cell cycle arrest and apoptosis induced by human Polo-like kinase 3 is mediated through perturbation of microtubule integrity. Mol Cell Biol 2002, 22: 3450–3459. 10.1128/MCB.22.10.3450-3459.2002
Ouyang B, Li W, Pan H, Meadows J, Hoffmann I, Dai W: The physical association and phosphorylation of Cdc25C protein phosphatase by Prk. Oncogene 1999, 18: 6029–6036. 10.1038/sj.onc.1202983
Bahassi el M, Hennigan RF, Myer DL, Stambrook PJ: Cdc25C phosphorylation on serine 191 by Plk3 promotes its nuclear translocation. Oncogene 2004, 23: 2658–2663. 10.1038/sj.onc.1207425
Lopez-Girona A, Furnari B, Mondesert O, Russell P: Nuclear localization of Cdc25 is regulated by DNA damage and a 14–3-3 protein. Nature 1999, 397: 172–175. 10.1038/16488
Zimmerman WC, Erikson RL: Finding Plk3. Cell Cycle 2007, 6: 1314–1318. 10.4161/cc.6.11.4275
Iida M, Matsuda M, Komatani H: Plk3 phosphorylates topoisomerase IIalpha at Thr(1342), a site that is not recognized by Plk1. Biochem J 2008, 411: 27–32. 10.1042/BJ20071394
Wang JC: Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol 2002, 3: 430–440. 10.1038/nrm831
Lopez-Sanchez I, Sanz-Garcia M, Lazo PA: Plk3 interacts with and specifically phosphorylates VRK1 in Ser342, a downstream target in a pathway that induces Golgi fragmentation. Mol Cell Biol 2009, 29: 1189–1201. 10.1128/MCB.01341-08
Ruan Q, Wang Q, Xie S, Fang Y, Darzynkiewicz Z, Guan K, Jhanwar-Uniyal M, Dai W: Polo-like kinase 3 is Golgi localized and involved in regulating Golgi fragmentation during the cell cycle. Exp Cell Res 2004, 294: 51–59. 10.1016/j.yexcr.2003.10.022
Xie S, Wang Q, Ruan Q, Liu T, Jhanwar-Uniyal M, Guan K, Dai W: MEK1-induced Golgi dynamics during cell cycle progression is partly mediated by Polo-like kinase-3. Oncogene 2004, 23: 3822–3829. 10.1038/sj.onc.1207479
Conn CW, Hennigan RF, Dai W, Sanchez Y, Stambrook PJ: Incomplete cytokinesis and induction of apoptosis by overexpression of the mammalian polo-like kinase, Plk3. Cancer Res 2000, 60: 6826–6831.
Li Z, Niu J, Uwagawa T, Peng B, Chiao PJ: Function of polo-like kinase 3 in NF-kappaB-mediated proapoptotic response. J Biol Chem 2005, 280: 16843–16850. 10.1074/jbc.M410119200
Xie S, Wang Q, Wu H, Cogswell J, Lu L, Jhanwar-Uniyal M, Dai W: Reactive oxygen species-induced phosphorylation of p53 on serine 20 is mediated in part by polo-like kinase-3. J Biol Chem 2001, 276: 36194–36199. 10.1074/jbc.M104157200
Kruse JP, Gu W: Modes of p53 regulation. Cell 2009, 137: 609–622. 10.1016/j.cell.2009.04.050
Dai W, Liu T, Wang Q, Rao CV, Reddy BS: Down-regulation of PLK3 gene expression by types and amount of dietary fat in rat colon tumors. Int J Oncol 2002, 20: 121–126.
Dai W, Yang Y, Jiang N: Plks as Novel Targets for Cancer Drug Design. In Cancer Drug Discovery and Development. Edited by: Dai W. Totowa: Humana Press; 2008:271–290.
Wiest J, Clark AM, Dai W: Intron/exon organization and polymorphisms of the PLK3/PRK gene in human lung carcinoma cell lines. Genes Chromosomes Cancer 2001, 32: 384–389. 10.1002/gcc.1204
Bockmuhl U, Wolf G, Schmidt S, Schwendel A, Jahnke V, Dietel M, Petersen I: Genomic alterations associated with malignancy in head and neck cancer. Head Neck 1998, 20: 145–151. 10.1002/(SICI)1097-0347(199803)20:2<145::AID-HED8>3.0.CO;2-2
Spurr NK, Blanton S, Bookstein R, Clarke R, Cottingham R, Daiger S, Drayna D, Faber P, Horrigan S, Kas K, et al.: Report and abstracts of the second international workshop on human chromosome 8 mapping 1994. Oxford, United Kingdom, September 16–18, 1994. Cytogenet Cell Genet 1995, 68: 147–164. 10.1159/000133908
Wang GL, Jiang BH, Rue EA, Semenza GL: Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995, 92: 5510–5514. 10.1073/pnas.92.12.5510
Cully M, You H, Levine AJ, Mak TW: Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer 2006, 6: 184–192. 10.1038/nrc1819
Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R: PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275: 1943–1947. 10.1126/science.275.5308.1943
Whitman M, Downes CP, Keeler M, Keller T, Cantley L: Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature 1988, 332: 644–646. 10.1038/332644a0
Liu P, Cheng H, Roberts TM, Zhao JJ: Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov 2009, 8: 627–644. 10.1038/nrd2926
Hamada K, Sasaki T, Koni PA, Natsui M, Kishimoto H, Sasaki J, Yajima N, Horie Y, Hasegawa G, Naito M, Miyazaki J, Suda T, Itoh H, Nakao K, Mak TW, Nakano T, Suzuki A: The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev 2005, 19: 2054–2065. 10.1101/gad.1308805
Jiang BH, Zheng JZ, Aoki M, Vogt PK: Phosphatidylinositol 3-kinase signaling mediates angiogenesis and expression of vascular endothelial growth factor in endothelial cells. Proc Natl Acad Sci USA 2000, 97: 1749–1753. 10.1073/pnas.040560897
Ma J, Sawai H, Ochi N, Matsuo Y, Xu D, Yasuda A, Takahashi H, Wakasugi T, Takeyama H: PTEN regulates angiogenesis through PI3K/Akt/VEGF signaling pathway in human pancreatic cancer cells. Mol Cell Biochem 2009, 331: 161–171. 10.1007/s11010-009-0154-x
Wen S, Stolarov J, Myers MP, Su JD, Wigler MH, Tonks NK, Durden DL: PTEN controls tumor-induced angiogenesis. Proc Natl Acad Sci USA 2001, 98: 4622–4627. 10.1073/pnas.081063798
Ueda S, Basaki Y, Yoshie M, Ogawa K, Sakisaka S, Kuwano M, Ono M: PTEN/Akt signaling through epidermal growth factor receptor is prerequisite for angiogenesis by hepatocellular carcinoma cells that is susceptible to inhibition by gefitinib. Cancer Res 2006, 66: 5346–5353. 10.1158/0008-5472.CAN-05-3684
Sutherland C, Leighton IA, Cohen P: Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J 1993,296(Pt 1):15–19.
Stambolic V, Woodgett JR: Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem J 1994,303(Pt 3):701–704.
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA: Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378: 785–789. 10.1038/378785a0
Gericke A, Munson M, Ross AH: Regulation of the PTEN phosphatase. Gene 2006, 374: 1–9.
Miller SJ, Lou DY, Seldin DC, Lane WS, Neel BG: Direct identification of PTEN phosphorylation sites. FEBS Lett 2002, 528: 145–153. 10.1016/S0014-5793(02)03274-X
Torres J, Pulido R: The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem 2001, 276: 993–998. 10.1074/jbc.M009134200
Al-Khouri AM, Ma Y, Togo SH, Williams S, Mustelin T: Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3beta. J Biol Chem 2005, 280: 35195–35202. 10.1074/jbc.M503045200
Mehenni H, Lin-Marq N, Buchet-Poyau K, Reymond A, Collart MA, Picard D, Antonarakis SE: LKB1 interacts with and phosphorylates PTEN: a functional link between two proteins involved in cancer predisposing syndromes. Hum Mol Genet 2005, 14: 2209–2219. 10.1093/hmg/ddi225
Valiente M, Andres-Pons A, Gomar B, Torres J, Gil A, Tapparel C, Antonarakis SE, Pulido R: Binding of PTEN to specific PDZ domains contributes to PTEN protein stability and phosphorylation by microtubule-associated serine/threonine kinases. J Biol Chem 2005, 280: 28936–28943. 10.1074/jbc.M504761200
Lu Y, Yu Q, Liu JH, Zhang J, Wang H, Koul D, McMurray JS, Fang X, Yung WK, Siminovitch KA, Mills GB: Src family protein-tyrosine kinases alter the function of PTEN to regulate phosphatidylinositol 3-kinase/AKT cascades. J Biol Chem 2003, 278: 40057–40066. 10.1074/jbc.M303621200
Li Z, Dong X, Wang Z, Liu W, Deng N, Ding Y, Tang L, Hla T, Zeng R, Li L, Wu D: Regulation of PTEN by Rho small GTPases. Nat Cell Biol 2005, 7: 399–404. 10.1038/ncb1236
Adey NB, Huang L, Ormonde PA, Baumgard ML, Pero R, Byreddy DV, Tavtigian SV, Bartel PL: Threonine phosphorylation of the MMAC1/PTEN PDZ binding domain both inhibits and stimulates PDZ binding. Cancer Res 2000, 60: 35–37.
Vazquez F, Grossman SR, Takahashi Y, Rokas MV, Nakamura N, Sellers WR: Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J Biol Chem 2001, 276: 48627–48630. 10.1074/jbc.C100556200
Tolkacheva T, Boddapati M, Sanfiz A, Tsuchida K, Kimmelman AC, Chan AM: Regulation of PTEN binding to MAGI-2 by two putative phosphorylation sites at threonine 382 and 383. Cancer Res 2001, 61: 4985–4989.
Odriozola L, Singh G, Hoang T, Chan AM: Regulation of PTEN activity by its carboxyl-terminal autoinhibitory domain. J Biol Chem 2007, 282: 23306–23315. 10.1074/jbc.M611240200
Rahdar M, Inoue T, Meyer T, Zhang J, Vazquez F, Devreotes PN: A phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. Proc Natl Acad Sci USA 2009, 106: 480–485. 10.1073/pnas.0811212106
Vazquez F, Ramaswamy S, Nakamura N, Sellers WR: Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol 2000, 20: 5010–5018. 10.1128/MCB.20.14.5010-5018.2000
Carracedo A, Alimonti A, Pandolfi PP: PTEN level in tumor suppression: how much is too little? Cancer Res 2011, 71: 629–633. 10.1158/0008-5472.CAN-10-2488
Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, Khoo AS, Roy-Burman P, Greenberg NM, Van Dyke T, Cordon-Cardo C, Pandolfi PP: Pten dose dictates cancer progression in the prostate. PLoS Biol 2003, 1: E59.
Kwabi-Addo B, Giri D, Schmidt K, Podsypanina K, Parsons R, Greenberg N, Ittmann M: Haploinsufficiency of the Pten tumor suppressor gene promotes prostate cancer progression. Proc Natl Acad Sci USA 2001, 98: 11563–11568. 10.1073/pnas.201167798
Casamayor A, Morrice NA, Alessi DR: Phosphorylation of Ser-241 is essential for the activity of 3-phosphoinositide-dependent protein kinase-1: identification of five sites of phosphorylation in vivo. Biochem J 1999,342(Pt 2):287–292.
Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, Wang J, Erdjument-Bromage H, Tempst P, Cordon-Cardo C, Pandolfi PP, Jiang X: NEDD4–1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell 2007, 128: 129–139. 10.1016/j.cell.2006.11.039
Wu W, Wang X, Zhang W, Reed W, Samet JM, Whang YE, Ghio AJ: Zinc-induced PTEN protein degradation through the proteasome pathway in human airway epithelial cells. J Biol Chem 2003, 278: 28258–28263. 10.1074/jbc.M303318200
Davies SP, Reddy H, Caivano M, Cohen P: Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 2000, 351: 95–105. 10.1042/0264-6021:3510095
Cohen P: Protein kinases-the major drug targets of the twenty-first century? Nat Rev Drug Discov 2002, 1: 309–315. 10.1038/nrd773
Noble ME, Endicott JA, Johnson LN: Protein kinase inhibitors: insights into drug design from structure. Science 2004, 303: 1800–1805. 10.1126/science.1095920
Strebhardt K: Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov 2010, 9: 643–660. 10.1038/nrd3184
Schoffski P: Polo-like kinase (PLK) inhibitors in preclinical and early clinical development in oncology. Oncologist 2009, 14: 559–570. 10.1634/theoncologist.2009-0010
Gleixner KV, Ferenc V, Peter B, Gruze A, Meyer RA, Hadzijusufovic E, Cerny-Reiterer S, Mayerhofer M, Pickl WF, Sillaber C, Valent P: Polo-like kinase 1 (Plk1) as a novel drug target in chronic myeloid leukemia: overriding imatinib resistance with the Plk1 inhibitor BI 2536. Cancer Res 2010, 70: 1513–23. 10.1158/0008-5472.CAN-09-2181
Acknowledgements
We thank co-workers in the laboratory for valuable discussions and suggestions. This study was supported in part by US Public Service Awards (CA113349 and ES019929), NIEHS Center grants (ES000260), and a key State project of China on iPS and stem cell research (2011ZX09102-010-04).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
DX: Designed and executed experiments and also involved in writing the manuscript. QW: Designed and performed experiments. YJ: Conceived experiments and interpreted data. YZ: Carried out experiments. EV-SM: Carried out experiments. IO: Data interpretation and discussion. WD: Direct the study; data interpretation, manuscript writing. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Xu, D., Wang, Q., Jiang, Y. et al. Roles of Polo-like kinase 3 in suppressing tumor angiogenesis. Exp Hematol Oncol 1, 5 (2012). https://doi.org/10.1186/2162-3619-1-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2162-3619-1-5