
Abstract
Background and aims
Smoking is the main risk factor for the development of chronic obstructive pulmonary disease (COPD) that has been recently defined as a systemic pulmonary inflammatory disease. However, the impact of smoking itself on systemic inflammation in COPD patients has not yet been well established. The aim of our study was to investigate the association between inflammatory markers and smoking status.
Materials and methods
We compared 202 current smokers, 61 ex-smokers and 57 never-smokers, all COPD patients. Assessments included medical history, spirometry, alpha-1 antitrypsin (AAT) genotyping, serum AAT, C-reactive protein (CRP), tumor necrosis factor (TNF)-α, and soluble tumor necrosis factor receptor (sTNFR)-1 and sTNFR-2 concentrations.
Results
AAT and CRP concentrations in smokers (1.75 ± 0.51 g/L and 14.4 [9.5-20.5] mg/L) and ex-smokers (1.69 ± 0.43 g/L and 12.3 [8.7-16.3] mg/L) were higher than in never-smokers (1.49 ± 0.38 g/L and 5.1 [2.5-8.7] mg/L; p < 0.05). sTNFR-1 level was higher in smokers than ex-smokers or never-smokers (241.2 pg/mL [145.3-349.4] vs. 213.7 pg/mL [147.1-280.3] and 205.2 pg/mL [125-275]; p < 0.05).
Conclusions
Our data confirm that smoking is associated with increased levels of AAT, CRP, and sTNFR-1 in COPD patients, an array of systemic inflammation markers that continue to be active even after smoking cessation.
Riassunto
Introduzione e obiettivi
Il fumo è la principale causa della broncopneumopatia cronica ostruttiva (BPCO), patologia che è stata recentemente definita come una malattia infiammatoria sistemica. Tuttavia, i dati relativi all'influenza del fumo sull’infiammazione sistemica per i malati di BPCO non sono sufficienti. Scopo della ricerca è quello di stabilire i collegamenti tra i marcatori dell'infiammazione e l'abitudine al fumo.
Materiali e metodi
Sono stati presi in esame 320 malati di BPCO: 202 fumatori, 61 ex fumatori, 57 non fumatori. Sono stati valutati: anamnesi, spirometria, genotipo di alfa-1 antitripsina (AAT), concentrazioni seriche di AAT, proteina C reattiva (CRP), fattore di necrosi tumorale alfa (TNF-α) e delle frazioni solubili dei recettori per il fattore di necrosi tumorale (sTNFR-1, sTNFR-2).
Risultati
I livelli di AAT e di CRP dei fumatori (1,75 ± 0,51 g/l e 14,4 mg/l [9,5–20,5]) e negli ex-fumatori (1,69 ± 0,43 g/l e 12,3 mg/l [8,7–16,3]) sono risultati più alti dei non fumatori (1,49 ± 0,38 g/l e 5,1 mg/l [2,5–8,7]; p < 0,05). I livelli di TNFR-1 sono risultati più alti nei fumatori rispetto agli ex-fumatori e ai non fumatori (241,2 pg/ml [145,3–349,4] versus 213,7 pg/ml [147,1–280,3] e 205,2 pg/ml [125–275]; p < 0,05).
Conclusioni
L’aumento delle concentrazioni di AAT e CRP nel siero dei pazienti fumatori ed ex-fumatori con BPCO, rispetto ai non fumatori, indica che l’infiammazione sistemica si prolunga anche dopo la cessazione del fumo, anche se le concentrazioni di sTNFR risultano inferiori dopo la cessazione.
Similar content being viewed by others


Introduction
Chronic obstructive pulmonary disease (COPD) is a prevalent and costly disease characterized by progressive airflow limitation related to an abnormal inflammatory response of the lungs to long-term tobacco smoke or inhalation of toxic gases [1]. The prevalence of COPD is appreciably higher in current or former heavy smokers aged > 40 years. Smoking may lead to COPD in 15-30% of those who smoke [2]. There is a growing body of evidence supporting the presence of systemic inflammation in patients with COPD [3–8]. Tumor necrosis factor α (TNF-α), a powerful proinflammatory cytokine primarily produced by activated macrophages, is thought to play a critical role in the pathogenesis of COPD by promoting and maintaining the expression and release of various proinflammatory mediators, which lead to tissue damage and remodeling [8, 9]. Lung inflammation is further amplified by oxidative stress and proteolytic damage by proteinases. Smoking itself may also cause a pro-tease-antiprotease imbalance in the lungs by reducing the functional activity of the major antiprotease - alpha-1 antitrypsin (AAT) - in the lung interstitium and alveolar lining fluid, and by increasing the amount of elastolytic proteases released in the lung [10]. Moreover, smoking increases the number of neutrophils, macrophages, and T cells, which release proinflammatory mediators into the lung and airways [11]. A number of studies suggest that AAT can also exhibit biological activities independently of the inhibition of serine proteases: namely, an immunoregulatory effect, inhibition of neutrophil superoxide production, and antimicrobial activity [12].
Little is known about the mechanism responsible for increased levels of inflammatory markers, especially TNF-α and its inhibitory soluble receptors, in the serum of COPD patients, and a direct impact of active smoking on these markers has not been well established. The aim of our study was to evaluate the concentrations of AAT, C-reactive protein (CRP), TNF-α, and soluble TNF receptors 1 and 2 (sTNFR-1 and sTNFR-2) in COPD patients in relation to smoking habit.
Materials and methods
Subjects
A total of 320 stable outpatients with COPD were examined: 202 current smokers, 61 ex-smokers (quit since at least 1 year), and 57 never-smokers followed up at the Department of Pulmonology and Immunology, Medical Academy, Lithuanian University of Health Sciences.
Patients met the following inclusion criteria: no use of oral corticosteroids or leukotrienes for at least 6 weeks prior to the study; none of the subjects showed signs of acute respiratory infection at least 1 month before the investigation; patients were free of systemic steroids for at least 1 month before the study; none of the patients had congenital AAT deficiency. All patients met the Global Initiative for Obstructive Lung Disease (GOLD) [1] recommended spirometric criteria for COPD (ratio of postbronchodilator forced expiratory volume in one second [FEV1] to forced vital capacity [FVC] less than 70%). When grouping the patients into smokers, ex-smokers, and never-smokers, we relied only on reported smoking status. Several studies [13] have reported trends of underestimation when smoking prevalence is based on self-report, and varying sensitivity levels for self-reported estimates depending on the population studied and the medium in which the biological sample is measured [13]. However, there are studies showing that reported smoking could be an acceptable assessment because prevalence estimates of smoking were similar to self-reports and cotinine assessment [14].
Smoking history was calculated in pack-years as the product of tobacco use (in years) and the average number of cigarettes smoked per day/20 (years x cig. per day/20). The study design was approved by the Regional Ethics Committee, and all studied subjects gave their informed consent.
Sample collection and evaluation
Blood samples were drawn in serum tubes, clotted at room temperature for 30-60 min, and centrifuged for 15 min at 4000 rpm. Serum samples were immediately frozen at -70°C for further analysis. AAT phenotyping was carried out by means of isoelectric focusing (LKB Multiphor II and LKB Macrodrive 5 Constant Power Supply, Amersham Pharmacia Biotech, Piscataway, NJ, USA). Serum AAT concentration was determined by nephelometry (Dade Behring Marburg GmbH, Germany) according to the manufacturer's instructions. TNF-α, sTNFR-1, and sTNFR-2 levels were determined using Duosit ELISA sets (R&D Systems, MN, USA; detection levels, 125, 15.6, and 25 pg/mL, respectively). Analysis of CRP in serum was performed using standard assays (Dade Behring, USA, minimum detection level less than 0.15 mg/L).
Statistical analysis
Descriptive statistics were used to tabulate the primary cohort database. Quantitative variables were expressed as means ± standard deviation (SD) or median (25th-75th percentile). Normally distributed data were compared using Student's t test and one-way ANOVA. The Mann-Whitney U and Kruskal-Wallis H tests were used for comparison of non normally distributed data to determine if they differed significantly. A p value of less than 0.05 was considered significant. Statistical analysis was performed using the SPSS 14.0 program.
Results
The majority of the patients were male (n = 250, 78.1%), mostly with moderate-to-severe COPD (Table 1). Mean age was 63.4 ± 11.9 years, and 82% of the patients were current or former smokers with an average smoking history of 22.7 ± 12.9 pack-years. Serum levels of biomarkers AAT, CRP, TNF-α, sTNFR-1, and sTNFR-2 are presented in Table 1. We found positive associations between active and former smoking habit and higher AAT and CRP levels. Figure 1 shows that AAT concentration was higher in smokers (1.75 ± 0.51 g/L) and ex-smokers (1.69 ± 0.43 g/L) than in never-smokers (1.49 ± 0.38 g/L) (p < 0.05). CRP concentration was also higher in smokers and ex-smokers than never-smokers (14.4 [9.5-20.5] mg/L and 12.3 [8.7-16.3] mg/L vs. 5.1 [2.5-8.7] mg/L; p < 0.05) (Figure 2).

Serum AAT concentration in COPD smokers, ex-smokers and never-smokers. Definition of abbreviation: AAT, alpha-1 antitrypsin. Data are presented as mean ± SD.

Serum CRP concentration in COPD smokers, ex-smokers and never-smokers. Definition of abbreviation: CRP, C-reactive protein. Data are presented as median (25th-75th percentile).
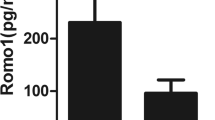
sTNFR-1 levels were higher in smokers than ex-smokers or never-smokers (241.2 [145.3-349.4] pg/mL vs. 213.7 [147.1-280.3] pg/mL and 205.2 [125-275] pg/mL; p < 0.05) (Figure 3).

Serum sTNFR-1 concentration in COPD smokers, ex-smokers and never-smokers. Definition of abbreviation: sTNFR-1, soluble tumor necrosis factor α receptor 1. Data are presented as median (25th-75th percentile).
No differences in TNF-α and sTNFR-2 concentrations were observed in relation to smoking status (p > 0.05) (Table 2).
Discussion
An important message of this study is that current smokers and ex-smokers had higher circulating AAT and CRP levels than never-smokers. These results suggest that smoking may be associated with higher AAT and CRP production in the liver of COPD patients and mechanisms related to systemic inflammation, which continues even after smoking cessation. Even in healthy individuals, positive associations between active smoking and AAT levels have been reported [15]. The amount of AAT that diffuses passively from the blood to the lungs increases during an inflammatory process, as occurs in COPD [16]. This may indicate an increased requirement of AAT to meet the need of overcoming the release of various enzymes from neutrophilic cells in the lungs, but its protective function may be overwhelmed by the high concentration of proteases [17]. Elevated AAT levels in smokers and ex-smokers reflect the dual role of AAT as a respiratory disease biomarker. The net impact of AAT on lung function seems to be a result of context-dependent (i.e. AAT genotype) and contrasting protective and inflammatory effects in the respiratory tract. On the one hand, elevated serum AAT can show a beneficial shift in the protease-antiprotease balance, the centrepiece of the pathophysiological pathway mediating the effect of severe congenital AAT deficiency on COPD. On the other hand, elevated serum AAT can also reflect a low-grade chronic inflammatory process in the lungs [18], which is considered a risk factor for COPD [19, 20].
CRP reflects the total systemic burden of inflammation in several disorders and has been shown to up-regulate the production of proinflammatory cyto kines [5, 7]. Gan and co-workers were the first to highlight the importance of high CRP levels in COPD patients, confirming the systemic inflammation in the stable phase of the disease [19]. Studies of circulating CRP in COPD have demonstrated that CRP levels are further elevated during exacerbations, and they were also found to be a mortality predictor [21]. Several previous studies have also shown higher serum CRP levels in COPD patients than in non-smoking controls [22, 23]. However, other studies did not find such differences in CRP levels [24]. These controversial data indicate that each patient has his/her own unique and different mechanism of disease development.
There is a consensus about the presence of small airway and lung parenchyma inflammation in smokers and COPD patients [25–27]. Local inflammation is characterized by increased numbers of inflammatory cells, such as neutrophils, lymphocytes, and macrophages, and higher TNF-α and IL-8 concentrations in smokers than healthy controls [25, 26, 28–30]. However, after smoking cessation, the inflammatory state changes only in asymptomatic ex-smokers, but not in COPD ex-smokers [31, 32]. Gamble et al. compared bronchial biopsy specimens from COPD current smokers and COPD ex-smokers and did not find any differences in cell counts or inflammatory markers, including TNF-α, between the groups [31]. TNF-α is an inflammatory cytokine important not only for COPD pathogenesis [3, 33], but also for the function of the main neutrophil protease inhibitor - AAT [12, 34]. We found no associations between TNF-α serum concentration and smoking status in our study. Some studies have reported elevated TNF-α levels in the sputum, bronchial biopsy specimens, and circulation of COPD patients as compared to controls [3, 19]. However, other authors did not find such associations [35].
On the contrary, in our study, sTNFR-1 level was higher in smokers than ex-smokers and never-smokers. These soluble receptors, which inhibit the inflammatory effect of TNF-α, are expressed and released from many different cells, causing also the elevation of their concentration in systemic circulation, where they can be detected [8, 36]. Our results show that elevated sTNFR-1 levels in smokers may show a systemic inflammatory response. In a study by Vernooy et al., circulating levels of sTNFR-1 and sTNFR-2 did not differ when comparing current smokers with ex-smokers in the COPD and control groups [27]. In another study, a steady decline in serum sTNFR-1 and sTNFR-2 levels was observed after smoking cessation [37].
Several authors suggest that circulating sTNFR levels reflect a systemic inflammatory state better than do individual short-lived cytokine levels and should be better predictors of inflammation than TNF [37]. Smoking cessation is the only clinical intervention associated with reduced FEV1 decline in COPD patients [38]. However, to date, no reduction in airway inflammation has been demonstrated when COPD ex-smokers are compared to COPD current smokers [32, 33]. We speculate that the reduced sTNFR-1 concentrations in ex-smokers and never-smokers compared to current smokers observed in our study may partially explain some of the systemic benefit associated with smoking cessation. However, we did not find any differences in sTNFR-2 levels in relation to smoking status. Despite the fact that TNF receptors are considered to be markers of a proinflammatory state inducible by cytokines such as TNF-α, they present different biological functions [39, 40].
We did not analyze local inflammation in our study, and some data suggest that local and systemic inflammation may be regulated differently [28]. Thus, the association among inflammatory markers is complex, and a better understanding of the interplay among various mediators will require appropriately designed studies in the future.
In conclusion, our data confirm that the impact of smoking status on systemic inflammation differs according to the type of inflammatory marker analyzed.
Higher levels of inflammatory markers in current smokers and ex-smokers indicate that in COPD patients inflammation continues for many years after smoking cessation.
Conflict of interests statement
None of the authors has any conflict of interest to declare in relation to the subject matter of this manuscript.
References
Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2006. Global strategy for the diagnosis, management, and prevention of COPD. Executive Summary. [http://www.goldcopd.org]
Willemse BW, Postma DS, Timens W, ten Hacken NH: The impact of smoking cessation on respiratory symptoms, lung function, airway hyperresponsiveness and inflammation. Eur Respir J. 2004, 23: 464-476. 10.1183/09031936.04.00012704.
Chung KF: Cytokines as targets in chronic obstructive pulmonary disease. Curr Drug Targets. 2006, 7: 675-681. 10.2174/138945006777435263.
Gan WQ, Man SF, Senthilselvan A, Sin DD: Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax. 2004, 59: 574-580. 10.1136/thx.2003.019588.
Garcia-Rio F, Miravitlles M, Soriano JB, Muñoz L, Duran-Tauleria E, Sánchez G, Sobradillo V, Ancochea J, EPI-SCAN Steering Committee: Systemic inflammation in chronic obstructive pulmonary disease: a population-based study. Respir Res. 2010, 11: 63-
Pinto-Plata VM, Livnat G, Girish M, Cabral H, Masdin P, Linacre P, Dew R, Kenney L, Celli BR: Systemic cytokines, clinical and physiological changes in patients hospitalized for exacerbation of COPD. Chest. 2007, 131: 37-43. 10.1378/chest.06-0668.
Karadag F, Kirdar S, Karul AB, Ceylan E: The value of C-reactive protein as a marker of systemic inflammation in stable chronic obstructive pulmonary disease. Eur J Intern Med. 2008, 19: 104-108. 10.1016/j.ejim.2007.04.026.
Bradley JR: TNF-mediated inflammatory disease. J Pathol. 2008, 214: 149-160. 10.1002/path.2287.
Mukhopadhyay S, Hoidal JR, Mukherjee TK: Role of TNFalpha in pulmonary pathophysiology. Respir Res. 2006, 7: 125-10.1186/1465-9921-7-125.
Carp H, Miller F, Hoidal JR, Janoff A: Potential mechanism of emphysema: alpha1-proteinase inhibitor recovered from lungs of cigarette smokers contains oxidized methionine and has decreased elastase inhibitory capacity. Proc Natl Acad Sci USA. 1982, 79: 2041-2045. 10.1073/pnas.79.6.2041.
van der Vaart H, Postma DS, Timens W, Hylkema MN, Willemse BW, Boezen HM, Vonk JM, de Reus DM, Kauffman HF, ten Hacken NH: Acute effects of cigarette smoking on inflammation in healthy intermittent smokers. Respir Res. 2005, 6: 22-10.1186/1465-9921-6-22.
Janciauskiene SM, Nita IM, Stevens T: Alpha1-antitrypsin, old dog, new tricks. Alpha1-antitrypsin exerts in vitro anti-inflammatory activity in human monocytes by elevating cAMP. J Biol Chem. 2007, 282: 8573-8582. 10.1074/jbc.M607976200.
Gorber SC, Schofield-Hurwitz S, Hardt J, Levasseur G, Tremblay M: The accuracy of self-reported smoking: a systematic review of the relationship between self-reported and cotinine-assessed smoking status. Nicotine Tob Res. 2009, 11: 12-24. 10.1093/ntr/ntn010.
Dolcini MM, Adler NE, Lee P, Bauman KE: An assessment of the validity of adolescent self-reported smoking using three biological indicators. Nicotine Tob Res. 2002, 5: 473-483.
Senn O, Russi EW, Schindler C, Imboden M, von Eckardstein A, Brändli O, Zemp E, Ackermann-Liebrich U, Berger W, Rochat T, Luisetti M, Probst-Hensch NM, SAPALDIA Team: Circulating alpha1-antitrypsin in the general population: determinants and association with lung function. Respir Res. 2008, 9: 35-10.1186/1465-9921-9-35.
Stockley RA, Burnett D: Alpha-1-antitrypsin and leukocyte elastase in infected and noninfected sputum. Am Rev Respir Dis. 1979, 120: 1081-1086.
Owen CA, Campbell EJ: Extracellular proteolysis: new paradigms for an old paradox. J Lab Clin Med. 1999, 134: 341-351. 10.1016/S0022-2143(99)90148-8.
Meyer KC, Rosenthal NS, Soergel P, Peterson K: Neutrophils and low-grade inflammation in the seemingly normal aging human lung. Mech Ageing Dev. 1998, 104: 169-181. 10.1016/S0047-6374(98)00065-7.
Gan WQ, Man SF, Senthilselvan A, Sin DD: Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax. 2004, 59: 574-580. 10.1136/thx.2003.019588.
Silverman EK, Province MA, Rao DC, Pierce JA, Campbell EJ: A family study of the variability of pulmonary function in alpha 1-antitrypsin deficiency. Quantitative phenotypes. Am Rev Respir Dis. 1990, 142: 1015-1021.
Sin DD, Man SF: Why are patients with chronic obstructive pulmonary disease at increased risk of cardiovascular diseases? The potential role of systemic inflammation in chronic obstructive pulmonary disease. Circulation. 2003, 107: 1514-1519. 10.1161/01.CIR.0000056767.69054.B3.
Daniels JM, Schoorl M, Snijders D, Knol DL, Lutter R, Jansen HM, Boersma WG: Procalcitonin versus C-reactive protein as predictive markers of response to antibiotic therapy in acute exacerbations of COPD. Chest. 2010, 138: 1108-1115. 10.1378/chest.09-2927.
Godoy I, Campana AO, Geraldo RR, Padovani CR, Paiva SA: Cytokines and dietary energy restriction in stable chronic obstructive pulmonary disease patients. Eur Respir J. 2003, 22: 920-925. 10.1183/09031936.03.00025303.
Dentener MA, Creutzberg EC, Schols AM, Mantovani A, van't Veer C, Buurman WA, Wouters EF: Systemic anti-inflammatory mediators in COPD: increase in soluble interleukin 1 receptor II during treatment of exacerbations. Thorax. 2001, 56: 721-726. 10.1136/thorax.56.9.721.
Baraldo S, Turato G, Badin C, Bazzan E, Beghé B, Zuin R, Calabrese F, Casoni G, Maestrelli P, Papi A, Fabbri LM, Saetta M: Neutrophilic infiltration within the airway smooth muscle in patients with COPD. Thorax. 2004, 59: 308-312. 10.1136/thx.2003.012146.
Battaglia S, Mauad T, van Schadewijk AM, Vignola AM, Rabe KF, Bellia V, Sterk PJ, Hiemstra PS: Differential distribution of inflammatory cells in large and small airways in smokers. J Clin Pathol. 2007, 60: 907-911.
Vernooy JH, Küçükaycan M, Jacobs JA, Chavannes NH, Buurman WA, Dentener MA, Wouters EF: Local and systemic inflammation in patients with chronic obstructive pulmonary disease: soluble tumor necrosis factor receptors are increased in sputum. Am J Respir Crit Care Med. 2002, 166: 1218-1224. 10.1164/rccm.2202023.
Quint JK, Wedzicha JA: The neutrophil in chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2007, 119: 1065-1071. 10.1016/j.jaci.2006.12.640.
Barceló B, Pons J, Ferrer JM, Sauleda J, Fuster A, Agustí AG: Phenotypic characterization of T-lymphocytes in COPD: abnormal CD4+CD25+ regulatory T-lymphocyte response to tobacco smoking. Eur Respir J. 2008, 31: 555-562. 10.1183/09031936.00010407.
Barnes PJ, Chowdhury B, Kharitonov SA, Magnussen H, Page CP, Postma D, Saetta M: Pulmonary biomarkers in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006, 174: 6-14. 10.1164/rccm.200510-1659PP.
Gamble E, Grootendorst DC, Hattotuwa K, O'Shaughnessy T, Ram FS, Qiu Y, Zhu J, Vignola AM, Kroegel C, Morell F, Pavord ID, Rabe KF, Jeffery PK, Barnes NC: Airway mucosal inflammation in COPD is similar in smokers and ex-smokers: a pooled analysis. Eur Respir J. 2007, 30: 467-471. 10.1183/09031936.00013006.
Willemse BW, ten Hacken NH, Rutgers B, Lesman-Leegte IG, Postma DS, Timens W: Effect of 1-year smoking cessation on airway inflammation in COPD and asymptomatic smokers. Eur Respir J. 2005, 26: 835-845. 10.1183/09031936.05.00108904.
Watt AP, Schock BC, Ennis M: Neutrophils and eosinophils: clinical implications of their appearance, presence and disappearance in asthma and COPD. Curr Drug Targets Inflamm Allergy. 2005, 4: 415-423. 10.2174/1568010054526313.
Libert C, Van Molle W, Brouckaert P, Fiers W: Alpha1 Antitrypsin inhibits the lethal response to TNF in mice. J Immunol. 1996, 157: 5126-5129.
Tanni SE, Pelegrino NR, Angeleli AY, Correa C, Godoy I: Smoking status and tumor necrosis factor-alpha mediated systemic inflammation in COPD patients. J Inflamm (Lond). 2010, 7: 29-10.1186/1476-9255-7-29.
Pinto-Plata V, Toso J, Lee K, Park D, Bilello J, Mullerova H, De Souza MM, Vessey R, Celli B: Profiling serum biomarkers in patients with COPD: associations with clinical parameters. Thorax. 2007, 62: 595-601. 10.1136/thx.2006.064428.
Reichert V, Xue X, Bartscherer D, Jacobsen D, Fardellone C, Folan P, Kohn N, Talwar A, Metz CN: A pilot study to examine the effects of smoking cessation on serum markers of inflammation in women at risk for cardiovascular disease. Chest. 2009, 136: 212-219. 10.1378/chest.08-2288.
Fletcher C, Peto R: The natural history of chronic airflow obstruction. Br Med J. 1997, 1: 1645-1648.
Croft M: The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. 2009, 9: 271-285. 10.1038/nri2526.
Horiuchi T, Mitoma H, Harashima S, Tsukamoto H, Shimoda T: Transmembrane TNF-alpha: structure, function and interaction with anti-TNF agents. Rheumatology. 2010, 49: 1215-1228. 10.1093/rheumatology/keq031.
Acknowledgements
The authors would like to thank all COPD patients who contributed to this research. This study was supported by the Scientific Foundation of Kaunas University of Medicine (project grant PAR8).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Serapinas, D., Narbekovas, A., Juskevicius, J. et al. Systemic inflammation in COPD in relation to smoking status. Multidiscip Respir Med 6, 214 (2011). https://doi.org/10.1186/2049-6958-6-4-214
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2049-6958-6-4-214