
Abstract
A preferential dysfunction/loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) accounts for the main motor symptoms of Parkinson’s disease (PD), the most common degenerative movement disorder. However, the neuronal loss is not stochastic, but rather displays regionally selectivity, indicating the existence of different DA subpopulations in the SNpc. To identify the underlying molecular determinants is thereby instrumental in understanding the pathophysiological mechanisms of PD-related neuron dysfunction/loss and offering new therapeutic targets. Recently, we have demonstrated that aldehyde dehydrogenase 1 (ALDH1A1) is one such molecular determinant that defines and protects an SNpc DA neuron subpopulation preferentially affected in PD. In this review, we provide further analysis and discussion on the roles of ALDH1A1 in the function and survival of SNpc DA neurons in both rodent and human brains. We also explore the feasibility of ALDH1A1 as a potential biomarker and therapeutic target for PD.
Similar content being viewed by others


Introduction
Parkinson’s disease (PD), clinically manifested with resting tremor, dyskinesia/akinesia, posture instability, rigidity, and other motor symptoms [1], results from a preferential dysfunction/loss of the substantia nigra pars compacta (SNpc) dopaminergic (DA) neurons [2]. As supporting evidence, dopamine replacement therapy using the dopamine precursor L-3, 4-dihydroxyphenylalanine (L-DOPA) has been broadly employed to alleviate the motor symptoms [3]. Although L-DOPA is the gold standard PD therapy, it cannot prevent the progressive loss of SNpc neurons and becomes less effective at the late stages of the disease [4]. In addition, some patients respond poorly to the administration of L-DOPA, while others develop dyskinesia [5]. To understand why the SNpc DA neurons are preferentially susceptible to degeneration and how to prevent it remain the most challenging questions in PD research and treatment. Here we discuss recent advances in the identification of key molecular determinants critical for the survival of a subpopulation of SNpc DA neurons selectively degenerated in PD.
SNpc regional selectivity in aging and PD
SNpc DA neurons are highly specialized and possess many distinct morphological and functional properties [6]. They have long, unmyelinated and highly ramified axons [7]; use highly reactive dopamine as the transmitter [8]; and, function as a pace-maker using calcium currents [6, 9]. SNpc DA neurons likely undertake tremendous stress to support their constant neural activities, to dispose cytotoxic dopamine metabolites, and to maintain calcium homeostasis, which likely render them more susceptible to aging, PD, and other risk factors [6, 9, 10].
Despite sharing many distinct features as mentioned above, SNpc DA neurons are not a homogeneous population of neurons [11–13]. They seem to organize into different subdivisions within the SNpc and display differential vulnerability during aging and PD processes [11–13]. Based on the pattern of neuronal loss in normal aging and PD, Fearnley and Lees divide human SNpc into six morphometric regions, including the medial part (DM), lateral part (DL) and pars lateralis (PL) in the dorsal tier, and the medial part (VM), intermediate part (VI) and lateral part (VL) in the ventral tier [12]. It appears that normal aging mainly affects the DA neurons distributed in the dorsal tier of SNpc, whereas PD causes additional and more severe loss of DA neurons in the ventral tier, especially the VL subpopulation [12]. Fearnley and Lees further postulate that SNpc DA neurons undergo a biphasic mode of degeneration in PD comprised of an age-dependent linear phase and a PD-induced accelerated phase of neuronal loss [12]. These earlier anatomical observations imply the existence of distinct molecular determinants that define and protect SNpc subpopulations selectively affected in PD. To identify the underlying molecular clues may not only shed light on the cause of SNpc DA neuronal loss in PD, but also provide new biomarkers and therapeutic targets for the treatment of the disease.
ALDH1A1 defines a subpopulation of SNpc DA neurons in both rodent and human brains
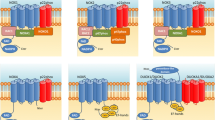
SNpc DA neurons express a selective set of genes encoding proteins critical for the synthesis, transport and degradation of dopamine, including tyrosine hydroxylase (TH), vesicular monoamine transporter 2 (VMAT2), dopamine transporter (DAT), and aldehyde dehydrogenase 1 (ALDH1A1) [14]. In contrast to a ubiquitous expression pattern of TH, VMAT2, and DAT in all SNpc DA neurons, ALDH1A1 appears to be expressed only in DA neurons residing at the ventral tier of rodent SNpc [15]. Moreover, a conserved topographic distribution of ALDH1A1-positive SNpc DA neurons is also observed in the ventral tier of human SNpc (Figure 1) [16]. Therefore, based on the expression of ALDH1A1, SNpc DA neurons can be divided into two subtypes that exhibit different susceptibility in PD (Figure 1) [16]. ALDH1A1 belongs to a large family of ALDH genes that consist of 19 members in the human genome [17]. Interestingly, among all ALDH genes only Aldh1a1 is predominantly and highly expressed by the SNpc DA neurons in the mouse CNS (Figure 2A) [16, 18]. The expression of Aldh1a1 mRNA was also much higher than any aldehyde reductase (AKR) genes, which may also involve with the oxidization of dopamine-3, 4-dihydroxyphenylacetaldehyde (DOPAL) (Figure 2B) [19]. Hence ALDH1A1 may possess some distinct characteristics critical for the function and survival of a subset of SNpc DA neurons preferentially degenerated in PD. Meanwhile, it is perhaps more accurate to pathologically characterize PD as caused by a selective loss of ALDH1A1-positive SNpc DA neurons.

Selective loss of ALDH1A1-positive subtype nigrostriatal DA neurons in PD. Cartoons illustrate the distribution of ALDH1A1–negative (ALDH1A1−, red) and –positive (ALDH1A1+, blue) DA neurons, as well as ALDH1A1+ neurons that lose ALDH1A1 expression (ALDH1A1+➔ ALDH1A1−, red with blue outline) in the SNpc of non-pathology control (NPC) and PD brains with mild and severe depigmentation. VL: ventral lateral, VM: ventral medial.

Expression of Aldh and Akr family genes in the mouse SNpc DA neurons. (A, B) RNA sequencing reveals the expression of Aldh (A) and Akt (B) family genes in SNpc DA neurons of 12-month-old wild type mice. Two independent sets of SNpc RNA samples were analyzed.
ALDH1A1 functions in periphery tissues and SNpc DA neurons
ALDH1A1 proteins exist as homotetramers in the cytosol, and oxidize various cytosolic aldehyde intermediates into the chemically more inert acidic forms [20]. The oxidative activity of ALDH1A1 requires the presence of the co-factor oxidized nicotinamide adenine dinucleotide (NAD+) [17]. ALDH1A1 has been involved in the metabolism of alcohol in the liver and retinol in the eye, brain, and other tissues [21]. ALDH1A1-deficiency has been indicated in alcohol-intolerance and cornea opacity [17]. On the other hand, an abnormal increase of ALDH1A1 has been observed in certain cancers, while ALDH1A1 inhibitors have been developed for cancer therapy [17, 22].
In the brain, ALDH1A1-mediated production of retinoid acid (RA) is required for the differentiation of DA neurons during embryonic development [23]. The expression of Aldh1a1 in the midbrain DA neurons is under the transcriptional control of pituitary homeobox 3 (Pitx3) and forkhead box protein A1/2 (Foxa1/2) [23, 24]. A lack of Pitx3 impairs the expression of Aldh1a1 and the terminal differentiation of midbrain DA neurons, whereas a supplement of RA in embryos rescues the developmental defects caused by Pitx3-deficiency [23]. Since Pitx3 and Foxa1/2 show rather ubiquitous expression pattern in the midbrain DA neurons, they may not be responsible for the selective expression of Aldh1a1 in the SNpc. It would be interesting to identify additional upstream transcription factors that regulate the expression of Aldh1a1 selectively in a subpopulation of DA neurons.
Additionally, ALDH1A1 also mediates the oxidation of DOPAL in DA neurons (Figure 3) [20]. Dopamine is produced in the cytosol before being sequestrated into the synaptic vesicles by VMAT2 (Figure 3) [25]. On the other hand, vesicular dopamine seems to constantly leak into the cytosol (Figure 3) [26]. Free cytosolic dopamine may undergo autoxidation to form cytotoxic quinones and other free radicals (Figure 3) [27]. To remove the cytosolic dopamine and its byproducts, monoamine oxidases (MAOs), ALDHs and AKRs are employed to degrade cytosolic dopamine and DOPAL into 3, 4-Dihydroxyphenylacetic acid (DOPAC) and other less reactive metabolites (Figure 3) [20, 26]. DOPAL is highly reactive and a lack of ALDH1A1 may lead to accumulation of DOPAL that has been shown to promote cytotoxic polymerization of PD-related α-synuclein and compromise the functions of proteins important in the activity and survival of SNpc DA neurons (Figure 3) [28]. In support of this notion, exposure of fungicide benomyl, an inhibitor of aldehyde dehydrogenase increases the risk of PD [29].

Dopamine metabolism and selective loss of SNpc DA neurons. Cartoon proposes the accumulation of cytosolic DOPAL triggers the degeneration of SNpc DA neurons in PD. Tyrosine hydroxylase (TH) and aromatic L-amino acid decarboxylase (AADC) mediate the synthesis of dopamine from tyrosine (Tyr) in the cytosol. Cytosolic dopamine is then immediately sequestered into the synaptic vesicles (SVs) by dopamine transporter VMAT2 for release. DAT mediates the reuptake of dopamine from extracellular space into the DA terminals. Leakage of dopamine from SVs also contributes to the cytosolic dopamine levels. MAO and ALDH1A1 are main enzymes for the degradation of cytosolic dopamine in DA neurons. A lack of ALDH1A1 may lead to a cytotoxic build-up of DOPAL which triggers the reactive oxygen species (ROS) production, protein adducts and α-synuclein aggregation, and eventually leads to cell death.
Given the importance of ALDH1A1 in dopamine metabolism, why ALDH1A1 is only expressed by a subset of DA neurons remains an intriguing question. In the absence of ALDH1A1, other ALDH and AKRs family proteins likely substitute its role in the oxidation of DOPAL and other cytosolic aldehyde intermediates. However, except for ALDH1A1, no other ALDHs or AKRs are particularly enriched in the SNpc DA neurons, or restricted to any subpopulations (Figure 2A, B) [16] (Allen Brain Atlas). We speculate the highly selective expression of ALDH1A1 in the ventral subpopulation of SNpc DA neurons may provide extra protection for these neurons that are preferentially vulnerable in PD [12].
ALDH1A1 contributes to the preferential loss of ventral SNpc DA neurons in PD
PD brains are featured with a more severe loss of ventral SNpc DA neurons [12]. One of the common molecular properties of these neurons is the expression of ALDH1A1 [16]. Correlatively, a more severe loss of ventral ALDH1A1-positive SNpc DA neurons has been observed in the PD cases (Figure 1) [16]. More interestingly, a significant increase of ALDH1A1-negative DA neurons in the ventral tier of SNpc is observed in the mild PD cases compared to the normal controls (Figure 1) [16]. A likely explanation of this observation is that PD may initially cause a reduction of ALDH1A1 expression in the ALDH1A1-positive DA neurons prior to the eventual neuronal loss. Reductions of ALDH1A1 mRNA and protein expression have also been reported in the SNpc of postmortem PD brains [30–32]. Moreover, in the α-synuclein transgenic mice both Aldh1a1 mRNA and protein levels are also decreased in DA neurons [16]. These findings suggest that ALDH1A1 itself is also a pathogenic target in PD. The reduction of ALDH1A1 expression in PD may weaken the protective function of ALDH1A1 in the ventral tier of SNpc, and predispose these neurons to degeneration at the later stages of disease. Therefore, a severe loss of ALDH1A1 expression may represent the turning point for ventral SNpc DA neurons that degenerate in PD. The expression level and activity of ALDH1A1 may serve as a useful biomarker to monitor the progression of the disease.
Mouse ALDH1A1-positive SNpc DA neurons are more resistant to α-synuclein-induced neurodegeneration
α-synuclein is a prominent genetic causal factor in the pathogenesis of PD [33–37]. A potential pathogenic interaction between cytosolic dopamine and α-synuclein has been implicated in the pathogenesis of PD [27, 38]. One of the key pathogenic mechanisms of α-synuclein in DA neurons is to form cytotoxic protein aggregates that may impair the synthesis, uptake, and degradation of dopamine [39–41]. The increased formation of cytotoxic dopamine quinones and DOPAL, on the other hand, may further promote α-synuclein aggregation through polymerization of monomeric α-synuclein [42, 43]. This pathogenic interplay between reactive dopamine derivatives and α-synuclein aggregation may form a vicious cycle that amplifies their detrimental effects to the DA neurons [42].
When the PD-related α-synuclein A53T missense mutation is introduced into the midbrain DA neurons, the resulting Pitx3–tTA::tetO–A53T bigenic mice develop profound motor disabilities and robust SNpc DA neuron loss [41]. Interestingly, the degenerated neurons are mainly distributed at the dorsal medial tier of SNpc that lack ALDH1A1 expression [16]. Noticeably, more cytotoxic α-synuclein aggregates are present in ALDH1A1-negative population of SNpc DA neurons in the mutant mice [16], suggesting that more DOPAL or other reactive dopamine intermediates may be present in these neurons to promote α-synuclein polymerization and aggregation [42, 43]. By contrast, the ventral ALDH1A1-postive SNpc DA neurons contain less α-synuclein aggregates and appear to resist α-synuclein-induced neuron loss during the aging process [16], thereby supporting the protective role of ALDH1A1 in these neurons. Correlatively, genetic deletion of Aldh1a1 gene exacerbates α-synuclein-induced SNpc DA neuronal loss in the Aldh1a1 knockout mice [16]. It is necessary to point out that the subtypes of the remaining SNpc DA neurons were not defined in the Aldh1a1-deficient mice due to a lack of molecular markers. Future studies will be required to identify additional molecular markers for different subtypes of SNpc DA neurons. To directly support the protective function of ALDH1A1 for DA neurons, over-expression of ALDH1A1 selectively ameliorates α-synuclein-induced cytotoxicity in the cultured DA neurons [16]. It will be interesting to examine the protective role of ALDH1A1 in vivo through the overexpression of ALDH1A1 or the use of selective activators. This proposed study may pave the way for establishing ALDH1A1 as an important therapeutic target for PD.
ALDH1A7 is highly homologous to ALDH1A1 and only exists in mouse but not human genome
There still lacks a pathologically more accurate PD mouse model that shows progressive loss of ALDH1A1-positive SNpc DA neurons. Why the mouse ALDH1A1-postive SNpc DA neurons are more resistant to α-synuclein-induced degeneration remains speculative [16]. Although ALDH1A1 appears to play an important role in the development and maintenance of SNpc DA neurons [16, 23], genetic deletion of Aldh1a1 fails to produce any overt motor symptoms or SNpc DA neuron loss in the Aldh1a1 knockout mice [44, 45]. Other ALDHs or AKRs may compensate for the loss of ALDH1A1 as shown in the Aldh1a1 and Aldh2 double knockout mice that develop mild but statistically significant loss of SNpc DA neurons [45].
Interestingly, when comparing the genomic organization of mouse and human ALDH1A1, mouse Aldh1a1 sits side-by-side with Aldh1a7 in chromosome 19, whereas no corresponding ALDH1A7 is found in the human genome (Figure 4A). Mouse ALDH1A7 proteins share 91% and 84% identical residues with mouse and human ALDH1A1, respectively, suggesting that ALDH1A1 and ALDH1A7 may have interchangeable functionalities (Figure 4B). The mouse Aldh1a1 and Aldh1a7 genes are likely derived from an ancestor gene through gene duplication, an event that seems not evolutionally conserved between mice and humans. In the midbrain lysates of Aldh1a1 homozygous knockout mice, around 80% reduction of ALDH1A1 protein expression was detected (Figure 4C). The remaining 20% of proteins likely reflect the expression of ALDH1A7, since the ALDH1A1 antibody possibly also recognizes ALDH1A7 due to the high homology shared by these two proteins. The presence of ALDH1A7 may thereby provide extra protection for the SNpc DA neurons in the mouse brains. Why mice but not human have ALDH1A7 is a mystery. However, ALDH1A7 may provide extra protection to the SNpc DA neurons in mouse brains.

ALDH1A7 is highly homologous to ALDH1A1 in the mouse genome. (A) Diagrams depict the genomic structures of human ALDH1A1 (hALDH1A1) and mouse Aldh1a1 (mAldh1a1) and Aldh1a7 (mAldh1a7). Arrows point to the direction of transcription. (B) Table shows the percentage of identical amino acids shared between hALDH1A1, mALDH1A1, and mALDH1A7 proteins. (C) Western blot shows the residual proteins recognized by an ALDH1A1 antibody in both the midbrain (MB) and striatum of Aldh1a1–/– mice.
ALDH1A1 as a biomarker and therapeutic target in PD
ALDH1A1 may exert its protective function to SNpc DA neurons via mitigating the cytotoxicity of DOPAL. A substantial reduction of ALDH1A1 expression and severe loss of ALDH1A1-positive SNpc DA have been observed in the postmortem PD brains [16, 30]. Collaboratively, studies of postmortem brains show a drop of DOPAC content in the putamen of PD patients, reflecting the reduced ALDH1A1 activity [14]. In addition, gene expression profiling of whole blood samples from 105 PD patients shows that ALDH1A1 mRNA together with other three genes are specific indicators for PD diagnosis since no such changes are found in control as well as Alzheimer’s cases [46]. Although the transcriptional regulation of ALDH1A1 may differ in SNpc DA neurons and blood cells, a similar systematic alteration of its expression might occur in both the CNS and periphery tissues. The levels of ALDH1A1 expression and activity either in the blood or CSF could serve as biomarkers for the diagnosis of PD.
The reduction of ALDH1A1 expression in PD might be employed as a compensatory mechanism to boost the release of dopamine in the remaining SNpc DA neurons via slowing down the turnover of dopamine. However, the undesired consequence of this approach is the resulting DOPAL-induced cytotoxicity, such as increased oxidative stress, protein adducts, and α-synuclein aggregation [47]. ALDH1A1 activation could be applied to suppress the toxic effects of DOPAL in the PD brains. Previous studies in cancer research have identified a number of intracellular signaling pathways that lead to an increase expression of ALDH1A1 in cancer cells [17]. However, whether SNpc DA neurons adopt the same pathways in regulating ALDH1A1 mRNA expression remains to be determined. In addition, it would be important to evaluate the impact of posttranslational modifications on the expression and function of ALDH1A1 proteins. The knowledge gained from these studies may help to design potential therapeutic interventions that boost the activity of ALDH1A1 in the PD brains.
On the other side, a variety of ALDH inhibitors have been developed to treat cancers, alcohol abuse and other disorders [17]. Among them, disulfiram, an alcohol-aversive drug [48], exhibits more potent inhibition of ALDH1A1 than ALDH2 and other ALDHs [49]. However, whether the administration of ALDH1A1 inhibitors may increase the risk of PD remains to determine. In addition, it would be interesting to learn if the experience obtained from designing ALDH1A1 inhibitors would help to produce ALDH1A1 specific activators for the treatment of PD.
Conclusions
Increasing evidence points out the significance of cytotoxic DOPAL and other dopamine metabolites in causing PD-related DA neurodegeneration [47]. ALDH1A1 is a key enzyme that irreversibly oxidizes DOPAL into less toxic DOPAC [20]. The presence of ALDH1A1 in a subpopulation of SNpc DA neurons not only defines a neuronal subtype selectively susceptible in PD, but also opens a new window to further characterize the connectivity and functionality of this important group of neurons in dopaminergic transmission [16]. Therefore, elucidation of the molecular and pathophysiological properties of these ALDH1A1-positive SNpc DA neurons may provide major advancement on our current understanding of the pathogenic mechanism of PD-related neuronal loss and lead to better treatment of the disease.
References
Parkinson J: An Essay On The Shaking Palsy. London: Sherwood, Nelly and Jones; 1817.
Lewis PD: Parkinsonism–neuropathology. Br Med J 1971, 3(5776):690-692. 10.1136/bmj.3.5776.690
Cotzias GC: L-Dopa for Parkinsonism. N Engl J Med 1968, 278(11):630.
Pankratz ND, Wojcieszek J, Foroud T: Parkinson Disease Overview. In GeneReviews. Edited by: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K. Seattle: University of Washington; 1993:1993-2014.
Mercuri NB, Bernardi G: The 'magic' of L-dopa: why is it the gold standard Parkinson's disease therapy? Trends Pharmacol Sci 2005, 26(7):341-344. 10.1016/j.tips.2005.05.002
Sulzer D, Surmeier DJ: Neuronal vulnerability, pathogenesis, and Parkinson's disease. Mov Disord 2013, 28(1):41-50. 10.1002/mds.25095
Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R, Kaneko T: Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci 2009, 29(2):444-453. 10.1523/JNEUROSCI.4029-08.2009
Schultz W: Multiple dopamine functions at different time courses. Annu Rev Neurosci 2007, 30: 259-288. 10.1146/annurev.neuro.28.061604.135722
Surmeier DJ: Calcium, ageing, and neuronal vulnerability in Parkinson's disease. Lancet Neurol 2007, 6(10):933-938. 10.1016/S1474-4422(07)70246-6
Surmeier DJ, Schumacker PT: Calcium, bioenergetics, and neuronal vulnerability in Parkinson's disease. J Biol Chem 2013, 288(15):10736-10741. 10.1074/jbc.R112.410530
Hirsch EC, Graybiel AM, Agid Y: Selective vulnerability of pigmented dopaminergic neurons in Parkinson's disease. Acta Neurol Scand Suppl 1989, 126: 19-22.
Fearnley JM, Lees AJ: Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain 1991, 114(Pt 5):2283-2301.
Damier P, Hirsch EC, Agid Y, Graybiel AM: The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson's disease. Brain 1999, 122(Pt 8):1437-1448.
Goldstein DS: Biomarkers, mechanisms, and potential prevention of catecholamine neuron loss in Parkinson disease. Adv Pharmacol 2013, 68: 235-272.
McCaffery P, Drager UC: High levels of a retinoic acid-generating dehydrogenase in the meso-telencephalic dopamine system. Proc Natl Acad Sci U S A 1994, 91(16):7772-7776. 10.1073/pnas.91.16.7772
Liu G, Yu J, Ding J, Xie C, Sun L, Rudenko I, Zheng W, Sastry N, Luo J, Rudow G, Troncoso JC, Cai H: Aldehyde dehydrogenase 1 defines and protects a nigrostriatal dopaminergic neuron subpopulation. J Clin Invest 2014, 124(7):3032-3046. 10.1172/JCI72176
Koppaka V, Thompson DC, Chen Y, Ellermann M, Nicolaou KC, Juvonen RO, Petersen D, Deitrich RA, Hurley TD, Vasiliou V: Aldehyde dehydrogenase inhibitors: a comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol Rev 2012, 64(3):520-539. 10.1124/pr.111.005538
Luo J, Sun L, Lin X, Liu G, Yu J, Parisiadou L, Xie C, Ding J, Cai H: A calcineurin- and NFAT-dependent pathway is involved in alpha-synuclein-induced degeneration of midbrain dopaminergic neurons. Hum Mol Genet 2014, 23(24):6567-6574. 10.1093/hmg/ddu377
Lamensdorf I, Eisenhofer G, Harvey-White J, Hayakawa Y, Kirk K, Kopin IJ: Metabolic stress in PC12 cells induces the formation of the endogenous dopaminergic neurotoxin, 3,4-dihydroxyphenylacetaldehyde. J Neurosci Res 2000, 60(4):552-558. 10.1002/(SICI)1097-4547(20000515)60:4<552::AID-JNR14>3.0.CO;2-U
Marchitti SA, Deitrich RA, Vasiliou V: Neurotoxicity and metabolism of the catecholamine-derived 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenylglycolaldehyde: the role of aldehyde dehydrogenase. Pharmacol Rev 2007, 59(2):125-150. 10.1124/pr.59.2.1
Vasiliou V, Pappa A, Petersen DR: Role of aldehyde dehydrogenases in endogenous and xenobiotic metabolism. Chem Biol Interact 2000, 129(1–2):1-19.
Tanei T, Morimoto K, Shimazu K, Kim SJ, Tanji Y, Taguchi T, Tamaki Y, Noguchi S: Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential Paclitaxel and epirubicin-based chemotherapy for breast cancers. Clin Cancer Res 2009, 15(12):4234-4241. 10.1158/1078-0432.CCR-08-1479
Jacobs FM, Smits SM, Noorlander CW, von Oerthel L, van der Linden AJ, Burbach JP, Smidt MP: Retinoic acid counteracts developmental defects in the substantia nigra caused by Pitx3 deficiency. Development 2007, 134(14):2673-2684. 10.1242/dev.02865
Domanskyi A, Alter H, Vogt MA, Gass P, Vinnikov IA: Transcription factors Foxa1 and Foxa2 are required for adult dopamine neurons maintenance. Front Cell Neurosci 2014, 8: 275.
Bernstein AI, Stout KA, Miller GW: The vesicular monoamine transporter 2: an underexplored pharmacological target. Neurochem Int 2014, 73: 89-97.
Goldstein DS, Sullivan P, Holmes C, Miller GW, Alter S, Strong R, Mash DC, Kopin IJ, Sharabi Y: Determinants of buildup of the toxic dopamine metabolite DOPAL in Parkinson's disease. J Neurochem 2013, 126(5):591-603. 10.1111/jnc.12345
Conway KA, Rochet JC, Bieganski RM, Lansbury PT Jr: Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science 2001, 294(5545):1346-1349. 10.1126/science.1063522
Rees JN, Florang VR, Eckert LL, Doorn JA: Protein reactivity of 3,4-dihydroxyphenylacetaldehyde, a toxic dopamine metabolite, is dependent on both the aldehyde and the catechol. Chem Res Toxicol 2009, 22(7):1256-1263. 10.1021/tx9000557
Fitzmaurice AG, Rhodes SL, Lulla A, Murphy NP, Lam HA, O'Donnell KC, Barnhill L, Casida JE, Cockburn M, Sagasti A, Stahl MC, Maidment NT, Ritz B, Bronstein JM: Aldehyde dehydrogenase inhibition as a pathogenic mechanism in Parkinson disease. Proc Natl Acad Sci U S A 2013, 110(2):636-641. 10.1073/pnas.1220399110
Galter D, Buervenich S, Carmine A, Anvret M, Olson L: ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson's disease and in the ventral tegmental area in schizophrenia. Neurobiol Dis 2003, 14(3):637-647. 10.1016/j.nbd.2003.09.001
Mandel SA, Fishman T, Youdim MB: Gene and protein signatures in sporadic Parkinson's disease and a novel genetic model of PD. Parkinsonism Relat Disord 2007, 13(Suppl 3):S242-247.
Werner CJ, Heyny-von Haussen R, Mall G, Wolf S: Proteome analysis of human substantia nigra in Parkinson's disease. Proteome Sci 2008, 6: 8. 10.1186/1477-5956-6-8
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M: Alpha-synuclein in Lewy bodies. Nature 1997, 388(6645):839-840. 10.1038/42166
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL: Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 1997, 276(5321):2045-2047. 10.1126/science.276.5321.2045
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K: Alpha-Synuclein locus triplication causes Parkinson's disease. Science 2003, 302(5646):841. 10.1126/science.1090278
Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Krüger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, et al.: Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet 2009, 41(12):1308-1312. 10.1038/ng.487
Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T: Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat Genet 2009, 41(12):1303-1307. 10.1038/ng.485
Sulzer D: Alpha-synuclein and cytosolic dopamine: stabilizing a bad situation. Nat Med 2001, 7(12):1280-1282. 10.1038/nm1201-1280
Kurz A, Double KL, Lastres-Becker I, Tozzi A, Tantucci M, Bockhart V, Bonin M, Garcia-Arencibia M, Nuber S, Schlaudraff F, Liss B, Fernández-Ruiz J, Gerlach M, Wüllner U, Lüddens H, Calabresi P, Auburger G, Gispert S: A53T-alpha-synuclein overexpression impairs dopamine signaling and striatal synaptic plasticity in old mice. PLoS One 2010, 5(7):e11464. 10.1371/journal.pone.0011464
Lam HA, Wu N, Cely I, Kelly RL, Hean S, Richter F, Magen I, Cepeda C, Ackerson LC, Walwyn W, Masliah E, Chesselet MF, Levine MS, Maidment NT: Elevated tonic extracellular dopamine concentration and altered dopamine modulation of synaptic activity precede dopamine loss in the striatum of mice overexpressing human alpha-synuclein. J Neurosci Res 2011, 89(7):1091-1102. 10.1002/jnr.22611
Lin X, Parisiadou L, Sgobio C, Liu G, Yu J, Sun L, Shim H, Gu XL, Luo J, Long CX, Ding J, Mateo Y, Sullivan PH, Wu LG, Goldstein DS, Lovinger D, Cai H: Conditional expression of Parkinson's disease-related mutant alpha-synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J Neurosci 2012, 32(27):9248-9264. 10.1523/JNEUROSCI.1731-12.2012
Burke WJ, Kumar VB, Pandey N, Panneton WM, Gan Q, Franko MW, O'Dell M, Li SW, Pan Y, Chung HD, Galvin JE: Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol 2008, 115(2):193-203. 10.1007/s00401-007-0303-9
Cremades N, Cohen SI, Deas E, Abramov AY, Chen AY, Orte A, Sandal M, Clarke RW, Dunne P, Aprile FA, Bertoncini CW, Wood NW, Knowles TP, Dobson CM, Klenerman D: Direct observation of the interconversion of normal and toxic forms of alpha-synuclein. Cell 2012, 149(5):1048-1059. 10.1016/j.cell.2012.03.037
Anderson DW, Schray RC, Duester G, Schneider JS: Functional significance of aldehyde dehydrogenase ALDH1A1 to the nigrostriatal dopamine system. Brain Res 2011, 1408: 81-87.
Wey MC, Fernandez E, Martinez PA, Sullivan P, Goldstein DS, Strong R: Neurodegeneration and motor dysfunction in mice lacking cytosolic and mitochondrial aldehyde dehydrogenases: implications for Parkinson's disease. PLoS One 2012, 7(2):e31522. 10.1371/journal.pone.0031522
Grunblatt E, Zehetmayer S, Jacob CP, Muller T, Jost WH, Riederer P: Pilot study: peripheral biomarkers for diagnosing sporadic Parkinson's disease. J Neural Transm 2010, 117(12):1387-1393. 10.1007/s00702-010-0509-1
Goldstein DS, Kopin IJ, Sharabi Y: Catecholamine autotoxicity. Implications for pharmacology and therapeutics of Parkinson disease and related disorders. Pharmacol Ther 2014, 144(3):268-282. 10.1016/j.pharmthera.2014.06.006
Bell RG, Smith HW: Preliminary report on clinical trials of antabuse. Can Med Assoc J 1949, 60(3):286-288.
Moore SA, Baker HM, Blythe TJ, Kitson KE, Kitson TM, Baker EN: Sheep liver cytosolic aldehyde dehydrogenase: the structure reveals the basis for the retinal specificity of class 1 aldehyde dehydrogenases. Structure 1998, 6(12):1541-1551. 10.1016/S0969-2126(98)00152-X
Acknowledgements
This work was supported by the Intramural Research Program of National Institute on Aging, National Institutes of Health (AG000959-07 and AG000945-03). We thank all members of Cai lab for very helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HC and GL complied the literatures and wrote the manuscript. GL contributed the data of ALDH1A1 expression in Figure 4. LS carried out the RNAseq experiments and edited the manuscript. JD performed the gene expression analyses and provided bioinformatics support shown in Figures 2 and 4. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Cai, H., Liu, G., Sun, L. et al. Aldehyde Dehydrogenase 1 making molecular inroads into the differential vulnerability of nigrostriatal dopaminergic neuron subtypes in Parkinson’s disease. Transl Neurodegener 3, 27 (2014). https://doi.org/10.1186/2047-9158-3-27
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2047-9158-3-27