
Abstract
Cerebral malaria (CM) is a life-threatening complication of falciparum malaria, associated with high mortality rates, as well as neurological impairment in surviving patients. Despite disease severity, the etiology of CM remains elusive. Interestingly, although the Plasmodium parasite is sequestered in cerebral microvessels, it does not enter the brain parenchyma: so how does Plasmodium induce neuronal dysfunction? Several independent research groups have suggested a mechanism in which increased blood–brain barrier (BBB) permeability might allow toxic molecules from the parasite or the host to enter the brain. However, the reported severity of BBB damage in CM is variable depending on the model system, ranging from mild impairment to full BBB breakdown. Moreover, the factors responsible for increased BBB permeability are still unknown. Here we review the prevailing theories on CM pathophysiology and discuss new evidence from animal and human CM models implicating BBB damage. Finally, we will review the newly-described role of matrix metalloproteinases (MMPs) and BBB integrity. MMPs comprise a family of proteolytic enzymes involved in modulating inflammatory response, disrupting tight junctions, and degrading sub-endothelial basal lamina. As such, MMPs represent potential innovative drug targets for CM.
Similar content being viewed by others
Introduction
Human malaria is a widespread infectious disease caused by Plasmodium protozoan parasites and is associated with high morbidity and mortality rates, resulting in 627,000 deaths among 207 million cases estimated in 2012 [1]. Human malaria is caused by five different Plasmodium species: P. falciparum, P. malariae, P. ovale, P. vivax and P. knowlesi. P. falciparum and P. vivax are the most common, correlating with the most severe forms of malaria and the highest death rate, whereas other Plasmodium species generally cause milder forms of malaria which are rarely fatal [1]. The majority of deaths occur among children under the age of five years living in sub-Saharan Africa, and in Southern/South-Eastern Asia and Central/Southern America where mortality mainly affects adults. Additionally, occasional cases are observed in non-immune adult travelers from developed countries returning from these areas. Despite the intense efforts made by the research community and the Global Eradication program [2], no effective vaccines or adjuvant therapies are available for complicated malaria. It is projected that in the next few years the dramatic issue of drug-resistant malaria could become a serious threat [3–5].
P. falciparum is unique in that it causes mature infected red blood cells (iRBCs) to sequester and adhere to microvascular beds in numerous organs. A paradigmatic complication of falciparum malaria is cerebral malaria (CM), which develops after iRBCs sequester in the microvasculature of the central nervous system (CNS). Unlike the other human malarial parasites which rarely cause neurological dysfunction, P. falciparum-induced CM often leads to death or severe neurological sequelae [6]. Curiously, P. falciparum appears to remain in the vascular space without ever entering the brain parenchyma, in contrast to other encephalitis-causing pathogens, such as Trypanosoma spp. or Toxoplasma gondii[7], thus raising question of how intravascular Plasmodium parasites are capable of inducing such a devastating neural dysfunction in CM.
Recent evidence suggests that a compromised integrity of the blood–brain barrier (BBB) results in a subsequent increase in BBB permeability which enables toxic soluble factors released either by host or parasite to cross this barrier and exert neurological effects. This review focuses on CM pathophysiology and novel insights from animal and human models into the role of BBB functional impairment in CM. Finally, we discuss the emerging role of host matrix metalloproteinases (MMPs), a family of proteolytic enzymes related to inflammation and BBB damage in CM, opening the possibility for discovery of new effective adjuvant therapies for CM.
Pathophysiology of cerebral malaria
CM appears as a diffuse encephalopathy commonly presenting with headache, agitation, frank psychosis, seizures and impaired consciousness, and occasionally with brainstem signs or focal neurological signs such as hemiplegia and cranial nerve palsies [8, 9]. According to the World Health Organization (WHO) clinical criteria, CM is defined as a potentially reversible, diffuse encephalopathy causing a Glasgow coma score of 11/15 or less, often associated with fitting, in the absence of other factors that could cause unconsciousness such as coexistent hypoglycemia or other CNS infections [10]. It is difficult to confirm diagnoses of CM in endemic areas because of overlapping infections such as bacterial meningitis in patients showing incidental malarial parasitaemia [11]. Children from areas endemic for malaria or non-immune adults traveling from developed countries are at higher risk for developing CM. On the contrary, CM is rarely encountered in > 10-year-old patients who have been exposed to P. falciparum since birth. Mortality ranges from 15–30%, and 11% of children display neurological deficits upon discharge [12].
The pathophysiological mechanisms underlying CM are not fully understood so far. As seen in Figure 1 and discussed in the next paragraphs, there are currently three distinct theories on the etiology of CM typical features: i) the mechanical hypothesis; ii) the permeability hypothesis; and iii) the humoral hypothesis [4, 9, 13–16]. It is possible that these theories are all pieces of that puzzle that need to be combined as they likely constitute more complementary than alternative models [6, 17].

Most commonly accepted hypotheses for pathophysiological mechanisms underlying clinical progress towards cerebral malaria (CM). The diagram summarizes the three distinct hypotheses on CM etiology and their typical features: i) the mechanical hypothesis is associated with iRBC cytoadherence and their reduced deformability, causing following anemia, rosette formation and microvascular obstruction; ii) the permeability hypothesis is based on BBB impairment and subsequent increase in vascular permeability, allowing toxic compounds to reach the brain parenchyma and causing neurological dysfunction; iii) the humoral hypothesis focuses on the enhanced production by the host of pro-inflammatory molecules, including cytokines and chemokines, and other soluble factors such as ROS, which are putatively responsible for inflammation, fever and coma during CM.
Mechanical hypothesis
The mechanical hypothesis proposes CM is caused by a mechanical obstruction of the cerebral microvasculature, with coma resulting from impaired brain perfusion [9, 14, 18]. Such a hypothesis was made after one of the first pathological studies on human CM showed that brain capillaries were packed with iRBCs [18]. In the mechanical hypothesis, specific interactions between iRBCs and vascular endothelium are thought to mediate sequestration of iRBCs within the brain resulting in removal from peripheral circulation [19–21]. The molecules involved in these interactions are parasite proteins expressed on iRBC surface, such as P. falciparum erythrocyte membrane protein-1 (PfEMP-1), and specific host receptors in the microvascular endothelium, including intracellular adhesion molecule-1 (ICAM-1), vascular cellular adhesion molecule-1 (VCAM-1), thrombospondin, CD36, and E-elastin [22–25].
Cytoadherence and decreased pliability are the main mechanisms underlying vascular obstruction [9, 17–21]. It is speculated that cytoadherence evolved as a mechanism for the parasite to evade triggering a host immune response and being cleared from the spleen. Cytoadherence is also beneficial for the parasite as to provide an optimal environment of low oxygen tension for parasite growth. Decreased deformability along with increased membrane stiffness and rigidity of iRBCs are due to changes in the cytoskeleton triggered by growing intracellular parasites. Cell deformability has been indicated as a predictor of anemia development [26], whereas cell rigidity correlates with a higher fatality rate [27]. Another phenomenon occurring along with iRBC sequestration is rosetting, characterized by iRBCs forming a flower-like cluster around a non-iRBC, making a tight rigid structure [28]. Rosetting is more frequent in patients with CM than in those with uncomplicated malaria. However, rosette formation has also been reported for other Plasmodium strains (P. vivax and P. ovale) which do not cause CM [29]. Since rosetting occurs in all manifestations of the disease, it is not associated with severity or clinical outcome of CM [30]. One question the mechanical hypothesis by itself does not explain is why most patients recovering from CM do not show any evidence of ischemic brain damage [12].
Permeability hypothesis
The permeability hypothesis proposes that BBB damage is the underlying mechanism of CM, such that a leaky BBB allows toxic compounds to enter the brain and cause neurological dysfunction [9, 13–15, 31]. Several animal CM models have confirmed that the BBB is disrupted and that cerebral edema is present in CM, although this is less evident in humans [15]. Nevertheless, iRBCs remain attached to endothelium, without entering the brain parenchyma [7, 9]. Interestingly, Adams and colleagues have suggested that iRBC cytoadherence might activate secondary signaling events similar to those occurring in leukocytes [32]. These secondary signaling events are thought to cause functional alterations in the BBB, which could allow toxic compounds to pass into the CNS. These events might be reversible, therefore explaining why neurological manifestations are just transient in most cases and why a large number of recovering patients lack neurological sequelae [32].
Enwonwu and colleagues implicated histamine as one of these toxic molecules that enters the brain parenchyma after BBB impairment and contributes to the neurological manifestions of CM [33–37]. The authors observed altered neural histidine uptake in children with severe falciparum malaria providing an explanation for the enhanced cerebral production of histamine [33]. They also found increased plasma levels of histamine in severe malaria patients, further supporting their hypothesis [34]. Moreover, the involvement of histamine in CM has also recently been confirmed in a murine model [35–37]. In this study, histidine decarboxylase-deficient mice were unable to synthesize free histamine and did not develop CM after infection with P. berghei ANKA. These mice displayed preserved BBB integrity, were void of iRBC aggregation in the brain vessels, and did not sequester CD4+ and CD8+ T cells [36]. Further investigation of histamine receptors revealed histamine-1-receptor (H1R) and histamine-2-receptor (H2R) are associated with severe malaria development [37], whereas histamine-3-receptor (H3R) has a neuroprotective role [36].
Humoral hypothesis
The humoral hypothesis is a natural extension of the permeability hypothesis. This hypothesis suggests that host factors such as leukocyte-derived cytokines and chemokines can enter the brain parenchyma after increased BBB permeability, thus causing CM symptoms such as fever and coma [9, 13, 14, 16, 38–40].
Effector cells including T cells, NK cells, and monocytes, along with inflammatory responses mediated by cytokines such as tumor necrosis factor-α (TNF-α), limphotoxin-α (LT-α), and interferon-γ (IFN-γ), are proposed to contribute to the development of murine CM [41–48]. However, the extent of their involvement and molecular mechanisms in human CM is still topic of debate [48, 49].
CD8+ T cells have been reported to initiate BBB tight junction disruption and promote CNS vascular permeability under neuroinflammatory conditions [50–52]. Consistently, CD8+ T cell sequestration in cerebral microvessels and subsequent brain infiltration have been demonstrated in murine CM [43, 44], where Plasmodium antigens can be cross-presented during infection by dendritic cells (DCs) [53, 54] and brain endothelial cells in association with MHC class I molecules [55]. Recent human studies support the idea that malaria antigens can be transferred to endothelial cells [56]. However, it is currently unknown whether Plasmodium-specific CD8+ T cells are involved in the pathogenesis of human CM [57]. Furthermore, lymphocyte infiltration into brain parenchyma remains to be investigated [49].
TNF-α relevance in CM is also unclear. TNF-α involvement in murine CM was first proposed in 1987 [58]. Since then there have been numerous studies investigating TNF-α levels in CM mice albeit the results are inconsistent. For example, some works confirmed the association of high TNF-α levels with murine CM [59–61], whereas others argued against such correlation, finding LT-α and IFN-γ as more suitable markers [44, 47, 62, 63]. To reconcile such discrepancies, it has been proposed that low concentrations of TNF-α could enhance parasite killing, whereas higher concentrations might be associated with increased incidence of murine CM [46]. However, data on TNF-α also appear inconclusive in human CM studies. Indeed, clinical studies tend to exclude any association between CM and increased plasma, serum or CSF levels of TNF-α [64–67], although a few works have proposed a correlation in two different Asian populations [68, 69]. Instead, in some of these studies, high CXCL10/IP-10 plasma levels and low angiogenic factors such as vascular endothelial growth factor (VEGF) and angiopoietin-1 (Ang-1) in children with CM, predicted subsequent mortality [65, 66, 68]. Moreover, a protective role for IL-12 has been proposed in human CM [70, 71].
Among soluble factors involved in CM, a critical role for nitric oxide (NO) has also been suggested. It was hypothesized that NO levels correlate with disease severity, since the sequestration of iRBCs might contribute to CM pathogenesis by causing hypoxia, which is related to enhanced production of cytokine-induced NO, compensatory vasodilatation, and subsequent brain volume increase [39]. However, activation of inducible NO synthase (NOS) might also serve protective functions, since NOS inhibits the side effects of brain indoleamine 2,3-dioxygenase (IDO) and quinolinic acid accumulation [72], although IDO systemic distribution is independent of malaria disease severity [73]. In a study performed on Tanzanian children infected with malaria, the plasma levels of NOS-suppressing IL-10 increased with disease severity, suggesting that a reduced NO production may contribute to CM [74]. Moreover, a genetic single nucleotide polymorphism found in the NOS2 promoter region causes elevated NO production and was significantly associated with protection against CM in Tanzanian and Kenyan children [75]. In line with these observations, Anstey and colleagues demonstrated that decreased NO production was associated with endothelial dysfunction in human CM [76, 77]. Similarly, van der Heyde and his group demonstrated that low NO bioavailability was associated with murine CM [78, 79]. Interestingly, prophylaxis with inhaled NO in CM-sensitive mice significantly reduced systemic inflammation and endothelial activation by lowering TNF-α, IFN-γ, monocyte chemotactic protein-1 (MCP-1), sICAM-1 and von Willebrand factor, and by increasing Ang-1 levels in peripheral blood [80]. The protective effect of exogenous NO on mouse CM appears associated with decreased brain vascular expression of inflammatory markers, resulting in attenuated endothelial junction damage and facilitating blood flow [81]. Lastly, treatment with exogenous L-arginine, the substrate for NOS, recently proved to be safe in a pilot study on CM patients, although effective doses still need to be optimized [82].
Furthermore, during malaria infection both host and parasite undergo strong oxidative stress, which leads to increased production of reactive oxygen species (ROS) and subsequent protein and lipid peroxidation [83, 84]. The coexistence of both parasite and erythrocyte is a matter of a delicate balance: low ROS concentrations seem to inhibit parasite growth, whereas larger amounts may damage vascular endothelial cells and increase vascular permeability [85]. Oxidative stress paradoxically has both a pathogenic and protective role in CM [86]. An anti-oxidant diet was shown to reduce BBB damage and counteract CM development in CM-sensitive mice [87], and anti-oxidant adjuvant therapy, provided at the initial stages of murine CM, prevented the development of persistent cognitive damage [88]. In contrast, NADPH-deficient mice were shown to develop CM despite the lack of ROS production, suggesting that ROS did not contribute to CM pathogenesis [89, 90]. To reconcile such an apparent inconsistency, Linares and colleagues have recently shown that glutathione peroxidase and heme oxygenase-1 up-regulation cooperate to suppress superoxide dismutase, catalase, heat shock protein-70 and thioredoxin-1 down-regulation effects in murine CM, counteracting oxidative damage and maintaining redox equilibrium [91]. In human CM, ROS have been associated with a pathogenic role thus far. In vitro, ROS inhibition was shown to protect brain endothelial cells against P. falciparum-induced apoptosis and to decrease iRBC cytoadherence through ICAM-1 down-regulation and iNOS induction [92, 93]. Consistently, in a recent clinical study performed on fifty Indian children with severe malaria, oxidative stress was associated with disease severity [94].
Blood–brain barrier impairment in cerebral malaria
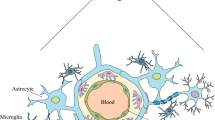
The BBB is one of three main barrier defences protecting the CNS. It is constituted of cerebral vascular endothelial cells, which do not form a rigid structure, but rather a dynamic interface with a range of physical, biochemical and immune properties and functions, built from effective inter-cellular junctions and cell-matrix adhesion molecules, enzymes, and trans-endothelial transport systems [95]. In particular, BBB integrity is dictated by tight junctions between adjacent endothelial cells, forming a network of strands composed by several proteins, including junctional adhesion molecules, claudins (mainly -1 and -5) and occludin, which interact with cellular actin through cytoplasmic proteins such as zonula occludens-1 (ZO-1) [96]. Figure 2 depicts the structure of neural inter-endothelial tight junctions, along with cell-matrix adhesion complexes including talin, filamin, tensin or α-actinin filaments associated with integrins. We will next discuss how the disruption of these molecules by host proteolytic enzymes such as MMPs could play a relevant role in CM pathophysiology.

Blood–brain barrier structure: cerebral microvascular inter-endothelial junctions (adherens and tight junctions) and cell-matrix adhesion molecules. Diagram showing the structures of CNS inter-endothelial junctions, including adherens junctions and tight junctions, and of cell-matrix adhesion complexes, including talin, filamin, tensin or α-actinin filaments associated with integrins in the extracellular matrix. The core of adherens junctions results after the interactions among transmembrane glycoproteins, such as VE-cadherin, whose cytoplasmic face is linked to the catenin family members, including p120-catenin, β-catenin, and α-catenin. Tight junctions are composed of a branching network of sealing strands, each of which is formed by extracellular domains of transmembrane proteins, claudins and occludin, joining one another directly. These transmembrane proteins associate with different peripheral membrane proteins such as ZO-1 located on the intracellular side of plasma membrane, anchoring the strands to the actin component of the cytoskeleton.
BBB functional integrity and permeability are generally assessed by evaluating the passage of molecules from the blood into the cerebral-spinal fluid (CSF). BBB permeability is determined by size and charge of the molecules, and the presence of specific BBB receptors to aid in the transport of certain molecules. The importance of BBB physiology and pathology has led to the development of several BBB models to better investigate the physiological, anatomical and functional characteristics [97]. However, once again the current experimental data on BBB status during CM are high variable among different model systems [98].
Phenotype of brain and non-brain endothelial cells co-cultured with Plasmodium iRBCs in vitro
As discussed below and summarized in Table 1, evidence showing differential phenotypes between neural and non-neural endothelial cells after co-culture with Plasmodium iRBCs comes from several in vitro studies [56, 93, 99–108].
First, the effects of P. falciparum infection were investigated in a BBB model of cultured primary porcine brain capillary endothelial cells (PBCECs) [99]. In this study, membrane-associated malaria antigens obtained from lysed P. falciparum schizont-iRBCs increased endothelial E-selectin and ICAM-1 expression, reduced the trans-endothelial electrical resistance (TEER), and promoted the disruption of tight junctions, indicative of increased BBB permeability.
Consistently in various types of human brain endothelium, including HMBEC primary cultures and HBEC-5i or hCMEC/D3 cell lines, iRBCs were also shown to increase ICAM-1 expression [93, 99, 104, 105], to reduce TEER [56, 101, 102], to alter tight junction expression and distribution [56, 105], and to enhance BBB permeability to 70-kDa dextran [101]. Interestingly, platelets were suggested to play a key role in iRBC-dependent increase in BBB permeability, releasing microparticles and causing cell apoptosis in TNF-α- and LT-α-activated HBEC-5i [101]. In hCMEC/D3 cells, iRBC-increased cell adhesion and paracellular permeability correlated with ZO-1 disorganization, but the latter effect appeared mediated by parasite-induced metabolic acidosis, independent from cytoadherence [105]. Moreover, differential global gene expression in HBMEC after interacting with iRBCs revealed significantly up-regulated transcripts related to immune and inflammatory responses, apoptosis, cell-cell signaling, signal transduction and nuclear factor-kB (NF-kB)-activation cascade [104]. After co-culturing with iRBCs, the mRNA expression of neural endothelial pro-inflammatory chemokines (IL-6, CXCL-8/IL-8, CXCL-1/GRO-α, CXCL-2/MIP-2α, and CCL-20/MIP-3α) increased more than 100-fold, highlighting the strong inflammatory component and the active role of the endothelium in CM pathogenesis [104]. Furthermore, in TNF-α-activated subcutaneous fat tissue-derived endothelial cells, a model comparable to cerebral endothelium, P. falciparum iRBCs induced several CM-specific effects, including up-regulation of ICAM-1, VCAM-1, and CD61, enhancement of microparticle, MCP-1 and IL-6 release, and higher caspase-3 activation [106]. Increased levels of inflammatory cytokines may have direct systemic effects and adversely affect the clinical outcome by increasing the cytoadherence of infected RBCs to venular endothelium through up-regulation of adhesion molecules, such as ICAM-1 [93].
To assess the specificity of these effects for human cerebral endothelium, additional comparative studies were also performed using non-neural endothelial cells. Interestingly, P. falciparum iRBCs did not affect the expression and distribution of tight junctions (as measured by claudin-5 and ZO-1) and did not induce pro-inflammatory response or cell death in human dermal or lung microvascular endothelium, although parasite sonicates did [103]. Additionally, the up-regulating effects of iRBCs on ICAM-1 expression observed in HBMEC were not reproduced in human umbilical vascular endothelial cells (HUVEC) from healthy donors [93]. An increase in ICAM-1, VCAM-1, and E-selectin mRNA was found in HUVEC from patients with different degrees of malaria (uncomplicated, severe, or CM) after co-culturing with iRBC-fed mononuclear cells, however such increase did not appear specific for CM. On the contrary, reduced mRNA levels of tight junction proteins (occludin, vinculin, and ZO-1) were strictly associated with CM [100].
Genetic differences between Plasmodium strains might also play a role in CM development. Indeed, it has been shown that different strains of P. falciparum display variable degrees of cytoadherence to HBEC-5i [107]. Additionally, P. berghei ANKA, a murine CM-associated Plasmodium strain, induces a higher VCAM-1-mediated cytoadherence compared to P. berghei K173 (non-CM strain) in either brain or lung mouse vascular endothelial cells [108].
Blood–brain barrier and in vivo animal models of cerebral malaria
Several in vivo animal models have reported alterations in BBB after exposure to Plasmodium parasites or malaria products such as hemozoin (Hz, malarial pigment) [47, 109–124]. As summarized in Table 2 and described below, these studies provide insightful findings regarding BBB breakdown in animal CM models.
The first animal studies on BBB permeability in malaria date back to 1968, when Migasena and Maegraith demonstrated the movement of albumin across the BBB in Macaca mulatta monkeys infected with P. knowlesi[109–111]. However, P. knowlesi does not induce CM. As such, the rhesus monkey infected with primate malaria parasites, P. coatneyi and P. fragile, is considered to be a more valid primate model to study in the context of severe malaria with cerebral involvement [112–114].
Of the four species of rodent malaria parasites (P. berghei, P. yoelii, P. chabaudi, P. vinckei), only a few P. berghei strains can induce experimental CM in mice, with the ANKA strain being the most widely studied. Symptoms of experimental CM in P. berghei ANKA-infected susceptible mice include paralysis, ataxia, head deviation, convulsion and coma [98]. In P. berghei K173-infected mice an excessive movement of water, albumin and other proteins into the brain, as well as severe brain edema, microthrombosis, sludging of mononuclear cells, arteriolar spasms, scattered disturbances of the microcirculation, and occasional proliferation of gliocytes were observed, suggesting a progressive deterioration of BBB integrity culminating in endothelial lesions and haemorrhages [31, 115–118]. Of note, mouse CM models present neurological signs (ataxia, hemiplegia and coma) similar to the clinical features reported in human CM [119].
In a recent work, Penet and colleagues presented the first in vivo magnetic resonance study of mouse CM, demonstrating BBB breakdown in CM. Multimodal magnetic resonance neuroimaging techniques (imaging, diffusion, perfusion, angiography, spectroscopy) of P. berghei ANKA-infected mice revealed vascular damage, including BBB disruption and haemorrhages, major edema formation, reduced brain perfusion and ischemic metabolic profile, with reduced high-energy phosphates and enhanced brain lactate. These data strongly point to the coexistence of inflammatory response and ischemic lesions [120].
Other recent works illustrated a complex strain-dependent relationship between leukocyte recruitment, BBB permeability and chemokine production. Major pathological consequences of malaria arise from inappropriate or excessive immune response mounted by the host in an attempt to eliminate the parasite. In P. berghei ANKA-infected mice, inflammation of the cerebral microvasculature and leukocyte recruitment were clearly evident and found to be driven by production of pro-inflammatory cytokines (IL-12, IFN-γ) and CM development [121]. On the other hand, P. berghei NK65-infected mice showed enhanced production of LT-α and several chemokines (CXCL-9/MIG, CCL-2/MCP-1, CCL-3/MIP-1α and CCL-5/RANTES), but no neurological symptoms [47, 122]. A complementary study performed on the same model proposed a concurrent role for Transforming Growth Factor-β (TGF-β) and TNF-α in promoting splenocyte apoptosis [123].
It should be noted that the cerebral microvascular tree contains two functionally distinct BBB: i) the physiological BBB, formed by capillaries 4–8 mm in diameter, consisting of a single layer of endothelia, gliovascular membrane, and astrocyte endfeet; and ii) the neuroimmunological BBB, formed by postcapillary venules 10–60 mm in diameter and encompassing two layers - the endothelium with its basement membrane and the glia limitans with associated astrocyte endfeet - separated by the perivascular space [125]. The physiological BBB serves as a tight diffusion barrier for small solutes while the neuroimmunological BBB permits transport of macromolecules and diapedesis of immune cells [125]. In a very recent study comparing different mouse models of experimental CM (P. berghei ANKA infection), human CM-like histopathology (P. yoelii 17XL) and non-CM (P. berghei NK65 and P. yoelii YM), Nacer and colleagues observed that the physiological BBB in the experimental CM model remained intact, whereas regulated fluid transport across the neuroimmunological BBB led to brain swelling, intracranial hypertension, coma, and ultimately death due to dysfunction of respiratory centers in pons and the medulla oblongata as a result of brain stem compression [124]. Thus, they proposed that CM may occur in two steps: 1) induction of coma based on regulated, preventable and reversible opening of the neuroimmunological BBB; and 2) endothelial death-associated haemorrhaging, which is difficult to reverse by treatment and eventually fatal [124]. A similar mechanism for neuroimmunological BBB opening in human CM would explain the reversibility of coma with treatment, the scarce traces of tissue necrosis in surviving patients, and the different neurological outcomes of patients despite similar clinical presentation [6, 8, 9, 13, 102].
Blood–brain barrier and human studies on cerebral malaria
BBB functional impairment during human CM has been investigated in several clinical and post mortem studies [126–143]. Table 3 summarizes the most relevant results. Here, the investigations on human CM patients were performed using albumin CSF/serum ratio as an indicator of BBB integrity [126–128], by post mortem immunohistochemical analysis [129–135], or through brain imaging techniques [136–144]. Interestingly, the BBB seems to be more impaired in children than in adults. Moreover, it appears that African and Asian patients display a different degree of BBB damage, with BBB breakdown being more likely to occur in African than Asian populations.
One of the first studies on Asian patients was conducted in Thailand [126]. In this work, albumin CSF/serum ratios were higher in CM patients than in controls, but it did not correlate with coma and mortality. Thus, the authors concluded that their data did not support the idea that cerebral edema might be the cause of coma. More than a decade later, albumin and Immunoglobulins G plasma/CSF ratios were found to be only mildly impaired in Vietnamese patients, suggesting only minimal degree of BBB breakdown in few CM cases [127]. Therein, human CM appeared to cause only subtle functional changes in BBB integrity, with minimal intra-parenchymal inflammatory response compared with other neurologic infections, such as cryptococcal, tubercular, and acute bacterial meningitis [127].
Regarding African populations, a study on Zairean children showed no difference in CSF albumin compared to controls [128]. However, in Malawian children with CM, the activation of endothelial cells and macrophages, along with the disruption of endothelial intercellular junctions in vessels containing sequestered iRBCs, and subtle but measurable changes in albumin CSF versus albumin serum levels were observed. Nevertheless, negligible leakage of plasma proteins was still apparent [129]. In Kenyan children with CM, protein and amino acid levels in paired plasma and CSF samples were measured, showing that BBB was mildly impaired in some children with severe falciparum malaria [130]. However, this impairment was not confined to CM, as it was also reported in children with prostration-associated malaria and, to a lesser extent, in children with malaria and seizures. Evidence of intrathecal immunoglobulin synthesis in children with malaria was also observed [130]. Finally, data obtained in a recent work performed on Malawian children are consistent with the proposed link between iRBCs sequestration and intravascular/perivascular pathology in fatal pediatric CM, resulting in myelin damage, axonal injury, and BBB breakdown; however, no Hz-laden monocyte extravasation was found [131].
Pathological studies on post mortem samples of CM patients showed cerebral edema and raised intracranial pressure in 50% of West African children [132] but not in South Asian adults [133, 134] or Malawian children [129]. Nevertheless, an important correlation between sequestration of iRBCs in the brain microvessels and the malaria-related encephalopathy was shown in Asian patients [133]. The adhesion of iRBCs to brain microvessels is mediated by specific receptors on the host endothelium, including ICAM-1, CD36 and CD31 [22–25]. Immunohistochemistry showed altered distribution of the cell junction proteins occludin, vinculin and ZO-1 in Vietnamese adults and Malawian children with CM [129, 135]. Sequestration of iRBCs in cerebral microvessels was significantly higher in the brains of patients with CM compared with non-CM patients in all parts of the brain (cerebrum, cerebellum, and medulla oblongata), and was quantitatively associated with pre mortem coma [129].
In recent years, several imaging studies have been also conducted on the brains of CM patients during disease progress or after recovery [136–144]. Using magnetic resonance or computed tomography, several common features implicating BBB damage have been observed, including cerebral edema, increased brain volume, ischemia and large vessel infarcts, hemorrhagic cortical lesions, focal and multifocal atrophy, and limited CSF circulation [136–139, 141–144]. Interestingly, magnetic resonance in a recently published case-report of a 37-year-old French patient with malaria travelling back from Equatorial Guinea, showed that he developed posterior reversible encephalopathy syndrome, which is characterized by diffuse symmetric signal-intensity abnormalities of white matter in the posterior circulation territory [140]. Since data from previous literature suggest one of the mechanisms of posterior reversible encephalopathy involves capillary leakage and acute disruption of the BBB, the authors concluded that this case-report supports the theory of BBB disruption as a key factor for CM development [140].
Blood–brain barrier impairment in cerebral malaria: some reflections upon the available studies
Clearly there is much discrepancy on the extent of BBB damage between animal and human models of CM. While several studies performed on mouse CM models suggest a strong BBB breakdown [115–124], data on increased BBB permeability in human CM are somehow less evident, generally suggesting the occurrence of only mild BBB impairment, characterized by a relevant degree of tight junction disruption, but lacking molecule exchange between serum and CSF [126–135].
In this context, it should be noted that the relevance of murine CM models for studying CM pathophysiology has been a topic of big debate in the recent years [49]. Being clearly an inflammatory syndrome with local vascular endothelial activation, murine CM displays obvious differences and some similarities to the clinical and pathological features of human CM, such as signs of vascular inflammation/damage [145]. A recurring issue concerns the degree of iRBC sequestration in the brain and other organs of P. berghei ANKA–infected mice. Although recent data find increased iRBC accumulation during murine CM in multiple organs including the brain [146], P. berghei infection is generally acknowledged to promote marked accumulation of leukocytes (particularly monocyte, macrophages and T cells), which is in stark contrast to human CM [147]. Thus, despite several processes shared either by murine or human CM, the changes in the endothelial cell microenvironment induced by cytoadherence and inflammation are not the same [104]. Additionally, mouse studies suggesting associations between high levels of cytokines and CM [58–61] have been recently challenged by works showing that high levels of pro-inflammatory cytokines such as TNF-α are poor indicators of human CM in African children [64–67]. Thus, future experimental studies on alternative animal models (non-human primates and other mouse models) are encouraged and urgently necessary to better understand the pathological processes underlying human infection [49].
Another interesting point emerging from clinical data is that the BBB appears more impaired in children than in adults [126–143]. Since CM often strikes children at a critical time in brain development, Hawkes and colleagues have nicely hypothesized that developmental changes in the cerebral vasculature may account for some of the differences in disease presentation and outcome between children and adults, including mortality, seizures and neurocognitive sequelae, rates of associated anemia and renal dysfunction, retinal vessel changes, frequency of ring haemorrhages, and inflammatory cell accumulation in brain microvessels [148]. To avoid any misunderstanding, it has been pointed out that the widespread belief amongst neurotoxicologists that BBB is immature or even absent in the newborn is contrasted by a substantial body of evidence supporting the concept of well-developed barrier mechanisms in the developing brain [149, 150]. For instance, inter-cellular tight junctions between cerebral endothelial cells and between choroid plexus epithelial cells are functionally effective as soon as they differentiate [150]. Nevertheless, the brain develops within an environment that is different from that of the rest of the body, and the developing brain possesses a number of unique features not generally present in the adult [149]. Interestingly, certain genes coding for influx/efflux proteins are expressed at much higher levels early in development than in the adult, and there is physiological evidence that these transport systems are functionally more active in the developing brain [150]. How such differences between the pediatric and adult BBB can affect CM pathogenesis and correlate with enhanced BBB permeability in pediatric CM is still unknown. Future research aimed at shedding light on this topic will certainly be useful.
Involvement of matrix metalloproteinases in cerebral malaria
In the last decade, experimental evidence implicated a specific family of host proteolytic enzymes known as MMPs in malaria pathogenesis [2, 151–154]. MMPs are either secreted or membrane-bound zinc-dependent proteases, and their role is also related to the inflammatory response and the BBB function [155–163]. Members of the MMP family are produced by a broad spectrum of specialized cells, including fibroblasts, endothelial cells, lymphocytes, monocytes, macrophages, smooth muscle cells, glial cells, and neurons [155, 158].
As detailed in Figure 3, the mammalian MMP family encompasses 25 members, categorized by different numbers or named depending on their matrix substrates. MMPs are evolutionarily conserved and tightly regulated. Conserved protein domains include an N-terminal signal peptide required for secretion, a cleavable pro-domain maintaining enzymatic latency, a catalytic domain, a Zn-binding domain, and (besides the minimal MMPs) a C-terminal hemopexin domain thought to be important for protein-protein interactions [155, 157, 160, 162, 163]. The active domain and the Zn-binding domain are essential for catalytic activity: upon pro-domain cleavage a Zn2+-ion becomes available to coordinate with a hydrolytic water molecule to enable nucleophilic attack of a substrate, and the enzyme is functionally active [164]. Additional MMP motifs include a gelatin-binding fibronectin-like domain, a serine-, threonine- and proline-rich collagen type V-like domain, a C-terminal transmembrane (TM) domain or GPI anchor, and in some cases a cytoplasmic domain [155, 157, 160, 162, 163]. MMP-2 and MMP-9 (also named gelatinase A and B) are further characterized by the presence of three head-to-tail cysteine-rich repeats within the catalytic domain reminiscent of the collagen-binding type II repeats of fibronectin and this domain is necessary for the binding and cleaving activities of these MMPs [161, 165, 166]. Six membrane-anchored MMPs (MT1- to MT6-MMPs) have a basic RX(K/R)R motif at the C-terminal end of their pro-domains. This motif is recognized and cleaved intracellularly by furin-like proteases. Four MT-MMPs (MT1- to MT4-MMPs) are anchored to the cell membrane through a type I TM domain while the other two MT-MMPs (MT5- and MT6-MMPs) are tethered to the membrane via a GPI moiety. An additional MT-MMP (MMP-23) has an N-terminal type II TM domain [162].

Nomenclature and structure of mammalian matrix metalloproteinases(MMPs). The mammalian MMP family encompasses 25 members, categorized by different numbers (standard MMP nomenclature) or named depending on their matrix substrates (enzyme nomenclature). Each MMP displays some conserved structural domains, including: i) an N-terminal signal peptide required for secretion; ii) a cleavable pro-domain maintaining enzymatic latency; iii) a catalytic and Zn-binding domain; and iv) a C-terminal hemopexin domain. Optional MMP motifs include a fibronectin-type domain, a vitronectin motif, a furine cleavage site, three head-to-tail cysteine-rich repeats, and (for MT-MMPs) a C-terminal transmembrane domain or GPI anchor occasionally associated with a cytoplasmic domain.
MMPs are regulated at multiple levels including transcription, translation, compartmentalization, secretion, activation, and inhibition by protein inhibitors. Most MMPs are found at low levels and not constitutively transcribed, but are expressed after external induction by pro-inflammatory molecules, growth factors, NO, cell-cell interactions, cell-matrix interactions, UV radiations [155, 157, 159–162]. Several signalling pathways and transcription factors are known to regulate MMP expression, including mitogen-activated protein kinases (MAPKs), NF-kB, and activator protein-1 (AP-1) [155, 165, 167]. After synthesis, MMPs are stored in inflammatory cell granules, which restrict their action [161]. Furthermore, MMPs are produced as inactive zymogens, referred to as pro-MMPs. Activation is achieved by various proteases (other activated MMPs and several serine proteases) or ROS that disrupt the interaction between the active site zinc atom in the catalytic domain and the conserved cysteine within the pro-domain. Pro-MMPs can be cleaved and activated through different mechanisms and in a context-specific manner. For example, pro-MMP-9 is activated through a proteolytic cascade sequentially involving plasminogen, MMP-3 and MMP-1 [165]. The activation of proMMP-2 requires previous formation of a pro-MMP-2/tissue inhibitor of metalloproteinase-2 (TIMP-2)/MT1-MMP (MMP-14) multimeric complex [166]. In addition to pro-MMP-2 activation, the binding of TIMP-2 to MT-1-MMP and MT-3-MMP slows down the autocatalytic turnover of these MT-MMPs, paradoxically enhancing surface proteolysis further by stabilizing the pool of active enzyme at the cell surface [160, 168]. Once MMPs have been released into the extracellular space or anchored to the membrane and activated, they are kept in check by their endogenous tissue inhibitors (four different forms, from TIMP-1 to TIMP-4). TIMPs inhibit MMP activity with relatively low selectivity in a 1:1 stoichiometric ratio. Interestingly, the ratio of MMP:TIMP can also influence activation mechanisms [157, 167].
MMPs were originally discovered in tadpoles as the agents responsible for tail resorption during frog metamorphosis. Thus, they were first characterized as proteases involved in degrading structural proteins comprising the extracellular matrix (ECM) and sub-endothelial basement membranes [155]. However, MMPs are now known to have more sophisticated processes than mere ECM turnover. MMPs can also cleave a growing spectrum of other substrates, including cytokines, chemokines, growth factors, hormones, chemotactic and adhesion molecules, membrane receptors, intercellular junctions, as well as other proteases, including some hemostasis-related molecules and MMPs themselves, protease inhibitors, clotting factors, and antimicrobial peptides [169–171]. MMP-dependent cleavage can serve to activate, inhibit, process, release, shed, or reveal cryptic codes in the substrates they act on. Therefore, the once formidable proteolytic potential of MMPs is now realized to serve essential roles in promoting or inhibiting cell survival, proliferation, migration, invasion, hemostasis and inflammation in either physiological or pathological processes [159, 167, 169, 170].
In physiology, MMPs are involved in diverse biological mechanisms ranging from wound repair to pregnancy [155, 157, 159, 167]. In pathology, MMP dysfunction has been implicated in cancer, cardio-vascular diseases, emphysema, acute renal failure, ophthalmic pathologies, neuroinflammation, neurodegenerative disorders, autoimmune diseases [156–160, 167, 169, 170] and, very recently, malaria [2, 151–154].
Matrix metalloproteinases and animal models
In vivo mouse models of CM have recently implicated MMP dysfunction in disease pathology, although it should be kept in mind that experimental CM presents important differences compared to human CM, such as leukocyte sequestration in cerebral microvessels and subsequent migration into brain parenchyma [49]. An excellent study performed by Van den Steen and his group comprehensively investigated mRNA expression levels of MMPs and protein release or pro-enzyme activation in five different organs (brain, lung, spleen, liver, and kidney) from CM-sensitive C57B1/6 mice infected with P. berghei ANKA (CM model) or P. berghei NK65 (non-CM model) and CM-resistant Balb/C mice infected with P. berghei ANKA (CM-resistant model) [172]. Importantly, they observed enhanced expression and activation of monocytic (CD11b+) MMP-9 in brains of CM mice [172] specific to CM, as suggested by comparison with non-CM models, such as lung pathology [173]. Additionally, tissue-specific increases in mRNA expression were found for several MMPs, including MMP-3, -4, -8, and -13 in spleen, MMP-8, -12, -13, and -14 in liver, and MMP-8 and -13 in brain. All of these increases were more pronounced in the CM model. In a CM-resistant model, MMP-3 expression was significantly enhanced, suggesting a protective role for this MMP in CM [172]. In another study, CM mice showed increased neural MMP-7 protein levels [151]. Interestingly, urokinase-type plasminogen activator (uPA) -/- or urokinase-type plasminogen activator receptor (uPAR) -/- knock-out mice with CM displayed enhanced survival and attenuated thrombocytopenia [174].
A parasite molecule, malarial pigment Hz - a lipid-bound ferriprotoporphyrin IX produced by P. falciparum after hemoglobin catabolism [175] - is proposed to play a role in experimental CM. Indeed, using a sensitive fluorometric method to determine Hz content in blood and tissue samples from mice infected with P. berghei NK65 (non-CM model) or ANKA (CM model), Sullivan and colleagues observed increased Hz levels in tissue correlating with the duration of infection, with neural Hz levels being higher in CM than non-CM mice, raising the possibility that Hz presence may be associated with cerebral pathology [176].
Interestingly, in vitro, Hz appears to play a major role in MMP dysfunction. Phagocytosis of Hz by RAW 264.7 rat macrophage cell line was shown to impair expression of several inflammatory molecules [177] and, after an early inhibitory peak, to increase the long-term mRNA expression of MMP-9 [178]. This effect was related to the lipid moiety of Hz, since lipid-free synthetic Hz (β-hematin) did not modulate MMP-9 expression. The Hz-dependent enhancement of MMP-9 transcription and protein release was mimicked by 4-hydroxy-2-nonenal (4-HNE) [178], a molecule generated by Hz from polyunsaturated fatty acids [179].
Matrix metalloproteinases and human studies
In vitro studies using human monocytes and endothelial cells [152, 180–195] provide convincing and homogeneous evidence for Hz-dependent mechanisms underlying aberrant MMP-9 function (see Figure 4). In a series of works performed with human adherent or immunopurified monocytes from peripheral blood, the phagocytosis of free Hz or Hz-containing trophozoites enhanced MMP-9 mRNA levels, protein expression, and activity [180–186]. This observation was also investigated using THP-1 monocyte cell line [187]. Hz-fed monocytes display increased total gelatinolytic activity [186] and invasiveness [180] caused by MMP-9 - but not MMP-2 [188] - enhancement.

Principal mechanisms underlying Hz-dependent dysregulation of matrix metalloproteinase-9 (MMP-9) and related molecules in human monocytes. The most relevant Hz-dependent mechanisms underlying aberrant MMP-9 function in malaria are based on current evidence from in vitro models of cultured human monocytes. After Hz phagocytosis, some lipoperoxidation products generated by Hz autocatalysis such as 15-HETE can promote early and late activation of PKC and p38 MAPK. These kinases have been associated with cytosolic I-kBα phosphorylation and degradation, resulting in subsequent nuclear translocation of NF-kB p50 and p65 subunits. Consequently, the transcription and protein expression of several pro-inflammatory molecules including TNF-α, IL-1β, and MIP-1α, and of some proteolytic enzymes or inhibitors such as lysozyme, pro-MMP-9, and TIMP-1 is enhanced. p38 MAPK can also promote monocyte degranulation, releasing pro-MMP-9, TIMP-1 and lysozyme into the extracellular environment. After secretion of these molecules, several proteolytic events can occur. Active MMP-9, generated from MMP-3 processing of the pro-enzyme, can further modulate TNF-α shedding from cell membrane in a similar manner as TACE, whereas ICE activates IL-1β after cleaving its pro-peptide. Soluble TNF-α, IL-1β, and MIP-1α have been shown to play a key role in mediating Hz effects on MMP-9, lysozyme, and TIMP-1 production, possibly generating some auto-enhancing loops. Hz-enhanced MMP-9 could favour CM development through complementary proteolytic activities (see Figure 5). On the other hand, TIMP-1 is primarily referred to as a MMP-9 inhibitor, thus TIMP-1 Hz-enhanced levels could supposedly be protective. However, several MMP-independent functions such as inhibition of cell apoptosis and growth have been recently described for TIMP-1. Thus, Hz-enhanced TIMP-1 protein may play a role in prolonged survival of impaired Hz-fed monocytes, in their altered maturation to dendritic cells and in their reduced ability to coordinate erythropoiesis. Finally, enhanced plasma levels of human lysozyme have been depicted as a risk factor for severe malaria.
Increased MMP-9 function in human monocytes appears to be mediated by Hz-dependent over-production of several pro-inflammatory molecules, including TNF-α [180], IL-1β [181], and CCL-3/MIP-1α [184]. Further investigation revealed increases in MMP-9 [181], TNF-α [189, 190] and IL-1β [181, 190], but not CCL-3/MIP-1α [183, 190], were dependent on the lipid moiety of Hz. These studies unveiled a major role for 15-HETE, a potent lipid peroxidation derivative generated by Hz autocatalysis. Hz was also causally related to increased TIMP-1 and lysozyme release from human adherent monocytes, two molecules stored in gelatinase granules along with MMP-9 [186, 190–192]. Further studies also showed that Hz-induced monocyte degranulation was mediated by TNF-α, IL-1β and MIP-1α/CCL-3 [191, 192] and dependent on Hz lipid moiety, suggesting a major role for 15-HETE [190]. The heme core of Hz was shown to bind MMP-9 hemopexin domain and to prime the activation of the zymogen by other MMPs, such as MMP-3 [193]. The mechanisms underlying Hz-dependent enhancement of MMP-9, TNF-α, IL-1β, CCL-3/MIP-1α, TIMP-1 and lysozyme appear to involve NF-kB activation, as suggested by results from parallel works performed with adherent monocytes from peripheral blood [182, 186, 190, 191] and THP-1 cell line [187]. In these works, Hz-induced enhancement of MMP-9, TNF-α, IL-1β, CCL-3/MIP-1α and TIMP-1, as well as total gelatinolytic and lysozyme activity were abrogated by using different NF-kB inhibitors showing anti-malarial properties (quercetin, artemisinin, parthenolide, and extracts from Punica granatum). Moreover, results from experiments with SB203580, a known inhibitor of p38 MAPK pathway suggest that concurrent activation of p38 MAPK pathway seems to be mandatory for Hz- and 15-HETE-dependent increased MMP-9 [185] and related molecules TNF-α, IL-1β, CCL-3/MIP-1α, TIMP-1 and lysozyme [186, 190, 191]. On the contrary, ERK and JNK MAPK pathways do not seem to be activated by Hz [185].
Additional evidence on Hz-dependent MMP dysregulation is also derived from studies using human endothelial cells. In the human microvascular endothelial cell line HMEC-1, either free Hz [194] or Hz-containing-iRBCs [195] induced the release of pro-MMP-9 and active MMP-9 proteins de novo without altering pro-MMP-2 basal levels. Interestingly, Hz also enhanced the protein levels of basal MMP-1 and MMP-3, two MMPs sequentially involved in pro-MMP-9 activation. Consequently, total gelatinolytic activity and cell invasion were increased. More evidence of Hz-triggered enhancement of MMP-9 protein release emerges from studies using human endothelial cells from large calibre vessels [152]. Similar to human monocytes, a role for the Hz lipid moiety also appears likely in endothelial cells, although the molecules responsible for these effects have not yet been identified [194].
Little evidence exists on the in vivo involvement of MMPs in human CM from organ autopsy studies or from fluid (blood, serum) analysis of patients with uncomplicated or severe malaria [65, 196–201]. The little data that does exist are somehow conflicting and still incomplete.
The accumulation of pro-angiogenic uPAR [196], MMP-1 and VEGF [197] in Durck granulomas, along with the lack of anti-angiogenic endostatin/collagen XVIII in ring haemorrhage areas [198] has been observed in the brains of patients with fatal CM, suggesting that the proMMP-9 proteolytic machinery is activated in areas of intense parasite sequestration and vascular damage. Consistently, microarray analysis performed on whole blood from Kenyan children with severe malaria showed P. falciparum activation of the human MMP-9 gene [199].
However, sera of Gabonese and Ghanaian children with uncomplicated or severe malaria did not display altered MMP-9 levels [65, 200]. Moreover, serum MMP-8 levels were elevated in Gabonese children with either uncomplicated malaria or CM [200], but not in Nigerian children with uncomplicated malaria [201]. Nonetheless, it has been argued that serum levels might not be an ideal source for reliable data concerning MMP levels, since MMP release from blood cells during sample processing might give a highly non-specific background result, thus hindering the ability to assess true concentrations of circulating MMPs [202].
Possible role of matrix metalloproteinases in pathophysiology and therapy of cerebral malaria
It is likely that MMPs play an active role in several steps during CM development as they can process a large repertoire of substrates [169–171], including pro-inflammatory molecules, tight junctions, and hemostatic factors likely involved in CM (see Figure 5 and Tables 4 and 5).

Multiple putative roles of matrix metalloproteinases (MMPs) in cerebral malaria (CM) according to their biochemical functions. MMPs could play an active role during CM development through several complementary mechanisms: i) by disrupting endothelial tight junctions after protein degradation of ZO-1, claudins and occludin, thus causing an increased BBB permeability; ii) by promoting TNF-α shedding, IL-1β activation and CXCL8/IL-8 potentiation after proteolytic cleavage of their pro-domains, therefore inducing exacerbated pro-inflammatory response; iii) by processing some CM-associated hemostatic factors such as tpA, upA, and PAI-1, thus increasing the risk for thrombotic events.
First, MMPs proteolytically cleave the pro-forms of many cytokines and chemokines reportedly enhanced in CM, including TNF-α, IL-1β, CXCL-8/IL-8. As a result of cleavage, these molecules are shed, activated or functionally potentiated, respectively. Thus, MMPs could contribute to the uncontrolled inflammatory response typical of CM. Consistently, an in vitro study using adherent monocytes demonstrated a Hz-dependent pathological auto-enhancing loop established between MMP-9 and TNF-α [180].
Moreover, a growing number of inter-endothelial tight junctions, including occludin, ZO-1, claudin-1 and claudin-5 are known MMP substrates (e.g. MMP-2, MMP-3, MMP-7 and MMP-9), which raises the possibility that MMP activity on these substrates can enhance the permeability of endothelial barriers by destroying these junctions [203–209]. Therefore, enhanced MMP levels in malaria might facilitate BBB leakage. This hypothesis is consistent with data showing that Hz-containing iRBCs reduced human BBB permeability in vitro[102], and that Hz and iRBCs enhanced MMP-1, MMP-3 and MMP-9 produced by human microvascular endothelium [194, 195].
Finally, MMPs can also influence hemostasis. Both tPA and uPA molecules, responsible for plasminogen conversion to plasmin, as well as tPA/uPA inhibitor PAI-1 can be processed by MMPs [210–212]. As a consequence, MMPs might affect fibrin degradation by plasmin, perhaps explaining thrombotic events that frequently occur during CM.
Interestingly, some molecules such as 4-aminoquinolines and artemisinins, which are currently used for primary therapy of uncomplicated malaria, have displayed MMP-inhibiting properties. Chloroquine treatment reduces MMP-9 serum levels in patients with systemic lupus erythematosus [213]. Artemisinin down-regulates MMP-2 levels in human melanoma cells [214] and MMP-1, MMP-2 and MMP-9 levels in mouse embryonic stem-cells derived from embryoid bodies [215]. Dihydroartemisinin inhibits MMP-2, MMP-9 and MMP-14 expression/activity in human fibrosarcoma cells [216] and MMP-9 expression in human umbilical vein endothelial cells [217]. Finally, artesunate down-regulates MMP-2 and MMP-7 expression in human non-small cell lung cancer [218]. However, it should be noticed that the drug concentrations used in such studies need to be verified and optimized for human clinical trials. This might explain why antimalarial drugs alone cannot prevent CM development.
It is intriguing to explore the idea of targeting MMPs with broad spectrum or specific MMP inhibitors as adjuvant therapy in CM. In the last two decades, a large number of synthetic MMP inhibitors have gone through clinical trials and largely failed as anti-cancer and anti-arthritis drugs due to serious long-term side effects, with only one currently commercially available [219–223]. Hopefully, using combinations of MMP inhibitors with antimalarials could justify lower therapeutic doses of both drugs, thereby reducing their potential side effects whilst still enhancing anti-MMP properties by drug synergy. To date, the effects of MMP inhibitors in CM remain scarce. In vitro, the use of a specific synthetic inhibitor of MMP-9 was shown to abrogate Hz-dependent increase of TNF-α in human monocytes, suggesting that MMP-9 inhibition might be useful to counteract pathological inflammation in CM [180]. However, MMP-9 knock-out mice infected with P. berghei ANKA did not display any protection from CM development, probably due to the redundant functions of other MMPs which might compensate for the loss of MMP-9 [172]. On the contrary, treatment with broad-spectrum MMP inhibitor BB-94 significantly improved survival of CM mice [172].
Future research aimed at determining the exact role (protective or detrimental) of each MMP during malaria infections will be highly informative. Unfortunately, with the exception of a few cases, specific inhibitors against individual MMPs are currently lacking [224–226]. Some metalloproteinases are also produced by malaria parasites, for example to perform hemoglobin degradation [227]. Therefore, MMP inhibitors may not only influence host but also parasitic pathways. Another issue to be taken in account is represented by the effects of MMPs on other organs than brain. However, it should be noted that the adverse effects of MMP inhibitors documented in other pathologies such as cancer were associated with long-term treatment [220], whereas the time course of drug administration in CM therapy should be reasonably shorter, possibly limiting the development of side effects. A detailed analysis of the role of each protease in physiology and pathology, along with the development of specific inhibitors, could yield novel insights to assess whether specific MMP inhibition might be considered as new adjuvant therapies.
Conclusion
As suggested by three complementary theories developed over the past century, CM might be a likely consequence of several concomitant phenomena, including iRBC sequestration in brain microvessels, enhanced BBB permeability, and release of pro-inflammatory molecules from host immune cells. Data from in vitro and in vivo studies suggest that a full BBB breakdown during CM is more likely to occur in mouse than in humans. In the latter case, the BBB appears only mildly impaired as a result of tight junction disruption. MMPs are host proteolytic enzymes involved in degradation of basement membranes, disruption of inter-endothelial tight junctions, and cleavage of a large spectrum of pro-inflammatory, membrane-bound and hemostasis-related molecules, and they may play a crucial role in CM. Further in-depth analysis of the involvement of MMPs in CM might help to design new adjuvant therapies. In this context, MMP inhibitors could prevent BBB leakage and reduce the exacerbated inflammatory response, thus reducing the high mortality rates of CM patients, along with the frequency of neurological sequelae in recovering patients.
Authors’ information
Manuela Polimeni holds a post-doc fellowship granted by Università di Torino along with Ministero Italiano dell’Università e della Ricerca (MIUR). Mauro Prato holds a professorship granted by Università di Torino along with Azienda Sanitaria Locale-19 (ASL-19) and Compagnia di San Paolo.
Abbreviations
- BBB:
-
Blood–brain barrier (BBB)
- CNS:
-
Central nervous system
- CM:
-
Cerebral malaria
- CSF:
-
Cerebral-spinal fluid
- ECM:
-
Extracellular matrix
- Hz:
-
Hemozoin
- HR:
-
Histamine receptor
- 15-HETE:
-
15-hydroxyeicosatetraenoic acid
- 4-HNE:
-
4-hydroxy-2-nonenal
- iRBCs:
-
Infected red blood cells
- IDO:
-
Indoleamine 2,3-dioxygenase
- IFN:
-
Interferon
- IL:
-
Interleukin
- ICAM-1:
-
Intracellular adhesion molecule-1
- MMP:
-
Matrix metalloproteinase
- MCP-1:
-
Monocyte chemotactic protein-1
- NO:
-
Nitric oxide
- NOS2:
-
Nitric oxide synthase type 2
- MAPK:
-
Mitogen-activated protein kinase
- NF-kB:
-
Nuclear factor-kB
- PfEMP-1:
-
P. falciparum erythrocyte membrane protein-1
- PBCEC:
-
Porcine brain capillary endothelial cell
- HBMEC:
-
Human brain microvascular endothelial cell
- HUVEC:
-
Human umbilical vascular endothelial cell
- ROS:
-
Reactive oxygen species
- TIMP:
-
Tissue inhibitor of metalloproteinase
- TEER:
-
Trans-endothelial electrical resistance
- TGF:
-
Transforming growth factor
- TNF:
-
Tumor necrosis factor
- VEGF:
-
Vascular endothelial growth factor
- uPA:
-
Urokinase-type plasminogen activator
- uPAR:
-
Urokinase-type plasminogen activator receptor
- VCAM-1:
-
Vascular cellular adhesion molecule-1
- ZO-1:
-
Zonula occludens-1.
References
World Health Organization (WHO): World Malaria Report. 2013, Geneva: World Health Organization
Khadjavi A, Giribaldi G, Prato M: From control to eradication of malaria: the end of being stuck in second gear?. Asian Pac J Trop Med. 2010, 3: 412-420. 10.1016/S1995-7645(10)60101-0.
World Health Organization Malaria Policy Advisory Committee and Secretariat: Malaria Policy Advisory Committee to the WHO: conclusions and recommendations of September 2012 meeting. Malar J. 2012, 11: 424-
Mohanty S, Patel DK, Pati SS, Mishra SK: Adjuvant therapy in cerebral malaria. Indian J Med Res. 2006, 124: 245-260.
John CC, Kutamba E, Mugarura K, Opoka RO: Adjunctive therapy for cerebral malaria and other severe forms of Plasmodium falciparum malaria. Expert Rev Anti Infect Ther. 2010, 8: 997-1008. 10.1586/eri.10.90.
Grau GE, Craig AG: Cerebral malaria pathogenesis: revisiting parasite and host contributions. Future Microbiol. 2012, 7: 291-302. 10.2217/fmb.11.155.
Elsheikha HM, Khan NA: Protozoa traversal of the blood–brain barrier to invade the central nervous system. FEMS Microbiol Rev. 2010, 34: 532-553.
Newton CR, Warrel DA: Neurological manifestations of falciparum malaria. Ann Neurol. 1998, 43: 695-702. 10.1002/ana.410430603.
Gitau EN, Newton CR: Review article: blood–brain barrier in falciparum malaria. Trop Med Int Health. 2005, 10: 285-292. 10.1111/j.1365-3156.2004.01366.x.
World Health Organization (WHO): Guidelines for the treatment of malaria. 2006, Geneva: World Health Organization
Berkley JA, Mwangi I, Mellington F, Mwarumba S, Marsh K: Cerebral malaria versus bacterial meningitis in children with impaired consciousness. Q J Med. 1999, 92: 151-157. 10.1093/qjmed/92.3.151.
Newton CR, Krishna S: Severe falciparum malaria in children: current understanding of pathophysiology and supportive treatment. Pharmacol Therap. 1998, 79: 1-53. 10.1016/S0163-7258(98)00008-4.
Coltel N, Combes V, Hunt NH, Grau GE: Cerebral malaria - a neurovascular pathology with many riddles still to be solved. Curr Neurovasc Res. 2004, 1: 91-110. 10.2174/1567202043480116.
Pino P, Taoufiq Z, Nitcheu J, Vouldoukis I, Mazier D: Blood–brain barrier breakdown during cerebral malaria: suicide or murder?. Thromb Haemost. 2005, 94: 336-340.
Medana IM, Turner GD: Human cerebral malaria and the blood–brain barrier. Int J Parasitol. 2006, 36: 5-
Clark IA, Budd AC, Alleva LM, Cowden WB: Human malarial disease: a consequence of inflammatory cytokine release. Malar J. 2006, 5: 85-10.1186/1475-2875-5-85.
van der Heyde HC, Nolan J, Combes V, Gramaglia I, Grau GE: A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 2006, 22: 503-508. 10.1016/j.pt.2006.09.002.
Thomson JG, Annecke S: Observations of the pathology of the central nervous system in malignant tertian malaria, with remarks on certain clinical phenomena. J Trop Med Hyg. 1926, 29: 313-346.
MacPherson GG, Warrell MJ, White NJ, Looareesuwan S, Warrell DA: Human cerebral malaria. A quantitative ultrastructural analysis of parasitized erythrocyte sequestration. Am J Pathol. 1985, 119: 385-401.
Pongponratn E, Riganti M, Punpoowong B, Aikawa M: Microvascular sequestration of parasitized erythrocytes in human falciparum malaria: a pathological study. Am J Trop Med Hyg. 1991, 44: 168-175.
Silamut K, Phu NH, Whitty C, Turner GD, Louwrier K, Mai NT: A quantitative analysis of the microvascular sequestration of malaria parasites in the human brain. Am J Pathol. 1999, 155: 395-410. 10.1016/S0002-9440(10)65136-X.
Udeinya IJ, Schmidt JA, Aikawa M, Miller LH, Green I: Falciparum malaria-infected erythrocytes specifically bind to cultured human endothelial cells. Science. 1981, 213: 555-557. 10.1126/science.7017935.
David PH, Hommel M, Miller LH, Udeinya IJ, Oligino LD: Parasite sequestration in Plasmodium falciparum malaria: spleen and antibody modulation of cytoadherence of infected erythrocytes. Proc Natl Acad Sci USA. 1983, 80: 5075-5079. 10.1073/pnas.80.16.5075.
Sherman IW, Eda S, Winograd E: Cytoadherence and sequestration in Plasmodium falciparum: defining the ties that bind. Microbes Infect. 2003, 5: 897-909. 10.1016/S1286-4579(03)00162-X.
Chakravorty SJ, Craig A: The role of ICAM-1 in Plasmodium falciparum cytoadherence. Eur J Cell Biol. 2005, 84: 15-27. 10.1016/j.ejcb.2004.09.002.
Dondorp AM, Angus BJ, Chotivanich K, Silamut K, Ruangveerayuth R, Hardeman MR: Red blood cell deformability as a predictor of anemia in severe falciparum malaria. Am J Trop Med Hyg. 1999, 60: 733-737.
Dondorp AM, Angus BJ, Hardeman MR, Chotivanich KT, Silamut K, Ruangveerayuth R: Prognostic significance of reduced red blood cell deformability in severe falciparum malaria. Am J Trop Med Hyg. 1997, 57: 507-511.
Udomsangpetch R, Webster HK, Pattanapanyasat K, Pitchayangkul S, Thaithong S: Cytoadherence characteristics of rosette-forming Plasmodium falciparum. Infect Immun. 1992, 60: 4483-4490.
Angus BJ, Thanikkul K, Silamut K, White NJ, Udomsangpetch R: Short report: Rosette formation in Plasmodium ovale infection. Am J Trop Med Hyg. 1996, 55: 560-561.
al-Yaman F, Genton B, Mokela D, Raiko A, Kati S, Rogerson S: Human cerebral malaria: lack of significant association between erythrocyte rosetting and disease severity. Trans R Soc Trop Med Hyg. 1995, 89: 55-58. 10.1016/0035-9203(95)90658-4.
Maegraith B, Fletcher A: The pathogenesis of mammalian malaria. Adv Parasitol. 1972, 10: 49-75.
Adams S, Brown H, Turner G: Breaking down the blood–brain barrier: signaling a path to cerebral malaria?. Trends Parasitol. 2002, 18: 360-366. 10.1016/S1471-4922(02)02353-X.
Enwonwu CO, Afolabi BM, Salako LA, Idigbe EO, al-Hassan H, Rabiu RA: Hyperphenylalaninaemia in children with falciparum malaria. QJM. 1999, 92: 495-503. 10.1093/qjmed/92.9.495.
Enwonwu CO, Afolabi BM, Salako LO, Idigbe EO, Bashirelah N: Increased plasma levels of histidine and histamine in falciparum malaria: relevance to severity of infection. J Neural Transm. 2000, 107: 1273-1287. 10.1007/s007020070017.
Beghdadi W, Porcherie A, Schneider BS, Dubayle D, Peronet R, Huerre M: Role of histamine and histamine receptors in the pathogenesis of malaria. Med Sci (Paris). 2009, 25: 377-381. 10.1051/medsci/2009254377.
Beghdadi W, Porcherie A, Schneider BS, Dubayle D, Peronet R, Huerre M: Inhibition of histamine-mediated signaling confers significant protection against severe malaria in mouse models of disease. J Exp Med. 2008, 205: 395-408. 10.1084/jem.20071548.
Beghdadi W, Porcherie A, Schneider BS, Morisset S, Dubayle D, Peronet R: Histamine H(3) receptor-mediated signaling protects mice from cerebral malaria. PLoS One. 2009, 4: 6004-10.1371/journal.pone.0006004.
Clark IA, Virelizier JL, Carswell EA, Wood PR: Possible importance of macrophage-derived mediators in acute malaria. Infect Immun. 1981, 32: 1058-1066.
Clark IA, Cowden WB: The pathophysiology of falciparum malaria. Pharmacol Ther. 2003, 99: 221-260. 10.1016/S0163-7258(03)00060-3.
Clark IA, Alleva LM, Budd AC, Cowden WB: Understanding the role of inflammatory cytokines in malaria and related diseases. Travel Med Infect Dis. 2008, 6: 67-81. 10.1016/j.tmaid.2007.07.002.
Hansen DS: Inflammatory responses associated with the induction of cerebral malaria: lessons from experimental murine models. PLoS Pathog. 2012, 8: e1003045-10.1371/journal.ppat.1003045.
Belnoue E, Kayibanda M, Vigario AM, Deschemin JC, van Rooijen N, Viguier M: On the pathogenic role of brain-sequestered alphabeta CD8+ T cells in experimental cerebral malaria. J Immunol. 2002, 169: 6369-6375.
Belnoue E, Potter SM, Rosa DS, Mauduit M, Grüner AC, Kayibanda M: Control of pathogenic CD8+ T cell migration to the brain by IFN-gamma during experimental cerebral malaria. Parasite Immunol. 2008, 30: 544-553. 10.1111/j.1365-3024.2008.01053.x.
Claser C, Malleret B, Gun SY, Wong AY, Chang ZW, Teo P: CD8+ T cells and IFN-γ mediate the time-dependent accumulation of infected red blood cells in deep organs during experimental cerebral malaria. PLoS One. 2011, 6: e18720-10.1371/journal.pone.0018720.
Hansen DS, Bernard NJ, Nie CQ, Schofield L: NK cells stimulate recruitment of CXCR3+ T cells to the brain during Plasmodium berghei-mediated cerebral malaria. J Immunol. 2007, 178: 5779-5788.
Gimenez F, Barraud de Lagerie S, Fernandez C, Pino P, Mazier D: Tumor necrosis factor alpha in the pathogenesis of cerebral malaria. Cell Mol Life Sci. 2003, 60: 1623-1635. 10.1007/s00018-003-2347-x.
Engwerda CR, Mynott TL, Sawhney S, De Souza JB, Bickle QD, Kaye PM: Locally up-regulated lymphotoxin alpha, not systemic tumor necrosis factor alpha, is the principle mediator of murine cerebral malaria. J Exp Med. 2002, 195: 1371-1377. 10.1084/jem.20020128.
Martins YC, Carvalho LJ, Daniel-Ribeiro CT: Challenges in the determination of early predictors of cerebral malaria: lessons from the human disease and the experimental murine models. Neuroimmunomodulation. 2009, 16: 134-145. 10.1159/000180268.
Craig AG, Grau GE, Janse C, Kazura JW, Milner D, Barnwell JW: The role of animal models for research on severe malaria. PLoS Pathog. 2012, 8: e1002401-10.1371/journal.ppat.1002401.
Suidan GL, Pirko I, Johnson AJ: A potential role for CD8+ T-cells as regulators of CNS vascular permeability. Neurol Res. 2006, 28: 250-255. 10.1179/016164106X98116.
Suidan GL, Mcdole JR, Chen Y, Pirko I, Johnson AJ: Induction of blood brain barrier tight junction protein alterations by CD8 T cells. PLoS One. 2008, 3: e3037-10.1371/journal.pone.0003037.
Suidan GL, Dickerson JW, Chen Y, McDole JR, Tripathi P, Pirko I: CD8 T cell-initiated vascular endothelial growth factor expression promotes central nervous system vascular permeability under neuroinflammatory conditions. J Immunol. 2010, 184: 1031-1040. 10.4049/jimmunol.0902773.
Lundie RJ, de Koning-Ward TF, Davey GM, Nie CQ, Hansen DS, Lau LS: Blood-stage Plasmodium infection induces CD8+ T lymphocytes to parasite-expressed antigens, largely regulated by CD8alpha + dendritic cells. Proc Natl Acad Sci USA. 2008, 105: 14509-14514. 10.1073/pnas.0806727105.
Miyakoda M, Kimura D, Yuda M, Chinzei Y, Shibata Y, Honma K, Yui K: Malaria-specific and nonspecific activation of CD8+ T cells during blood stage of Plasmodium berghei infection. J Immunol. 2008, 181: 1420-1428.
Howland SW, Poh CM, Gun SY, Claser C, Malleret B, Shastri N: Brain microvessel cross-presentation is a hallmark of experimental cerebral malaria. EMBO Mol Med. 2013, 5: 916-931.
Jambou R, Combes V, Jambou MJ, Weksler BB, Couraud PO, Grau GE: Plasmodium falciparum adhesion on human brain microvascular endothelial cells involves transmigration-like cup formation and induces opening of intercellular junctions. PLoS Pathog. 2010, 6: e1001021-10.1371/journal.ppat.1001021.
Yui K: Cross-presentation of malaria antigen by brain microvessels: why CD8(+) T cells are critical for the pathogenesis of experimental cerebral malaria. EMBO Mol Med. 2013, 5: 899-901.
Grau GE, Fajardo LF, Piguet PF, Allet B, Lambert PH, Vassalli P: Tumor necrosis factor (cachectin) as an essential mediator in murine cerebral malaria. Science. 1987, 237: 1210-1212. 10.1126/science.3306918.
Rudin W, Eugster HP, Bordmann G, Bonato J, Müller M, Yamage M, Ryffel B: Resistance to cerebral malaria in tumor necrosis factor-alpha/beta-deficient mice is associated with a reduction of intercellular adhesion molecule-1 up-regulation and T helper type 1 response. Am J Pathol. 1997, 150: 257-266.
de Miranda AS, Lacerda-Queiroz N, de Carvalho Vilela M, Rodrigues DH, Rachid MA, Quevedo J, Teixeira AL: Anxiety-like behavior and proinflammatory cytokine levels in the brain of C57BL/6 mice infected with Plasmodium berghei (strain ANKA). Neurosci Lett. 2011, 491: 202-206. 10.1016/j.neulet.2011.01.038.
Wu JJ, Chen G, Liu J, Wang T, Zheng W, Cao YM: Natural regulatory T cells mediate the development of cerebral malaria by modifying the pro-inflammatory response. Parasitol Int. 2010, 59: 232-241. 10.1016/j.parint.2010.02.007.
Parekh SB, Bubb WA, Hunt NH, Rae C: Brain metabolic markers reflect susceptibility status in cytokine gene knockout mice with murine cerebral malaria. Int J Parasitol. 2006, 36: 1409-1418. 10.1016/j.ijpara.2006.07.004.
Amante FH, Haque A, Stanley AC, Rivera Fde L, Randall LM, Wilson YA: Immune-mediated mechanisms of parasite tissue sequestration during experimental cerebral malaria. J Immunol. 2010, 185: 3632-3642. 10.4049/jimmunol.1000944.
Esamai F, Ernerudh J, Janols H, Welin S, Ekerfelt C, Mining S, Forsberg P: Cerebral malaria in children: serum and cerebrospinal fluid TNF-alpha and TGF-beta levels and their relationship to clinical outcome. J Trop Pediatr. 2003, 49: 216-223. 10.1093/tropej/49.4.216.
Armah HB, Wilson NO, Sarfo BY, Powell MD, Bond VC, Anderson W: Cerebrospinal fluid and serum biomarkers of cerebral malaria mortality in Ghanaian children. Malar J. 2007, 6: 147-10.1186/1475-2875-6-147.
Jain V, Armah HB, Tongren JE, Ned RM, Wilson NO, Crawford S: Plasma IP-10, apoptotic and angiogenic factors associated with fatal cerebral malaria in India. Malar J. 2008, 7: 83-10.1186/1475-2875-7-83.
Thuma PE, van Dijk J, Bucala R, Debebe Z, Nekhai S, Kuddo T: Distinct clinical and immunologic profiles in severe malarial anemia and cerebral malaria in Zambia. J Infect Dis. 2011, 203: 211-219. 10.1093/infdis/jiq041.
Lovegrove FE, Tangpukdee N, Opoka RO, Lafferty EI, Rajwans N, Hawkes M: Serum angiopoietin-1 and -2 levels discriminate cerebral malaria from uncomplicated malaria and predict clinical outcome in African children. PLoS One. 2009, 4: e4912-10.1371/journal.pone.0004912.
Prakash D, Fesel C, Jain R, Cazenave PA, Mishra GC, Pied S: Clusters of cytokines determine malaria severity in Plasmodium falciparum-infected patients from endemic areas of Central India. J Infect Dis. 2006, 194: 198-207. 10.1086/504720.
Stevenson MM, Su Z, Sam H, Mohan K: Modulation of host responses to blood-stage malaria by interleukin-12: from therapy to adjuvant activity. Microbes Infect. 2001, 3: 49-59. 10.1016/S1286-4579(00)01354-X.
Marquet S, Doumbo O, Cabantous S, Poudiougou B, Argiro L, Safeukui I: A functional promoter variant in IL12B predisposes to cerebral malaria. Hum Mol Genet. 2008, 17: 2190-2195. 10.1093/hmg/ddn118.
Sanni LA: The role of cerebral oedema in the pathogenesis of cerebral malaria. Redox Rep. 2001, 6: 137-142. 10.1179/135100001101536238.
Hansen AM, Driussi C, Turner V, Takikawa O, Hunt NH: Tissue distribution of indoleamine 2,3-dioxygenase in normal and malaria-infected tissue. Redox Rep. 2000, 5: 112-115. 10.1179/135100000101535384.
Anstey NM, Weinberg JB, Hassanali MY, Mwaikambo ED, Manyenga D, Misukonis MA: Nitric oxide in Tanzanian children with malaria: inverse relationship between malaria severity and nitric oxide production/nitric oxide synthase type 2 expression. J Exp Med. 1996, 184: 557-567. 10.1084/jem.184.2.557.
Hobbs MR, Udhayakumar V, Levesque MC, Booth J, Roberts JM, Tkachuk AN: A new NOS2 promoter polymorphism associated with increased nitric oxide production and protection from severe malaria in Tanzanian and Kenyan children. Lancet. 2002, 360: 1468-1475. 10.1016/S0140-6736(02)11474-7.
Lopansri BK, Anstey NM, Weinberg JB, Stoddard GJ, Hobbs MR, Levesque MC: Low plasma arginine concentrations in children with cerebral malaria and decreased nitric oxide production. Lancet. 2003, 361: 676-678. 10.1016/S0140-6736(03)12564-0.
Yeo TW, Lampah DA, Gitawati R, Tjitra E, Kenangalem E, McNeil YR: Impaired nitric oxide bioavailability and L-arginine reversible endothelial dysfunction in adults with falciparum malaria. J Exp Med. 2007, 204: 2693-2704. 10.1084/jem.20070819.
Sobolewski P, Gramaglia I, Frangos J, Intaglietta M, van der Heyde HC: Nitric oxide bioavailability in malaria. Trends Parasitol. 2005, 21: 415-422. 10.1016/j.pt.2005.07.002.
Gramaglia I, Sobolewski P, Meays D, Contreras R, Nolan JP, Frangos JA: Low nitric oxide bioavailability contributes to the genesis of experimental cerebral malaria. Nat Med. 2006, 12: 1417-1422.
Serghides L, Kim H, Lu Z, Kain DC, Miller C, Francis RC: Inhaled nitric oxide reduces endothelial activation and parasite accumulation in the brain, and enhances survival in experimental cerebral malaria. PLoS One. 2011, 6: e27714-10.1371/journal.pone.0027714.
Zanini GM, Cabrales P, Barkho W, Frangos JA, Carvalho LJ: Exogenous nitric oxide decreases brain vascular inflammation, leakage and venular resistance during Plasmodium berghei ANKA infection in mice. J Neuroinflamm. 2011, 8: 66-10.1186/1742-2094-8-66.
Yeo TW, Lampah DA, Rooslamiati I, Gitawati R, Tjitra E, Kenangalem E: A randomized pilot study of L-arginine infusion in severe falciparum malaria: preliminary safety, efficacy and pharmacokinetics. PLoS One. 2013, 8: e69587-10.1371/journal.pone.0069587.
Das BS, Mohanty S, Mishra SK, Patnaik JK, Satpathy SK, Mohanty D, Bose TK: Increased cerebrospinal fluid protein and lipid peroxidation products in patients with cerebral malaria. Trans R Soc Trop Med Hyg. 1991, 85: 733-734. 10.1016/0035-9203(91)90436-3.
Das BS, Patnaik JK, Mohanty S, Mishra SK, Mohanty D, Satpathy SK: Plasma antioxidants and lipid peroxidation products in falciparum malaria. Am J Trop Med Hyg. 1993, 49: 720-725.
Mishra NC, Kabilan L, Sharma A: Oxidative stress and malaria-infected erythrocytes. Indian J Malariol. 1994, 31: 77-87.
Postma NS, Mommers EC, Eling WM, Zuidema J: Oxidative stress in malaria; implications for prevention and therapy. Pharm World Sci. 1996, 18: 121-129. 10.1007/BF00717727.
Thumwood CM, Hunt NH, Cowden WB, Clark IA: Antioxidants can prevent cerebral malaria in Plasmodium berghei-infected mice. Br J Exp Pathol. 1989, 70: 293-303.
Reis PA, Comim CM, Hermani F, Silva B, Barichello T, Portella AC: Cognitive dysfunction is sustained after rescue therapy in experimental cerebral malaria, and is reduced by additive antioxidant therapy. PLoS Pathog. 2010, 6: e1000963-10.1371/journal.ppat.1000963.
Sanni LA, Fu S, Dean RT, Bloomfield G, Stocker R, Chaudhri G: Are reactive oxygen species involved in the pathogenesis of murine cerebral malaria?. J Infect Dis. 1999, 179: 217-222. 10.1086/314552.
Potter SM, Mitchell AJ, Cowden WB, Sanni LA, Dinauer M, de Haan JB: Phagocyte-derived reactive oxygen species do not influence the progression of murine blood-stage malaria infections. Infect Immun. 2005, 73: 4941-4947. 10.1128/IAI.73.8.4941-4947.2005.
Linares M, Marín-García P, Martínez-Chacón G, Pérez-Benavente S, Puyet A, Diez A, Bautista JM: Glutathione peroxidase contributes with heme oxygenase-1 to redox balance in mouse brain during the course of cerebral malaria. Biochim Biophys Acta. 1832, 2013: 2009-2018.
Taoufiq Z, Pino P, Dugas N, Conti M, Tefit M, Mazier D, Vouldoukis I: Transient supplementation of superoxide dismutase protects endothelial cells against Plasmodium falciparum-induced oxidative stress. Mol Biochem Parasitol. 2006, 150: 166-173. 10.1016/j.molbiopara.2006.07.008.
Tripathi AK, Sullivan DJ, Stins MF: Plasmodium falciparum-infected erythrocytes increase intercellular adhesion molecule 1 expression on brain endothelium through NF-kappaB. Infect Immun. 2006, 74: 3262-3270. 10.1128/IAI.01625-05.
Narsaria N, Mohanty C, Das BK, Mishra SP, Prasad R: Oxidative stress in children with severe malaria. J Trop Pediatr. 2012, 58: 147-150. 10.1093/tropej/fmr043.
Abbott NJ, Friedman A: Overview and introduction: the blood–brain barrier in health and disease. Epilepsia. 2012, 53 (Suppl 6): 1-6.
Luissint AC, Artus C, Glacial F, Ganeshamoorthy K, Couraud PO: Tight junctions at the blood brain barrier: physiological architecture and disease-associated dysregulation. Fluids Barriers CNS. 2012, 9: 23-10.1186/2045-8118-9-23.
Cucullo L, Aumayr B, Rapp E, Janigro D: Drug delivery and in vitro models of the blood–brain barrier. Curr Opin Drug Discov Devel. 2005, 8: 89-99.
Renia L, Howland SW, Claser C, Gruner AC, Suwanarusk R, Hui Teo T, Russell B, Ng LF: Cerebral malaria – Mysteries at the blood-brain barrier. Virulence. 2012, 3: 193-201. 10.4161/viru.19013.
Treeratanapiboon L, Psathaki K, Wegener J, Looareesuwan S, Galla HJ, Udomsangpetch R: In vitro study of malaria parasite induced disruption of blood–brain barrier. Biochem Biophys Res Commun. 2005, 335: 810-818. 10.1016/j.bbrc.2005.07.151.
Susomboon P, Maneerat Y, Dekumyoy P, Kalambaheti T, Iwagami M, Komaki-Yasuda K: Down-regulation of tight junction mRNAs in human endothelial cells co-cultured with Plasmodium falciparum-infected erythrocytes. Parasitol Int. 2006, 55: 107-112. 10.1016/j.parint.2005.11.054.
Wassmer SC, Combes V, Candal FJ, Juhan-Vague I, Grau GE: Platelets potentiate brain endothelial alterations induced by Plasmodium falciparum. Infect Immun. 2006, 74: 645-653. 10.1128/IAI.74.1.645-653.2006.
Tripathi AK, Sullivan DJ, Stins MF: Plasmodium falciparum-infected erythrocytes decrease the integrity of human blood–brain barrier endothelial cell monolayers. J Infect Dis. 2007, 195: 942-950. 10.1086/512083.
Gillrie MR, Krishnegowda G, Lee K, Buret AG, Robbins SM, Looareesuwan S: Src-family kinase dependent disruption of endothelial barrier function by Plasmodium falciparum merozoite proteins. Blood. 2007, 110: 3426-3435. 10.1182/blood-2007-04-084582.
Tripathi AK, Sha W, Shulaev V, Stins MF, Sullivan DJ: Plasmodium falciparum-infected erythrocytes induce NF-kappaB regulated inflammatory pathways in human cerebral endothelium. Blood. 2009, 114: 4243-4252. 10.1182/blood-2009-06-226415.
Zougbédé S, Miller F, Ravassard P, Rebollo A, Cicéron L, Couraud PO: Metabolic acidosis induced by Plasmodium falciparum intraerythrocytic stages alters blood–brain barrier integrity. J Cereb Blood Flow Metab. 2011, 31: 514-526. 10.1038/jcbfm.2010.121.
Wassmer SC, Moxon CA, Taylor T, Grau GE, Molyneux ME, Craig AG: Vascular endothelial cells cultured from patients with cerebral or uncomplicated malaria exhibit differential reactivity to TNF. Cell Microbiol. 2011, 13: 198-209. 10.1111/j.1462-5822.2010.01528.x.
Claessens A, Rowe JA: Selection of Plasmodium falciparum parasites for cytoadhesion to human brain endothelial cells. J Vis Exp. 2012, 59: e3122-
El-Assaad F, Wheway J, Mitchell AJ, Lou J, Hunt NH, Combes V, Grau GE: Cytoadherence of Plasmodium berghei-infected red blood cells to murine brain and lung microvascular endothelial cells in vitro. Infect Immun. 2013, 81: 3984-3991. 10.1128/IAI.00428-13.
Migasena P, Maegraith BG: The movement of radioactive albumin (I131 R.H.S.A.) from blood into C.S.F. and vice versa by dilution method in normal and Plasmodium knowlesi infected Macaca mulatta. Med J Malaya. 1968, 22: 250-
Migasena P, Maegraith BG: Factor affecting on the movement of protein across the blood: brain: C.S.F. barriers in Plasmodium knowlesi infected Macaca mulatta. Med J Malaya. 1968, 22: 251-
Migasena P, Maegraith BG: The movement of fluorescent isothiocyanate (F.I.T.C.) labelled human albumin from blood into brain tissue examined by fluorescent technique in normal and Plasmodium knowlesi infected Macaca mulatta. Med J Malaya. 1968, 22: 251-
Fujioka H, Millet P, Maeno Y, Nakazawa S, Ito Y, Howard RJ: A nonhuman primate model for human cerebral malaria: rhesus monkeys experimentally infected with Plasmodium fragile. Exp Parasitol. 1994, 78: 371-376. 10.1006/expr.1994.1040.
Moreno A, Cabrera-Mora M, Garcia A, Orkin J, Strobert E, Barnwell JW: Plasmodium coatneyi in rhesus macaques replicates the multisystemic dysfunction of severe malaria in humans. Infect Immun. 2013, 81: 1889-1904. 10.1128/IAI.00027-13.
Tongren JE, Yang C, Collins WE, Sullivan JS, Lal AA, Xiao L: Expression of proinflammatory cytokines in four regions of the brain in Macaque mulatta (rhesus) monkeys infected with Plasmodium coatneyi. Am J Trop Med Hyg. 2000, 62: 530-534.
Thumwood CM, Hunt NH, Clark IA, Cowden WB: Breakdown of the blood–brain barrier in murine cerebral malaria. Parasitology. 1988, 96: 579-589. 10.1017/S0031182000080203.
Polder T, Jerusalem C, Eling W: Topographical distribution of the cerebral lesions in mice infected with Plasmodium berghei. Tropenmed Parasitol. 1983, 34: 235-243.
Polder TW, Eling WM, Kubat K, Jerusalem CR: Histochemistry of cerebral lesions in mice infected with Plasmodium berghei. Trop Med Parasitol. 1988, 39: 277-283.
Polder TW, Eling WM, Curfs JH, Jerusalem CR, Wijers-Rouw M: Ultrastructural changes in the blood–brain barrier of mice infected with Plasmodium berghei. Acta Leiden. 1992, 60: 31-46.
Neill AL, Hunt NH: Pathology of fatal and resolving Plasmodium berghei cerebral malaria in mice. Parasitology. 1992, 105: 165-175. 10.1017/S0031182000074072.
Penet MF, Viola A, Confort-Gouny S, Le Fur Y, Duhamel G, Kober F: Imaging experimental cerebral malaria in vivo: significant role of ischemic brain oedema. J Neurosci. 2005, 25: 7352-7358. 10.1523/JNEUROSCI.1002-05.2005.
Schmidt KE, Schumak B, Specht S, Dubben B, Limmer A, Hoerauf A: Induction of pro-inflammatory mediators in Plasmodium berghei infected BALB/c mice breaks blood–brain-barrier and leads to cerebral malaria in an IL-12 dependent manner. Microbes Infect. 2011, 13: 828-836. 10.1016/j.micinf.2011.04.006.
Lacerda-Queiroz N, Lima OC, Carneiro CM, Vilela MC, Teixeira AL, Teixeira-Carvalho A: Plasmodium berghei NK65 induces cerebral leukocyte recruitment in vivo: an intravital microscopic study. Acta Trop. 2011, 120: 31-39. 10.1016/j.actatropica.2011.04.020.
Keswani T, Bhattacharyya A: Splenocyte apoptosis in Plasmodium berghei ANKA infection: possible role of TNF-α and TGF-β. Parasite Immunol. 2013, 35: 73-90. 10.1111/pim.12005.
Nacer A, Movila A, Baer K, Mikolajczak SA, Kappe SH, Frevert U: Neuroimmunological blood brain barrier opening in experimental cerebral malaria. PLoS Pathog. 2012, 8: e1002982-10.1371/journal.ppat.1002982.
Owens T, Bechmann I, Engelhardt B: Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol. 2008, 67: 1113-1121. 10.1097/NEN.0b013e31818f9ca8.
Warrell DA, Looareesuwan S, Phillips RE, White NJ, Warrell MJ, Chapel HM: Function of the blood-cerebrospinal fluid barrier in human cerebral malaria: rejection of the permeability hypothesis. Am J Trop Med Hyg. 1986, 35: 882-889.
Brown HC, Chau TT, Mai NT, Day NP, Sinh DX, White NJ: Blood–brain barrier function in cerebral malaria and CNS infections in Vietnam. Neurology. 2000, 55: 104-111. 10.1212/WNL.55.1.104.
Badibanga B, Dayal R, Depierreux M, Pidival G, Lambert PH: Principle immunological factors and the blood–brain barrier in cerebral malaria in children in endemic countries (Zaïre). Ann Soc Belg Med Trop. 1986, 66: 23-37.
Brown H, Rogerson S, Taylor T, Tembo M, Mwenechanya J, Molyneux M: Blood–brain barrier function in cerebral malaria in Malawian children. Am J Trop Med Hyg. 2001, 64: 207-213.
Mturi N, Keir G, Maclennan CA, Ross A, Willis AC, Elford BC: Cerebrospinal Fluid Studies in Kenyan Children with Severe Falciparum Malaria. Open Trop Med J. 2008, 1: 56-62. 10.2174/1874315300801010056.
Dorovini-Zis K, Schmidt K, Huynh H, Fu W, Whitten RO, Milner D: The neuropathology of fatal cerebral malaria in Malawian children. Am J Pathol. 2011, 178: 2146-2158. 10.1016/j.ajpath.2011.01.016.
Walker O, Salako LA, Sowunmi A, Thomas JO, Sodeine O, Bondi FS: Prognostic risk factors and post mortem findings in cerebral malaria in children. Trans R Soc Trop Med Hyg. 1992, 86: 491-493. 10.1016/0035-9203(92)90082-N.
Pongponratn E, Turner GD, Day NP, Phu NH, Simpson JA, Stepniewska K: An ultrastructural study of the brain in fatal Plasmodium falciparum malaria. Am J Trop Med Hyg. 2003, 69: 345-359.
Medana IM, Day NP, Sachanonta N, Mai NT, Dondorp AM, Pongponratn E: Coma in fatal adult human malaria is not caused by cerebral oedema. Malar J. 2011, 10: 267-10.1186/1475-2875-10-267.
Brown H, Hien TT, Day N, Mai NT, Chuong LV, Chau TT: Evidence of blood–brain barrier dysfunction in human cerebral malaria. Neuropathol Appl Neurobiol. 1999, 25: 331-340. 10.1046/j.1365-2990.1999.00188.x.
Newton CR, Peshu N, Kendall B, Kirkham FJ, Sowunmi A, Waruiru C: Brain swelling and ischaemia in Kenyans with cerebral malaria. Arch Dis Child. 1994, 70: 281-287. 10.1136/adc.70.4.281.
Cordoliani YS, Sarrazin JL, Felten D, Caumes E, Leveque C, Fisch A: MR of cerebral malaria. AJNR Am J Neuroradiol. 1998, 19: 871-874.
Ngoungou EB, Dulac O, Poudiougou B, Druet-Cabanac M, Dicko A, Mamadou Traore A: Epilepsy as a consequence of cerebral malaria in area in which malaria is endemic in Mali, West Africa. Epilepsia. 2006, 47: 873-879.
Lacout A, Guidoux C, Carlier RY: Posterior reversible encephalopathy syndrome in neuro-malaria. Indian J Radiol Imaging. 2010, 20: 198-201. 10.4103/0971-3026.69357.
Potchen MJ, Birbeck GL, Demarco JK, Kampondeni SD, Beare N, Molyneux ME, Taylor TE: Neuroimaging findings in children with retinopathy-confirmed cerebral malaria. Eur J Radiol. 2010, 74: 262-268. 10.1016/j.ejrad.2009.02.010.
Rasalkar DD, Paunipagar BK, Sanghvi D, Sonawane BD, Loniker P: Magnetic resonance imaging in cerebral malaria: a report of four cases. Br J Radiol. 2011, 84: 380-385. 10.1259/bjr/85759874.
Potchen MJ, Kampondeni SD, Seydel KB, Birbeck GL, Hammond CA, Bradley WG: Acute brain MRI findings in 120 Malawian children with cerebral malaria: new insights into an ancient disease. AJNR Am J Neuroradiol. 2012, 33: 1740-1746. 10.3174/ajnr.A3035.
Kampondeni SD, Potchen MJ, Beare NA, Seydel KB, Glover SJ, Taylor TE, Birbeck GL: MRI findings in a cohort of brain injured survivors of pediatric cerebral malaria. Am J Trop Med Hyg. 2013, 88: 542-546. 10.4269/ajtmh.12-0538.
Frevert U, Nacer A, Cabrera M, Movila A, Leberl M: Imaging Plasmodium immunobiology in the liver, brain, and lung. Parasitol Int. 2014, 63: 171-186. 10.1016/j.parint.2013.09.013.
Taylor TE, Fu WJ, Carr RA, Whitten RO, Mueller JS, Fosiko NG: Differentiating the pathologies of cerebral malaria by postmortem parasite counts. Nat Med. 2004, 10: 143-145. 10.1038/nm986.
Riley EM, Couper KN, Helmby H, Hafalla JC, de Souza JB, Langhorne J: Neuropathogenesis of human and murine malaria. Trends Parasitol. 2010, 26: 277-278. 10.1016/j.pt.2010.03.002.
Cabrales P, Zanini GM, Meays D, Frangos JA, Carvalho LJ: Murine cerebral malaria is associated with a vasospasm-like microcirculatory dysfunction, and survival upon rescue treatment is markedly increased by nimodipine. Am J Pathol. 2010, 176: 1306-1315. 10.2353/ajpath.2010.090691.
Hawkes M, Elphinstone RE, Conroy AL, Kain KC: Contrasting pediatric and adult cerebral malaria: the role of the endothelial barrier. Virulence. 2013, 4: 543-555. 10.4161/viru.25949.
Saunders NR, Habgood MD, Dziegielewska KM: Barrier mechanisms in the brain, II. Immature brain. Clin Exp Pharmacol Physiol. 1999, 26: 85-91. 10.1046/j.1440-1681.1999.02987.x.
Ek CJ, Dziegielewska KM, Habgood MD, Saunders NR: Barriers in the developing brain and Neurotoxicology. Neurotoxicology. 2012, 33: 586-604. 10.1016/j.neuro.2011.12.009.
Szklarczyk A, Stins M, Milward EA, Ryu H, Fitzsimmons C, Sullivan D: Glial activation and matrix metalloproteinase release in cerebral malaria. J Neurovirol. 2007, 13: 2-10. 10.1080/13550280701258084.
Prato M, Giribaldi G: Matrix metalloproteinase-9 and haemozoin: wedding rings for human host and Plasmodium falciparum parasite in complicated malaria. J Trop Med. 2011, 2011: 628435-
Geurts N, Opdenakker G, Van den Steen PE: Matrix metalloproteinases as therapeutic targets in protozoan parasitic infections. Pharmacol Ther. 2012, 133: 257-279. 10.1016/j.pharmthera.2011.11.008.
Piña-Vázquez C, Reyes-López M, Ortíz-Estrada G, de la Garza M, Serrano-Luna J: Host-parasite interaction: parasite-derived and -induced proteases that degrade human extracellular matrix. J Parasitol Res. 2012, 2012: 748206-
Nagase H, Woessner JF: Matrix metalloproteinases. J Biol Chem. 1999, 274: 21491-21494. 10.1074/jbc.274.31.21491.
Rosenberg GA: Matrix metalloproteinases in neuroinflammation. Glia. 2002, 39: 279-291. 10.1002/glia.10108.
Woessner JF: MMPs and TIMPs–an historical perspective. Mol Biotechnol. 2002, 22: 33-49. 10.1385/MB:22:1:033.
Rosenberg GA, Yang Y: Vasogenic oedema due to tight junction disruption by matrix metalloproteinases in cerebral ischemia. Neurosurg Focus. 2007, 22: E4-
Klein T, Bischoff R: Physiology and pathophysiology of matrix metalloproteases. Amino Acids. 2011, 41: 271-290. 10.1007/s00726-010-0689-x.
Peng WJ, Yan JW, Wan YN, Wang BX, Tao JH, Yang GJ: Matrix metalloproteinases: a review of their structure and role in systemic sclerosis. J Clin Immunol. 2012, 32: 1409-1414. 10.1007/s10875-012-9735-7.
Mannello F, Medda V: Nuclear localization of matrix metalloproteinases. Prog Histochem Cytochem. 2012, 47: 27-58. 10.1016/j.proghi.2011.12.002.
Marco M, Fortin C, Fulop T: Membrane-type matrix metalloproteinases: key mediators of leukocyte function. J Leukoc Biol. 2013, 94: 237-246. 10.1189/jlb.0612267.
Siefert SA, Sarkar R: Matrix metalloproteinases in vascular physiology and disease. Vascular. 2012, 20: 210-216. 10.1258/vasc.2011.201202.
Van Wart HE, Birkedal-Hansen H: The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc Natl Acad Sci USA. 1990, 87: 5578-5582. 10.1073/pnas.87.14.5578.
Van den Steen PE, Dubois B, Nelissen I, Rudd PM, Dwek RA, Opdenakker G: Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9). Crit Rev Biochem Mol Biol. 2002, 37: 375-536. 10.1080/10409230290771546.
Nagase H: Cell surface activation of progelatinase A (proMMP-2) and cell migration. Cell Res. 1998, 8: 179-186. 10.1038/cr.1998.18.
Nagase H, Visse R, Murphy G: Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006, 69: 562-573. 10.1016/j.cardiores.2005.12.002.
Zhao H, Bernardo MM, Osenkowski P, Sohail A, Pei D, Nagase H: Differential inhibition of membrane type 3 (MT3)-matrix metalloproteinase (MMP) and MT1-MMP by tissue inhibitor of metalloproteinase (TIMP)-2 and TIMP-3 rgulates pro-MMP-2 activation. J Biol Chem. 2004, 279: 8592-8601. 10.1074/jbc.M308708200.
Cauwe B, Van den Steen PE, Opdenakker G: The biochemical, biological, and pathological kaleidoscope of cell surface substrates processed by matrix metalloproteinases. Crit Rev Biochem Mol Biol. 2007, 42: 113-185. 10.1080/10409230701340019.
Van Lint P, Libert C: Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J Leukoc Biol. 2007, 82: 1375-1381. 10.1189/jlb.0607338.
Butler GS, Overall CM: Updated biological roles for matrix metalloproteinases and new “intracellular” substrates revealed by degradomics. Biochemistry. 2009, 48: 10830-10845. 10.1021/bi901656f.
Van den Steen PE, Van Aelst I, Starckx S, Maskos K, Opdenakker G, Pagenstecher A: Matrix metalloproteinases, tissue inhibitors of MMPs and TACE in experimental cerebral malaria. Lab Invest. 2006, 86: 873-888. 10.1038/labinvest.3700454.
Deroost K, Tyberghein A, Lays N, Noppen S, Schwarzer E, Vanstreels E: Hemozoin Induces Lung Inflammation and Correlates with Malaria-Associated Acute Respiratory Distress Syndrome. Am J Respir Cell Mol Biol. 2013, 48: 589-600. 10.1165/rcmb.2012-0450OC.
Piguet PF, Da Laperrousaz C, Vesin C, Tacchini-Cottier F, Senaldi G, Grau GE: Delayed mortality and attenuated thrombocytopenia associated with severe malaria in urokinase- and urokinase receptor-deficient mice. Infect Immun. 2000, 68: 3822-3829. 10.1128/IAI.68.7.3822-3829.2000.
Egan TJ: Recent advances in understanding the mechanism of hemozoin (malaria pigment) formation. J Inorg Biochem. 2008, 102: 1288-1299. 10.1016/j.jinorgbio.2007.12.004.
Sullivan AD, Ittarat I, Meshnick SR: Patterns of haemozoin accumulation in tissue. Parasitology. 1996, 112 (Pt 3): 285-294.
Carney CK, Schrimpe AC, Halfpenny K, Harry RS, Miller CM, Broncel M: The basis of the immunomodulatory activity of malaria pigment (hemozoin). J Biol Inorg Chem. 2006, 11: 917-929. 10.1007/s00775-006-0147-0.
Schrimpe AC, Wright DW: Comparative analysis of gene expression changes mediated by individual constituents of hemozoin. Chem Res Toxicol. 2009, 22: 433-445. 10.1021/tx8002752.
Schrimpe AC, Wright DW: Differential gene expression mediated by 15-hydroxyeicosatetraenoic acid in LPS-stimulated RAW 264.7 cells. Malar J. 2009, 8: 195-10.1186/1475-2875-8-195.
Prato M, Giribaldi G, Polimeni M, Gallo V, Arese P: Phagocytosis of hemozoin enhances matrix metalloproteinase-9 activity and TNF-alpha production in human monocytes: role of matrix metalloproteinases in the pathogenesis of falciparum malaria. J Immunol. 2005, 175: 6436-6442.
Prato M, Gallo V, Giribaldi G, Arese P: Phagocytosis of haemozoin (malarial pigment) enhances metalloproteinase-9 activity in human adherent monocytes: role of IL-1beta and 15-HETE. Malar J. 2008, 7: 157-10.1186/1475-2875-7-157.
Prato M, Gallo V, Giribaldi G, Aldieri E, Arese P: Role of the NF-κB transcription pathway in the haemozoin- and 15-HETE-mediated activation of matrix metalloproteinase-9 in human adherent monocytes. Cell Microbiol. 2010, 12: 1780-1791. 10.1111/j.1462-5822.2010.01508.x.
Giribaldi G, Prato M, Ulliers D, Gallo V, Schwarzer E, Akide-Ndunge OB: Involvement of inflammatory chemokines in survival of human monocytes fed with malarial pigment. Infect Immun. 2010, 78: 4912-4921. 10.1128/IAI.00455-10.
Giribaldi G, Valente E, Khadjavi A, Polimeni M, Prato M: Macrophage inflammatory protein-1alpha mediates matrix metalloproteinase-9 enhancement in human adherent monocytes fed with malarial pigment. Asian Pac J Trop Med. 2011, 4: 925-930. 10.1016/S1995-7645(11)60220-4.
Khadjavi A, Valente E, Giribaldi G, Prato M: Involvement of p38 MAPK in natural haemozoin- and 15-HETE-dependent MMP-9 enhancement in human adherent monocytes. Cell Biochem Funct. 2014, 32: 5-15. 10.1002/cbf.2963.
Polimeni M, Valente E, Ulliers D, Opdenakker G, Van den Steen PE, Giribaldi G, Prato M: Natural Haemozoin Induces Expression and Release of Human Monocyte Tissue Inhibitor of Metalloproteinase-1. PLoS ONE. 2013, 8: e71468-10.1371/journal.pone.0071468.
Dell’Agli M, Galli GV, Bulgari M, Basilico N, Romeo S, Bhattacharya D: Ellagitannins of the fruit rind of pomegranate (Punica granatum) antagonize in vitro the host inflammatory response mechanisms involved in the onset of malaria. Malar J. 2010, 9: 208-10.1186/1475-2875-9-208.
Prato M: Malarial pigment does not induce MMP-2 and TIMP-2 protein release by human monocytes. Asian Pac J Trop Med. 2011, 4: 756-10.1016/S1995-7645(11)60187-9.
Prato M, Gallo V, Arese P: Higher production of tumor necrosis factor alpha in hemozoin-fed human adherent monocytes is dependent on lipidic component of malarial pigment: new evidences on cytokine regulation in P. falciparum malaria. Asian Pac J Trop Med. 2010, 3: 85-89. 10.1016/S1995-7645(10)60041-7.
Polimeni M, Valente E, Aldieri E, Khadjavi A, Giribaldi G, Prato M: Role of 15-hydroxyeicosatetraenoic acid in hemozoin-induced lysozyme release from human adherent monocytes. Biofactors. 2013, 39: 304-314. 10.1002/biof.1071.
Polimeni M, Valente E, Aldieri E, Khadjavi A, Giribaldi G, Prato M: Haemozoin induces early cytokine-mediated lysozyme release from human monocytes through p38 MAPK- and NF-kappaB-dependent mechanisms. PLoS One. 2012, 7: e39497-10.1371/journal.pone.0039497.
Prato M, Giribaldi G, Arese P: Hemozoin triggers tumor necrosis factor alpha-mediated release of lysozyme by human adherent monocytes: new evidences on leukocyte degranulation in P. falciparum malaria. Asian Pac J Trop Med. 2009, 2 (3): 35-40.
Geurts N, Martens E, Van Aelst I, Proost P, Ulliers D, Opdenakker G, Van den Steen PE: Beta-hematin interaction with the hemopexin domain of gelatinase B/MMP-9 provokes autocatalytic processing of the propeptide, thereby priming activation by MMP-3. Biochemistry. 2008, 47: 2689-2699. 10.1021/bi702260q.
Prato M, D’Alessandro S, Van den Steen PE, Opdenakker G, Arese P, Taramelli D, Basilico N: Natural haemozoin modulates matrix metalloproteinases and induces morphological changes in human microvascular endothelium. Cell Microbiol. 2011, 13: 1275-1285. 10.1111/j.1462-5822.2011.01620.x.
D’Alessandro S, Basilico N, Prato M: Effects of Plasmodium falciparum-infected erythrocytes on matrix metalloproteinase-9 regulation in human microvascular endothelial cells. Asian Pac J Trop Med. 2013, 6: 195-199. 10.1016/S1995-7645(13)60022-X.
Fauser S, Deininger MH, Kremsner PG, Magdolen V, Luther T, Meyermann R: Lesion associated expression of urokinase-type plasminogen activator receptor (uPAR, CD87) in human cerebral malaria. J Neuroimmunol. 2000, 111: 234-240. 10.1016/S0165-5728(00)00368-4.
Deininger MH, Winkler S, Kremsner PG, Meyermann R, Schluesener HJ: Angiogenic proteins in brains of patients who died with cerebral malaria. J Neuroimmunol. 2003, 142: 101-111. 10.1016/S0165-5728(03)00250-9.
Deininger MH, Fimmen B, Kremsner PG, Meyermann R, Schluesener HJ: Accumulation of endostatin/collagenXVIII in brains of patients who died with cerebral malaria. J Neuroimmunol. 2002, 131: 216-221. 10.1016/S0165-5728(02)00276-X.
Griffiths MJ, Shafi MJ, Popper SJ, Hemingway CA, Kortok MM, Wathen A: Genomewide analysis of the host response to malaria in Kenyan children. J Infect Dis. 2005, 191: 1599-1611. 10.1086/429297.
Dietmann A, Helbok R, Lackner P, Issifou S, Lell B, Matsiegui PB: Matrix metalloproteinases and their tissue inhibitors (TIMPs) in Plasmodium falciparum malaria: serum levels of TIMP-1 are associated with disease severity. J Infect Dis. 2008, 197: 1614-1620. 10.1086/587943.
Noone C, Parkinson M, Dowling DJ, Aldridge A, Kirwan P, Molloy SF: Plasma cytokines, chemokines and cellular immune responses in pre-school Nigerian children infected with Plasmodium falciparum. Malar J. 2013, 12: 5-10.1186/1475-2875-12-5.
Jung K: Measurement of matrix metalloproteinases and their tissue inhibitors in serum produces doubtful results. J Infect Dis. 2008, 198: 1722-1723. 10.1086/593070.
Giebel SJ, Menicucci G, McGuire PG, Das A: Matrix metalloproteinases in early diabetic retinopathy and their role in alteration of the blood-retinal barrier. Lab Invest. 2005, 85: 597-607. 10.1038/labinvest.3700251.
Gurney KJ, Estrada EY, Rosenberg GA: Blood–brain barrier disruption by stromelysin-1 facilitates neutrophil infiltration in neuroinflammation. Neurobiol Dis. 2006, 23: 87-96. 10.1016/j.nbd.2006.02.006.
Gorodeski GI: Estrogen decrease in tight junctional resistance involves matrix-metalloproteinase-7-mediated remodeling of occludin. Endocrinology. 2007, 148: 218-231. 10.1210/en.2006-1120.
Chiu PS, Lai SC: Matrix metalloproteinase-9 leads to claudin-5 degradation via the NF-κB pathway in BALB/c mice with eosinophilic meningoencephalitis caused by Angiostrongylus cantonensis. PLoS One. 2013, 8: e53370-10.1371/journal.pone.0053370.
Liu J, Jin X, Liu KJ, Liu W: Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood–brain barrier damage in early ischemic stroke stage. J Neurosci. 2012, 32: 3044-3057. 10.1523/JNEUROSCI.6409-11.2012.
Feng S, Cen J, Huang Y, Shen H, Yao L, Wang Y, Chen Z: Matrix metalloproteinase-2 and -9 secreted by leukemic cells increase the permeabilità of blood-brain barrier by disrupting tight junction proteins. PLoS One. 2011, 6: e20599-10.1371/journal.pone.0020599. Erratum in: PLoS One 2011; 6(8)
Qiu LB, Zhou Y, Wang Q, Yang LL, Liu HQ, Xu SL, Qi YH, Ding GR, Guo GZ: Synthetic gelatinases inhibitor attenuates electromagnetic pulse-induced blood–brain barrier disruption by inhibiting gelatinases-mediated ZO-1 degradation in rats. Toxicology. 2011, 285: 31-38. 10.1016/j.tox.2011.03.019.
Lijnen HR: Elements of the fibrinolytic system. Ann N Y Acad Sci. 2001, 936: 226-236.
Lijnen HR: Matrix metalloproteinases and cellular fibrinolytic activity. Biochemistry. 2002, 67: 92-98.
Santos-Martínez MJ, Medina C, Jurasz P, Radomski MW: Role of metalloproteinases in platelet function. Thromb Res. 2008, 121: 535-542. 10.1016/j.thromres.2007.06.002.
Lesiak A, Narbutt J, Sysa-Jedrzejowska A, Lukamowicz J, McCauliffe DP, Wózniacka A: Effect of chloroquine phosphate treatment on serum MMP-9 and TIMP-1 levels in patients with systemic lupus erythematosus. Lupus. 2010, 19: 683-688. 10.1177/0961203309356455.
Buommino E, Baroni A, Canozo N, Petrazzuolo M, Nicoletti R, Vozza A, Tufano MA: Artemisinin reduces human melanoma cell migration by down-regulating alpha V beta 3 integrin and reducing metalloproteinase 2 production. Invest New Drugs. 2009, 27: 412-418. 10.1007/s10637-008-9188-2.
Wartenberg M, Wolf S, Budde P, Grünheck F, Acker H, Hescheler J, Wartenberg G, Sauer H: The antimalaria agent artemisinin exerts antiangiogenic effects in mouse embryonic stem cell-derived embryoid bodies. Lab Invest. 2003, 83: 1647-1655. 10.1097/01.LAB.0000098424.38003.FF.
Hwang YP, Yun HJ, Kim HG, Han EH, Lee GW, Jeong HG: Suppression of PMA-induced tumor cell invasion by dihydroartemisinin via inhibition of PKCalpha/Raf/MAPKs and NF-kappaB/AP-1-dependent mechanisms. Biochem Pharmacol. 2010, 79: 1714-1726. 10.1016/j.bcp.2010.02.003.
Wang SJ, Sun B, Cheng ZX, Zhou HX, Gao Y, Kong R, Chen H, Jiang HC, Pan SH, Xue DB, Bai XW: Dihydroartemisinin inhibits angiogenesis in pancreatic cancer by targeting the NF-κB pathway. Cancer Chemother Pharmacol. 2011, 68: 1421-1430. 10.1007/s00280-011-1643-7.
Rasheed SA, Efferth T, Asangani IA, Allgayer H: First evidence that the antimalarial drug artesunate inhibits invasion and in vivo metastasis in lung cancer by targeting essential extracellular proteases. Int J Cancer. 2010, 127: 1475-1485. 10.1002/ijc.25315.
Peterson JT: The importance of estimating the therapeutic index in the development of matrix metalloproteinase inhibitors. Cardiovasc Res. 2006, 69: 677-687. 10.1016/j.cardiores.2005.11.032.
Konstantinopoulos PA, Karamouzis MV, Papatsoris AG, Papavassiliou AG: Matrix metalloproteinase inhibitors as anticancer agents. Int J Biochem Cell Biol. 2008, 40: 1156-1168. 10.1016/j.biocel.2007.11.007.
Dormán G, Cseh S, Hajdú I, Barna L, Kónya D, Kupai K: Matrix metalloproteinase inhibitors: a critical appraisal of design principles and proposed therapeutic utility. Drugs. 2010, 70: 949-964. 10.2165/11318390-000000000-00000.
Murphy G, Nagase H: Progress in matrix metalloproteinase research. Mol Aspects Med. 2008, 29: 290-308. 10.1016/j.mam.2008.05.002.
Hu J, Van den Steen PE, Sang QX, Opdenakker G: Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat Rev Drug Discov. 2007, 6: 480-498. 10.1038/nrd2308.
Pirard B: Insight into the structural determinants for selective inhibition of matrix metalloproteinases. Drug Discov Today. 2007, 12: 640-646. 10.1016/j.drudis.2007.06.003.
Devel L, Czarny B, Beau F, Georgiadis D, Stura E, Dive V: Third generation of matrix metalloprotease inhibitors: gain in selectivity by targeting the depth of the S1′ cavity. Biochimie. 2010, 92: 1501-1508. 10.1016/j.biochi.2010.07.017.
Fingleton B: MMPs as therapeutic targets–still a viable option?. Semin Cell Dev Biol. 2008, 19: 61-68. 10.1016/j.semcdb.2007.06.006.
Murata CE, Goldberg DE: Plasmodium falciparum falcilysin: a metalloprotease with dual specificity. J Biol Chem. 2003, 278: 38022-38028. 10.1074/jbc.M306842200.
Acknowledgements
Thanks are due to Mrs. Franca Squazza for financial support and to Ms. Mary Lynn Dear for help with English editing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Authors contributed equally in the process of creating the manuscript. Both authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Polimeni, M., Prato, M. Host matrix metalloproteinases in cerebral malaria: new kids on the block against blood–brain barrier integrity?. Fluids Barriers CNS 11, 1 (2014). https://doi.org/10.1186/2045-8118-11-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2045-8118-11-1