
Abstract
Laminin-211 is a cell-adhesion molecule that is strongly expressed in the basement membrane of skeletal muscle. By binding to the cell surface receptors dystroglycan and integrin α7β1, laminin-211 is believed to protect the muscle fiber from damage under the constant stress of contractions, and to influence signal transmission events. The importance of laminin-211 in skeletal muscle is evident from merosin-deficient congenital muscular dystrophy type 1A (MDC1A), in which absence of the α2 chain of laminin-211 leads to skeletal muscle dysfunction. MDC1A is the commonest form of congenital muscular dystrophy in the European population. Severe hypotonia, progressive muscle weakness and wasting, joint contractures and consequent impeded motion characterize this incurable disorder, which causes great difficulty in daily life and often leads to premature death. Mice with laminin α2 chain deficiency have analogous phenotypes, and are reliable models for studies of disease mechanisms and potential therapeutic approaches. In this review, we introduce laminin-211 and describe its structure, expression pattern in developing and adult muscle and its receptor interactions. We will also discuss the molecular pathogenesis of MDC1A and advances toward the development of treatment.
Similar content being viewed by others
Introduction
The basement membrane is a thin scaffold of specific extracellular protein networks associated with various cell types, including muscle fibers. This specialized framework of extracellular matrix (ECM) provides important functional cues to cells. Laminins comprise a family of glycoproteins that are major components of all basement membranes [1]. Occurrence of a laminin molecule in hydra, one of the oldest multicellular organisms, indicates that laminins existed already 600 million years ago [2]. Laminins are large (400-900 kDa) heterotrimeric molecules composed of one α, one β and one γ subunit in a cruciform or T-shaped appearance. To date, five α, three β and three γ chains have been characterized. They represent the products of distinct genes that evolved by duplication and recombination of ancestral α, β and γ genes, hence they share sequence similarity. Currently, the trimers are named according to the composition of the α, β and γ chains and more than 15 different laminin isoforms, with various arrangements of laminin subunits, have been identified [3–5]. The first laminin isoform, laminin-111, was discovered more than 30 years ago in the Engelbreth-Holm-Swarm tumor [6]. Subsequently, laminin-211 (composed of α2, β1 and γ1 chains) (Figure 1) was isolated from placenta and was originally called merosin [7]. It is now well established that laminin-211 is the main laminin isoform in skeletal muscle [8, 9], and identification of laminin α2 chain mutations in a severe form of congenital muscular dystrophy (merosin-deficient congenital muscular dystrophy; MDC1A) showed the importance of laminin-211 for normal muscle function [10].

Scheme of laminin-211 heterotrimeric structure. Laminin α2 chain is depicted in red, β1 in green and γ1 in blue. Laminin α2 chain consists of: the N terminal globular domain (LN); tandem rod domains of epidermal growth factor (LEa, LEb, LEc), separating the LN, L4a and L4b globular domains; the laminin coiled-coil (LCC) domain that tangles with the LCC domains of the β1 and γ1 chains; and the C-terminal laminin globular (LG) domains.
Laminin α2 chain gene and protein
The LAMA2 gene is located on chromosome 6q22-23 in humans and on chromosome 10 in mice [10–12]. The gene is composed of 65 exons that encode a protein with a predicted molecular mass of 390 kDa. However, it is cleaved by a furin-like convertase into a 300 kDa N-terminal segment and a 80 kDa C-terminal segment, which remain non-covalently associated [13–15]. Whether this proteolytic processing has functional consequences in muscle in vivo is not known. The laminin α2 chain has a similar domain organization to that of the other laminin chains, with several globular and rod-like regions. Domains LN, L4a and L4b form globular structures separated by rod-like spacers of LE domains (epidermal growth factor-like repeats), followed by a coiled-coil domain, and finally, the C-terminal end is composed of five homologous laminin globular (LG) domains (LG1 to LG5) (Figure 1) [3]. Key biologic activities have been mapped to several of these domains. The LN domain is essential for laminin polymerization into supramolecular networks and consequently for incorporation into basement membranes [16], and mutations in this domain reduce the ability of polymer formation [17]. The coiled-coil domain is involved in the formation of laminin heterotrimers, and the laminin α2 chain can assemble with the β1, γ1, β2 and γ3 chains to form laminins 211, 221 and 213 [5]. The laminin α2 LG domains at the C-terminus bind cellular receptors (dystroglycan and integrin α7β1) [15, 18], and such interactions are required for adhesion, basement-membrane assembly and downstream signaling events [19, 20].
Laminin-211 and other laminins in developing muscle
Myogenesis is a complex multistep process, but it has been found that muscle morphogenesis is strongly guided by ECM cues [21]. There is robust evidence that laminins are important for synaptogenesis [22–26], but their precise function in myogenesis is still not known. Each immature murine somite is surrounded by a laminin-111-rich basement membrane [27]. While entering the myotome during somite differentiation, muscle progenitors begin to form the myotomal basement membrane that separates the myotome and sclerotome [28], and laminin-111 seems to be fundamental for the initiation of its assembly, at least in mice [29]. After the initial myogenic events, formation of primary and secondary myotubes takes place. Basement-membrane remodeling and differential expression of laminin subunits is tightly correlated with these events. During the first fusion events in mice at embryonic day (E)11, laminin-211 and laminin-511 are the major heterotrimers of the newly formed basement membrane (the laminin α1 chain is still present at E11.5, but it is largely restricted to the ends of myotubes) [9]. Just before the fusion of secondary myotubes (at E14), expression of the laminin α4 chain increases dramatically, and it is deposited throughout the secondary myotube basement membrane by E15 [9]. In developing human muscle, the laminin α2 chain is present from around the seventh week of gestation, reaching maximum expression levels at week 21 [30, 31], and the laminin α4 chain is strongly expressed at week 16 [32]. Additionally, the laminin α5 subunit was shown to be a major laminin α chain during myogenesis in humans, whereas the laminin α1 subunit was detected only in the developing myotendinous junction (MTJ) [31, 33]. It is noteworthy that the laminin composition is also modified during development of specialized muscle sites, such as the neuromuscular junction (NMJ) and the MTJ [9, 31, 34].
Further changes om the laminin array in muscle basement membrane occur perinatally both in human and mouse as myotubes mature into myofibers. The levels of laminin α4 and α5 subunits markedly decrease at birth and are not detectable at the sarcolemma by the end of the first postnatal week [9, 32, 35]. Thus, the laminin α2 subunit is the only laminin α chain expressed in the extrasynaptic basement membrane. Interestingly, in vitro studies with myogenic cell lines found that both the laminin α1 and α2 chains possess myogenic properties, performing both shared and specific tasks in myogenesis [36, 37].
Although several laminins are expressed in a distinct manner during myogenesis, none of the laminin α chains seems to be essential for this event [23, 38, 39]. Myogenesis occurs normally in patients and mice lacking laminin α2 subunit [11, 12, 40–43], even though myofibers are smaller at birth in patients with MDC1A [40, 41]. It is possible that laminin α4 and/or α5 could compensate for absence of the laminin α2 chain in developing muscle, and studies of muscles devoid of several α chains would be therefore be interesting. Laminins containing the α2 chain are instead crucial in adult muscle, and this topic will be discussed in more detail in later sections.
Laminin-211 and other laminins in mature muscle
The basement membrane surrounding the mature muscle cell (Figure 2A,B) contains laminins 211 and 221 [8, 9, 44] (Figure 2C). To form a strongly crosslinked basement membrane, which provides significant structural support to muscle cells [45], laminins 211 and 221 bind to each other and to other matrix proteins including nidogens (which in turn (connect) the laminin network to the collagen network), fibulins and agrin [18, 46]. There are also structurally and functionally specialized basement membranes within the skeletal muscle compartment (Figure 2B,C). The sites of muscle contact with motor nerves (the NMJ) encompass three basement membranes, each with a distinct laminin expression pattern; the extrasynaptic basement membrane, the basement membrane of the synaptic cleft, and the Schwann cell basement membrane (Figure 2B). The basement membrane within the synaptic cleft contains laminins 221, 421 and 521 at the sites of concentration of acetylcholine receptors (primary clefts), whereas the sites of concentration of sodium channels (folds of secondary clefts) include laminins 221 and 421 [9]. Basement membranes surrounding Schwann cells in the peripheral nervous system include mainly laminins 211, 411 [47] and probably 221 and 421 [48]. There is also a specialized junction where muscle abuts tendon (the MTJ) and laminins 211 and 221 are strongly expressed at this site [49, 9] (Figure 2C).

Basement membrane (BMs) of the neuromuscular system. (A) Electron microscopy of BMs in normal diaphragm muscle (SM, skeletal muscle). Red arrow = BM, blue arrow = sarcolemma, yellow arrowhead = sarcomere (cross-section), green arrowhead = collagen fibrils in the interstitial matrix (ECM). (B) Scheme of the motor innervation with distinct BMs. The BMs comprise a continuous layer around muscle fibers and Schwann cells of the peripheral nervous system (red layer), and there is a different composition of laminin chains and other extracellular matrix components at the neuromuscular junction (NMJ) (yellow layer). Muscle fibers and satellite cells are surrounded by an extrasynaptic BM containing mainly laminin-211. At the NMJ, laminins 221, 421 and 521 are the prominent laminin isoforms. Schwann cells at the NMJ and myelinating Schwann cells (blue = myelin) that frame the axon of the motor nerve are surrounded by a BM with slightly different laminin composition (laminins 211 and 411 as well as 221 and 421). (C) Laminin α2 chain expression in BMs of the neuromuscular system (immunofluorescent staining with the 4H8-2 antibody generated against the N-terminal domain of the laminin α2 chain). The laminin α2 subunit (red staining) forms the backbone of BMs that surround muscle fibers (extrasynaptic BMs) (purple arrow),underlie the NMJ (yellow) (the co-localization of laminin α2 chain (red staining) and acetylcholine receptors (green staining with α-bungarotoxin) is also shown), surround Schwann cells of the intramuscular peripheral nerve (blue arrow), surround muscle spindles (white arrowhead) and underlie the myotendinous junction (MTJ) (white arrows). Inset shows a higher magnification of the MTJ.
Laminin-211 in the sarcolemmal basement membrane is extremely important for maintenance and stabilization of differentiated muscle [37, 50], and absence of the laminin α2 chain leads to muscular dystrophy in humans and mice [10–12, 40–43]. Subtle NMJ defects have also been reported in laminin α2 chain-deficient mice [51], but it is possible that these arise from muscle abnormalities caused by the dystrophic process. The laminin α4 and β2 chains, by contrast, have important roles in the NMJ. Mice devoid of the laminin α4 and β2 chainshave abnormal neuromuscular (synapses) [22, 23, 26], and laminin β2 chain deficiency in humans (Pierson syndrome) is characterized by muscular and neurologic defects in addition to kidney failure [52].
Laminin receptors in skeletal muscle
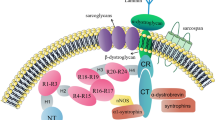
Transmembrane receptors that interact with laminin networks, connecting them to the cytoskeleton and intracellular signaling pathways, trigger the biologic functions of laminins. The two major laminin-211/221 receptors in skeletal muscle are dystroglycan and integrin α7β1 (Figure 3). Dystroglycan is a highly glycosylated, ubiquitously expressed protein that consists of two subunits, α-dystroglycan and β-dystroglycan [53]. In muscle, it forms the backbone of the multisubunit dystrophin-glycoprotein complex (DGC) linking laminin-211 to the intracellular components dystrophin and actin [54, 55]. It was recently found that a phosphorylated O-mannosyl glycan on α-dystroglycan is required for laminin binding [56], which occurs through the LG domains of the laminin α2 chain [18]. Both laminin α2 LG1 to 3 and LG4 to 5 bind α-dystroglycan strongly (whereas individual modules do not bind, except for weak interactions with the LG3 domain) [14, 18]. The crystal structure has been solved for the laminin α2 LG4 to 5 domain, and the α-dystroglycan binding site in the α2 LG4 to 5 domain has been defined [57]. Binding to α-dystroglycan is calcium-dependent [55]. The structure of the mouse laminin α2 LG4 to 5 domain showed that the two calcium ions, implicated in dystroglycan binding, are located in LG4 and LG5, respectively, and the extensive basic surface region between the calcium sites is proposed to bind α-dystroglycan [57].

Laminin-211 receptors in muscle and their binding sites on the laminin α2 chain. Laminin α2 subunit binds dystroglycan and integrin α7β1 via the laminin globular (LG) domains. LG1 to 3 and 4 to 5 bind α-dystroglycan, whereas only LG1 to 3 binds integrin α7β1. Glycosylation of α-dystroglycan is important for laminin binding.
Integrin α7β1 is the second transmembrane unit that links laminin-211 to the cytoskeleton [58–60] and binding occurs through the laminin α2 LG1 to3 domain with involvement of the coiled-coil domain [14, 15]. However, the adaptor molecules that connect integrin α7β1 to the cytoskeleton remain to be identified [61], although talin [62] and integrin-linked kinase [63] are likely candidates. We also recently identified a novel integrin α7β1 interacting protein (Cib2), whose expression in muscle is dependent on the presence of the laminin α2 chain [64].
The significance of the laminin receptors for normal muscle function is emphasized by the fact that mutations in DGC components and post-translational defects in dystroglycan processing and mutations in the integrin α7 gene causes various forms of muscular dystrophy and myopathy [65, 66]. Hence, there is strong evidence that both receptors contribute to linking laminin-211 to the cytoskeleton and mediate the effects of laminin-211 on muscle integrity and function. It has been shown that the two systems act synergistically [67, 68], but separate roles have also been delineated [69, 70]. Both dystroglycan and integrin α7β1 contribute to force production, but only dystroglycan is involved in anchoring the basement membrane to the sarcolemma [69]. Furthermore, different muscles may have different requirements for the laminin-dystroglycan interaction as it may not be crucial in diaphragm but important in limb muscle [70]. Nevertheless, many of the downstream events of the laminin-211-receptor interaction remain to be elucidated. Several signaling pathways may be affected, but the importance of each of those pathways in skeletal muscle is not obvious [71–76].
Finally, it should be noted that laminin-211 also binds other cell-surface receptors, although dystroglycan and integrin α7β1 may be considered as the major laminin-211 receptors in skeletal muscle. These other receptors include the syndecans and sulfated glycolipids [18, 77]. Interestingly, sulfatides have been proposed to anchor laminin-211 by binding to its LG domains to initiate basement-membrane assembly and to engage the activation of receptors (dystroglycan and β1 integrins), at least in Schwann cells [20].
Congenital muscular dystrophy type 1A
Laminin α2 chain-deficient muscular dystrophy (MDC1A), showing autosomal recessive inheritance, was recognized as a particular form of congenital muscular dystrophy in 1994 when Tomé et al., found specific absence of the laminin α2 chain in patients [41]. Shortly after, the first causative mutations in the LAMA2 gene were identified, and a number of mutations have subsequently been reported [10, 78, 79]. It is now clear that complete laminin α2 chain deficiency leads to a severe phenotype, whereas partial deficiency may lead to a severe or a milder phenotype [80]. The estimated prevalence of congenital muscular dystrophy is around 7 × 10-6 [81], and MDC1A accounts for approximately 40% of cases of congenital muscular dystrophy in Europe [78, 79]. The clinical features of MDC1A include profound muscle hypotonia at birth and generalized muscle weakness accompanied by contractures that mostly affect the elbows, hips knees and ankles, along with scoliosis, kyphosis, increased creatine kinase levels, and delayed motor milestones (Figure 4). Patients may achieve unsupported sitting, but very few attain ambulation. Common serious complications of MDC1A include respiratory failure and feeding difficulties. Importantly, treatment with noninvasive ventilation and gastrostomy can greatly improve health. However, respiratory-tract infection is the commonest cause of death, which may occur in the first decade of life or anytime subsequently [78, 79]. As the laminin α2 chain is also expressed in the central nervous system (CNS), peripheral nervous system and heart [8], these tissues are also affected to various degrees in MDC1A. Most patients (after 1 year of age) display white-matter abnormalities, which are readily detected with magnetic resonance imaging, but these changes do not seem to be associated with any particular functional impairment. Structural brain changes have been reported in some patients, and epilepsy may be present. Moreover, patients have decreased peripheral nerve conduction velocity because of myelination defects. Severe heart failure is rare in MDC1A, but left ventricular dysfunction has been reported in about 30% of patients [78, 79, 82]. No treatment is currently available for this devastating disease.

Clinical features of a patient with merosin-deficient congenital muscular dystrophy (MDC1A) with complete laminin α2 chain deficiency. (A) Facial weakness with an open mouth and reduced facial expression. Patient has developed bilateral elbow-flexion contractures, a fairly common sign in patients with MDC1A. (B) Bilateral knee-flexion contractures and lumbar hyperlordosis. (C) Truncal weakness and neck-flexion weakness (lack of head control) when the patient is pulled up from a lying position. Informed consent was obtained from the patient's parents for publication of the photographs.
Mouse models for laminin α2 chain deficiency
A number of mouse models for laminin α2 chain deficiency exist, and in general they adequately model human disease. In addition, they confirm the relationship between laminin α2 chain expression and severity of disease [83] (Table 1 and references therein). The dy/dy mouse expresses reduced levels of an apparently normal laminin α2 chain, but the causative mutation remains to be identified. The dy/dy mouse displays moderate muscular dystrophy, peripheral neuropathy, heart fibrosis and defects in CNS myelination [11, 12, 84]. The dy2J/dy2Jmouse harbors a mutation in the N-terminal (LN) domain, which leads to abnormal splicing and slightly reduced expression of a laminin α2 chain lacking this domain. These mice display a relatively mild muscular dystrophy and peripheral neuropathy [85, 86]. Two further mouse models have been generated by homologous recombination: the dyW/dyWmouse expresses small amounts of a truncated α2 chain lacking the LN domain whereas the dy3K/dy3Kmouse (Figure 5) is completely deficient in the laminin α2 chain. Both dyW/dyWand dy3K/dy3Kmice develop severe muscular dystrophy and die within a few weeks of age. They also exhibit pronounced hind-leg lameness [42, 43, 83]. Nevertheless, it seems that the phenotype of the dy3K/dy3Kmouse is more severe than that of the dyW/dyWmouse, and it could be that the residual laminin α2 chain expression in dyW/dyWmuscle is beneficial. The most recently described mouse model is the dynmj417/dynmj417mouse, in which a single point mutation in the LN domain leads to normal levels of a mutated laminin α2 chain and mild muscular dystrophy [87].

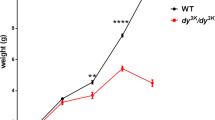
A dy3K/ dy3Klaminin α2 chain-deficient mouse with a littermate (both 5 weeks old). Laminin α2 chain knockout mice have severe muscle wasting and growth retardation, resulting in dramatically decreased weight. Hind-limb paralysis (depicted with arrow), a result of peripheral neuropathy, is often seen before death.
Although it can be debated whether mice are reliable as preclinical models for human disease, analyses of the various laminin α2 chain-deficient mouse models have led to a significant improvement in our understanding of development of MDC1A. More importantly, they have been valuable tools for the development of novel therapeutic approaches for laminin α2 chain deficiency.
Pathogenesis of MDC1A muscle
Although the primary defect in MDC1A is known to be loss of the laminin α2 chain, the secondary molecular mechanisms ultimately leading to muscle degeneration have yet to be determined. Absence of laminin α2 from skeletal muscle gives rise to a marked dystrophic pattern with muscle fiber-size variation (with atrophy predominance), central nucleation and extensive fibrosis (Figure 6) [79]. Further features typical of MDC1A include disrupted basement membranes [11] and increased apoptosis [88, 89]. It has been suggested that the laminin α2 chain confers a structural link (by binding to dystroglycan) from the ECM to the cytoskeleton, and that such linkage stabilizes the muscle-cell membrane and protects it from contraction-induced damage [90, 91]. However, this hypothesis was challenged by Hall et al. [92]. Using a zebrafish laminin α2-chain mutant, they suggested that damage to the muscle fiber occurs by mechanically induced fiber detachment in the absence of sarcolemma rupture, and that detached fibers undergo apoptosis. Nevertheless, cell membranes are ruptured to some extent in animals with complete laminin α2 chain deficiency [70] and it has been proposed that laminin α2 chain binding to α-dystroglycan strengthens the sarcolemmal integrity [69]. The downstream signaling events leading to apoptosis remain to be deciphered, but recent data suggest that it includes glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-Siah1-CBP/p300-p53 signaling [93]. However, there are relatively few apoptotic fibers in laminin α2 chain-deficient muscles [89, 93], hence there must be other mechanisms underlying the muscle wasting seen in MDC1A. Both the ubiquitin-proteasome system and the autophagy-lysosome pathway play key roles in protein degradation in skeletal muscle cells [94]. Interestingly, we recently found that increased proteasomal degradation is a feature of dy3K/dy3Kmuscle [95], and preliminary unpublished data indicate that there might also be excessive autophagy in dy3K/dy3Kmuscle. Defects in both of these degradative systems have also been found in other muscular dystrophies. For example, Duchenne muscular dystrophy pathogenesis may involve proteasomal degradation of dystrophin and the DGC [96], and autophagy is impaired (but not increased as in MDC1A) in collagen VI-deficient muscular dystrophy [97].

Dystrophic features of laminin α2 chain muscle. Hematoxylin and eosin staining of triceps and diaphragm cross-sections from a 3.5-week-old dy3K/dy3Kmouse reveals fiber-size variability with predominance of small atrophic fibers. Regenerating fibers with centrally located nucleus and wide-spread fibrosis (green arrows) are also hallmarks of laminin α2 chain-deficient muscular dystrophy. Inset shows a higher magnification of dy3K/dy3Ktriceps.
At the molecular level, absence of the laminin α2 chain affects the expression and localization of several other laminin chains and cell-surface receptors. In particular, expression of the laminin β2 chain is severely reduced from the sarcolemmal basement membranes in laminin α2 chain-deficient muscle [44]. Conversely, laminin α4 (and α5 chain to some extent) is increased at this site [9, 98]. However, it does not seem to compensate for the absence of the laminin α2 chain, presumably because the laminin α4 chain cannot bind α-dystroglycan [99], or possibly because that the laminin α4 chain is not upregulated in sufficient amounts. In extraocular muscles, which have a number of differences from other skeletal muscles, the laminin α4 chain is strongly expressed in the basement membrane adjoining the sarcolemma, and its expression is further enhanced in the extraocular muscle of dy3K/dy3Kanimals. Interestingly, laminin α2 chain-deficient extraocular muscles are spared from dystrophic changes, and it has been hypothesized that binding of the laminin α4 chain to integrin α7β1 may protect the extraocular muscles from damage [100, 101].
Changes in the expression of laminin-211 receptors might also contribute to the pathology of MDC1A. A dramatic decrease of integrin α7 subunit in muscle from laminin α2 chain-deficient patients and mice [60, 102, 103], and a striking impairment of its deposition at the sarcolemma [104], has been noted, suggesting that integrin α7β1 signaling is abolished. By contrast, expression of β-dystroglycan at the sarcolemma, is upregulated in laminin α2 chain-deficient mouse muscle [104, 105]. However, conflicting data have been reported on α-dystroglycan expression, with its production either not found to be significantly affected by laminin α2 chain deficiency [60, 103] or shown to be moderately increased [104]. Moll et al. and Jimenez-Mallebrera et al. reported severe reduction of α-dystroglycan core protein [105, 106]. The precise physiological outcomes of receptor alterations remain largely unknown, but altogether they point towards a central position of laminin-211 in regulating the expression of α7β1 and dystroglycan.
Amelioration of disease in mice
Several strategies to combat disease in MDC1A mouse models have been explored during the past decade. Because the basement membrane is affected in MDC1A, many of these approaches have targeted the expression of ECM proteins. Transgenic expression of the laminin α1 and α2 chains, mini-agrin, and cytotoxic T cell GalNac transferase have been found to compensate for laminin α2 chain deficiency in mice [43, 105, 107–109]. DyW/dyWmice bred to overexpress linker molecules (for example, mini-agrin, full-length agrin, agrin-perlecan fusion protein) between the laminin α4 chain and dystroglycan have a prolonged lifespan and significantly improved muscle tissue. Importantly, it was also found that mini-agrin can slow down the progression of disease at every stage [105, 107, 110]. The effects of laminin α1 chain overexpression in the neuromuscular system have also been extensively studied, and it has been shown that dy3K/dy3Kmice overexpressing the laminin α1 chain have a near-normal lifespan and display considerably improved muscle, heart and nerve morphology and function [48, 108, 111] (Figure 7; see Additional file 1). The reduction in muscle fibrosis was particularly marked in these animals. Because transgenic expression of laminin α1 chain reconstituted integrin α7 at the sarcolemma [104], we reasoned that the laminin α1 chain mediated reduction of laminin α2 chain deficient muscular dystrophy mainly involves integrin α7β1 (which also binds laminin α1 chain with high affinity [112]). Consistent with this notion, a laminin α1 chain devoid of the dystroglycan binding site but retaining the integrin α7β1 binding domain significantly increased the lifespan of dy3K/dy3Kmice and partially rescued dystrophic muscles, in particular the diaphragm [70]. However, subtle muscle defects (that is, residual degeneration) were seen in old animals overexpressing full-length laminin α1 chain [111]. Hence, it is possible that laminin α1 chain interactions with integrin α7β1 and dystroglycan might trigger different signaling cascades to those triggered by the laminin α2 chain.

A 2-year-old laminin α2 chain-deficient mouse ( dy3K/ dy3K) overexpressing laminin α1 chain, with a wild-type littermate. Laminin α1 chain overexpression resulted in a remarkable improvement of overall health and prevention of muscular dystrophy that persisted throughout life.
Additional file 1: Supplemental video 1. A 2-year-old laminin α2 chain-deficient mouse ( dy3K/ dy3K) overexpressing laminin α1 chain together with a wild-type littermate. The rescue mouse is denoted with a blue pointer at the beginning of the f. Both mice were placed in a new cage. The dy3KLMα1 mouse is as active as wild-type littermate; it explores the cage and often stands up on its hind limbs. (MPEG 4 MB)
Despite significant therapeutic benefits in mice, it is important to realize that these transgenic approaches are not clinically feasible. Therefore, adenoassociated virus-mediated gene transfer of mini-agrin was tested in dyW/dyWand dy/dy mice. Notably, systemic gene delivery of mini-agrin improved the overall phenotype and muscle function in treated animals [113].
Several approaches aimed at assuaging the secondary defects in MDC1A, instead of targeting the primary deficiency, have also been undertaken. As increased apoptosis had been suggested to contribute to the pathology of MDC1A, Miller et al. caused either inactivation of the proapoptotic protein Bax or overexpression of the antiapoptotic protein Bcl-2 in dyW/dyWanimals [114, 115]; both of these genetic interventions improved the health of the animals. Overexpression of Bcl-2 had no major effect in dystrophin-deficient mice, indicating that Bcl-2-mediated apoptosis is a more significant contributor to the pathogenesis of MDC1A than that of Duchenne muscular dystrophy [115]. The same group also recently explored the use of anti-apoptotic pharmacologic treatment. Interestingly, treatment with minocycline or doxycycline increased the lifespan of dyW/dyWanimals and lessened muscle pathology [116]. Similarly, treatment with omigapil, which inhibits GAPDH-Siah1-mediated apoptosis, ameliorated the pathological features in dyW/dyWanimals [93]. Recently, it was also established that mitochondria isolated from dyW/dyWmuscle are swollen. This is a typical feature of abnormal opening of the permeability transition pore caused by a strong increase in intracellular calcium (which may be detrimental for the muscle cell). Persistent opening may cause mitochondrial rupture and subsequent cell death. Laminin α2 chain-deficient dyW/dyWmice devoid of cyclophilin-D, which is a regulatory protein of the permeability transition pore, displayed reduced muscular dystrophy pathology [117]. Additionally, because enhanced proteasomal degradation is a feature of laminin α2 chain-deficient muscle [95], we hypothesized that inhibition of the proteasome would lessen the myopathology, and indeed, treatment with the proteasome inhibitor MG-132 significantly improved the lifespan and muscle morphology of dy3K/dy3Kmice [95].
Finally, cell therapy has been evaluated in mouse models of MDC1A. Myoblast and CD90-positive cell transplantation led to laminin α2 chain expression in dy/dy and dy3K/dy3Kmice, respectively, but no further improvement in the animals, was reported [118, 119]. However, bone-marrow transplantation improved life span, growth rate, muscle strength and importantly, respiratory function of dy/dy animals [120].
Altogether, considering that laminin α2 chain deficiency seems to affect different cellular events, combinatorial treatment strategies (for example, apoptosis and proteasome inhibitors together with replacement therapy) may be relevant for MDC1A. Moreover, bearing in mind that MDC1A is associated with peripheral neuropathy, therapies that also alleviate the neurologic dysfunction should be favored. Previous studies found that motor nerve pathology could not be prevented by muscle-specific expression of laminin α2 chain [43] and mini-agrin [105], whereas ubiquitous expression of the laminin α1 chain significantly reduced peripheral neuropathy [48]. In addition, inactivation of Bax s [114] and treatment with doxycycline [116] were reported to be beneficial for the condition of motor neuron.
Conclusion
A great deal is known about the structure and function of laminin-211, and advances concerning the development of future therapies have been made for murine laminin α2 chain-deficient muscular dystrophy. Absence of the laminin α2 chain does not only affect skeletal muscle but also several non-muscle tissues. Analysis of these organs has been hampered by the relatively early death of the animals. It would therefore be informative to analyze the non-muscle organs in animals that have been rescued from the muscle defects or to generate mice with a tissue-specific disruption of the laminin α2 chain. Furthermore, the targeted genetic elimination of individual laminin domains (in particular the LG domains) would be valuable to understand their role in vivo. Finally, elucidation of laminin α2 chain-induced signal transduction pathways is an important task. Such studies would be helpful to further clarify the details of laminin α2 chain function to design future treatment for MDC1A.
Authors' information
KIG is a post-doctoral student at the Department of Experimental Medical Science, University of Lund, with a PhD in cell and molecular biology, specializing in preclinical studies of laminins and muscle disease. MD is a professor in muscle biology at the Department of Experimental Medical Science, University of Lund, with a PhD in animal physiology, specializing in preclinical studies of laminins and muscle disease.
Abbreviations
- CBP:
-
cAMP response element binding protein (CREB) binding protein.
References
Miner JH, Yurchenco PD: Laminin functions in tissue morphogenesis. Annu Rev Cell Dev Biol 2004, 20: 255-284. 10.1146/annurev.cellbio.20.010403.094555
Sarass MP Jr, Yan L, Grens A, Zhang X, Agbas A, Huff JK, St John PL, Abrahamson DR: Cloning and biological function of laminin in Hydra vulgaris. Dev Biol 1994, 164: 312-324. 10.1006/dbio.1994.1201
Aumailley M, Bruckner-Tuderman L, Carter WG, Deutzmann R, Edgar D, Ekblom P, Engel J, Engvall E, Hohenester E, Jones JC, Kleinman HK, Marinkovich MP, Martin GR, Mayer U, Meneguzzi G, Miner JH, Miyazaki K, Patarroyo M, Paulsson M, Quaranta V, Sanes JR, Sasaki T, Sekiguchi K, Sorokin LM, Talts JF, Tryggvason K, Uitto J, Virtanen I, von der Mark K, Wewer UM, Yamada Y, Yurchenco PD: A simplified laminin nomenclature. Matrix Biol 2005, 24: 326-332. 10.1016/j.matbio.2005.05.006
Tzu J, Marinkovich MP: Bridging structure with function: structural, regulatory, and developmental roles of laminins. Int J Biochem Cell Biol 2008, 40: 199-214. 10.1016/j.biocel.2007.07.015
McDonald PR, Lustig A, Steinmetz MO, Kammerer RA: Laminin chain assembly is regulated by specific coiled-coil interactions. J Struct Biol 2010, 170: 398-405. 10.1016/j.jsb.2010.02.004
Timpl R, Rohde H, Robey PG, Rennard SI, Foidart JM, Martin GM: Laminin- a glycoprotein from basement membranes. J Biol Chem 1979, 254: 9933-9937.
Ehrig K, Leivo I, Argraves WS, Ruoslahti E, Engvall E: Merosin, a tissue-specific basement membrane protein, is a laminin-like protein. Proc Natl Acad Sci USA 1991, 87: 3264-3268. 10.1073/pnas.87.9.3264
Leivo I, Engvall E: Merosin, a protein specific for basement membranes of Schwann cells, striated muscle, and trophoblast, is expressed late in nerve and muscle development. Proc Natl Acad Sci USA 1988, 85: 1544-1588. 10.1073/pnas.85.5.1544
Patton BL, Miner JH, Chiu AY, Sanes JR: Distribution and functions of laminins in the neuromuscular system of developing, adult, and mutant mice. J Cell Biol 1997, 139: 1507-1521. 10.1083/jcb.139.6.1507
Helbling-Leclerc A, Zhang X, Topaloglu H, Cruaud C, Tesson F, Weissenbach J, Tomé FMS, Schwartz K, Fardeau M, Tryggvason K, Guicheney P: Mutations in the laminin α2 chain gene (LAMA2) cause merosin-deficient muscular dystrophy. Nat Genet 1995, 11: 216-218. 10.1038/ng1095-216
Xu H, Christmas P, Wu XR, Wewer UM, Engvall E: Defective muscle basement membrane and lack of M-laminin in the dystrophic dy/dy mouse. Proc Natl Acad Sci USA 1994, 91: 5572-5576. 10.1073/pnas.91.12.5572
Sunada Y, Bernier SM, Utani A, Yamada Y, Campbell KP: Deficiency of merosin in dystrophic dy mice and genetic linkage of laminin M chain to dy locus. J Biol Chem 1994, 269: 13279-13732.
Paulsson M, Saladin K, Engvall E: Structure of laminin variants: The 300-kDa chains of murine and bovine heart laminin are related to the human placenta merosin heavy chain and replace the a chain in some laminin variants. J Biol Chem 1991, 266: 17545-17551.
Talts JF, Mann K, Yamada Y, Timpl R: Structural analysis and proteolytic processing of recombinant G domain of mouse laminin α2 chain. FEBS Lett 1998, 426: 71-76. 10.1016/S0014-5793(98)00312-3
Smirnov SP, McDearmon EL, Li S, Ervasti JM, Tryggvason K, Yurchenco PD: Contributions of the LG modules and furin processing to laminin-2 functions. J Biol Chem 2002, 277: 18928-18937. 10.1074/jbc.M201880200
Yurchenco PD, O'Rear JJ: Basement membrane assembly. Methods Enzymol 1994, 245: 489-518. full_text
Colognato H, Yurchenco PD: The laminin α2 expressed by dystrophic dy(2J) mice is defective in its ability to form polymers. Curr Biol 1999, 9: 1327-1330. 10.1016/S0960-9822(00)80056-1
Talts JF, Andac Z, Gohring W, Brancaccio A, Timpl R: Binding of the G domains of laminin α1 and α2 chains and perlecan to heparin, sulfatides, α-dystroglycan and several extracellular matrix proteins. EMBO J 1999, 18: 863-870. 10.1093/emboj/18.4.863
Talts JF, Timpl R: Mutation of a basic sequence in the laminin α2LG3 module leads to a lack of proteolytic processing and has different effects on β1 integrin-mediated cell adhesion and α-dystroglycan binding. FEBS Lett 1999, 458: 319-23. 10.1016/S0014-5793(99)01180-1
Li S, Liquari P, McKee KK, Harrison D, Patel R, Lee S, Yurchenco PD: Laminin-sulfatide binding initiates basement membrane assembly and enables receptor signaling in Schwann cells and fibroblasts. J Cell Biol 2005, 169: 179-189. 10.1083/jcb.200501098
Sanes JR: The extracellular matrix. In Myology. Volume 1. Edited by: Engel A, Franzini-Armstrong C. New York: McGraw-Hill; 2004:471-488.
Noakes PG, Gautam M, Mudd J, Sanes JR, Merlie JP: Aberrant differentiation of neuromuscular junctions in mice lacking s-laminin/laminin β2. Nature 1995, 374: 258-262. 10.1038/374258a0
Patton BL, Cunningham JM, Thyboll J, Kortesmaa J, Westerblad H, Edstrom L, Tryggvason K, Sanes JR: Properly formed but improperly localized synaptic specializations in the absence of laminin α4. Nat Neurosci 2001, 4: 597-604. 10.1038/88414
Nishimune H, Sanes JR, Carlson SS: A synaptic laminin-calcium channel interaction organizes active zones in motor nerve terminals. Nature 2004, 432: 580-587. 10.1038/nature03112
Nishimune H, Valdez G, Jarad G, Moulson CL, Müller U, Miner JH, Sanes JR: Laminins promote postsynaptic maturation by an autocrine mechanism at the neuromuscular junction. J Cell Biol 2008, 182: 1201-1215. 10.1083/jcb.200805095
Miner JH, Go G, Cunningham J, Patton BL, Jarad G: Transgenic isolation of skeletal muscle and kidney defects in laminin β2 mutant mice: implications for Pierson syndrome. Development 2006, 133: 967-975. 10.1242/dev.02270
Tiger CF, Gullberg D: Absence of laminin α1 chain in the skeletal muscle of dystrophic dy/dy mice. Muscle Nerve 1997, 12: 1515-1524. 10.1002/(SICI)1097-4598(199712)20:12<1515::AID-MUS6>3.0.CO;2-B
Tosney KW, Dehnbostel DB, Erickson CA: Neural crest prefer the myotome's basal lamina over the sclerotome as a substratum. Dev Biol 1994, 163: 389-406. 10.1006/dbio.1994.1157
Anderson C, Thorsteinsdottir S, Borycki AG: Sonic hedgehog-dependent synthesis of laminin α1 controls basement membrane assembly in the myotome. Development 2009, 136: 3495-3504. 10.1242/dev.036087
Sewry CA, Chevallay M, Tomé FM: Expression of laminin subunits in human fetal skeletal muscle. Histochem J 1995, 27: 497-504.
Pedrosa-Domellöf F, Tiger CF, Virtanen I, Thornell LE, Gullberg D: Laminin chains in developing and adult human myotendinous junctions. J Histochem Cytochem 2000, 48: 201-210.
Petäjäniemi N, Korhonen M, Kortesmaa J, Tryggvason K, Sekiguchi K, Fujiwara H, Sorokin L, Thornell LE, Wondimu Z, Assefa D, Patarroyo M, Virtanen I: Localization of laminin α4-chain in developing and adult human tissues. J Histochem Cytochem 2002, 50: 1113-1130.
Tiger CF, Champliaud MF, Pedrosa-Domellöf F, Thornell LF, Ekblom P, Gullberg D: Presence of laminin α5 chain and lack of laminin α1 chain during human muscle development and in muscular dystrophies. J Biol Chem 1997, 272: 28590-28595. 10.1074/jbc.272.45.28590
Gullberg D, Tiger CF, Velling T: Laminins during muscle development and in muscular dystrophies. Cell Mol Life Sci 1999, 30: 442-460. 10.1007/s000180050444
Patton BL, Connoll AM, Martin PT, Cunningham JM, Mehta S, Pestronk A, Miner JH, Sanes JR: Distribution of ten laminin chains in dystrophic and regenerating muscles. Neuromuscul Disord 1999, 9: 423-433. 10.1016/S0960-8966(99)00033-4
Schuler F, Sorokin LM: Expression of laminin isoforms in myogenic cells in vitro and in vivo. J Cell Sci 1995, 108: 3795-3805.
Vachon PH, Loechel F, Xu H, Wewer UM, Engvall E: Merosin and laminin in myogenesis; specific requirements for merosin in myotubal stability and survival. J Cell Biol 1996, 134: 1483-1497. 10.1083/jcb.134.6.1483
Miner JH, Cunningham J, Sanes JR: Roles for laminin in embryogenesis: exencephaly, syndactyly, and placentopathy in mice lacking the laminin α5 chain. J Cell Biol 1998, 143: 1713-1723. 10.1083/jcb.143.6.1713
Edwards MM, Mammadova-Bach E, Alpy F, Klein A, Hicks WL, Roux M, Simon-Assmann P, Smith RS, Orend G, Wu J, Peachey NS, Naggert JK, Lefebvre O, Nishina PM: Mutations in Lama1 disrupt retinal vascular development and inner limiting membrane formation. J Biol Chem 2010, 285: 7697-7711. 10.1074/jbc.M109.069575
Hayashi YK, Engvall E, Arikawa-Hirasawa E, Goto K, Koga R, Nonaka I, Sugita H, Arahata K: Abnormal localization of laminin subunits in muscular dystrophies. J Neurol Sci 1993, 119: 53-64. 10.1016/0022-510X(93)90191-Z
Tomé FM, Evangelista T, Leclerc A, Sunada Y, Manole E, Estournet B, Barois A, Campbell KP, Fardeau M: Congenital muscular dystrophy with merosin deficiency. CR Acad Sci III 1994, 317: 351-357.
Miyagoe Y, Hanaoka K, Nonaka I, Hayasaka M, Nabeshima Y, Arahata K, Nabeshima Y, Takeda S: Laminin α2 chain-null mutant mice by targeted disruption of the Lama2 gene: a new model of merosin (laminin 2)-deficient congenital muscular dystrophy. FEBS Lett 1997, 415: 33-39. 10.1016/S0014-5793(97)01007-7
Kuang W, Xu H, Vachon PH, Engvall E: Merosin-deficient congenital muscular dystrophy. Partial genetic correction in two mouse models. J Clin Invest 1998, 102: 844-852. 10.1172/JCI3705
Cohn RD, Herrmann R, Wewer UM, Voit T: Changes of laminin β2 chain expression in congenital muscular dystrophy. Neuromuscul Disord 1997, 7: 373-378. 10.1016/S0960-8966(97)00072-2
Sanes JR: The basement membrane/basal lamina of skeletal muscle. J Biol Chem 2003, 278: 12601-12604. 10.1074/jbc.R200027200
Denzer AJ, Brandenberger R, Gesemann M, Chiquet M, Ruegg MA: Agrin binds to the nerve-muscle basal lamina via laminin. J Cell Biol 1997, 137: 671-683. 10.1083/jcb.137.3.671
Feltri ML, Wrabetz L: Laminins and their receptors in Schwann cells and hereditary neuropathies. J Peripher Nerv Syst 2005, 10: 128-143. 10.1111/j.1085-9489.2005.0010204.x
Gawlik KI, Li JY, Petersen Å, Durbeej M: Laminin α1 chain improves laminin α2 chain deficient neuropathy. Hum Mol Genet 2006, 15: 2690-2700. 10.1093/hmg/ddl201
Engvall E, Earwicker D, Haaparanta T, Ruoslahti E, Sanes JR: Distribution and isolation of four laminin variants; tissue restricted distribution of heterotrimers assembled from five different subunits. Cell Regul 1990, 10: 731-40.
Kuang W, Xu H, Vachon PH, Engvall E: Disruption of the lama2 gene in embryonic stem cells: laminin α2 is necessary for the sustenance of mature muscle cells. Exp Cell Res 1998, 241: 117-125. 10.1006/excr.1998.4025
Desaki J, Matsuda S, Sakanaka M: Morphological changes of neuromuscular junctions in the dystrophic (dy) mouse: a scanning and transmission electron microscopic study. J Electron Microsc (Tokyo) 1995, 44: 59-65.
Gubler MC: Inherited diseases of the glomerular basement membrane. Nat Clin Pract Nephrol 2008, 4: 24-37. 10.1038/ncpneph0671
Ibraghimov-Beskrovnaya O, Milatovich A, Ozcelik T, Yang B, Koepnick K, Francke U, Campbell KP: Human dystroglycan: skeletal muscle cDNA, genomic structure, origin of tissue specific isoforms and chromosomal localization. Hum Mol Genet 1993, 2: 1651-1657. 10.1093/hmg/2.10.1651
Campbell KP, Kahl SD: Association of dystrophin and an integral membrane glycoprotein. Nature 1989, 338: 259-262. 10.1038/338259a0
Ervasti JM, Campbell KP: A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol 1993, 122: 809-823. 10.1083/jcb.122.4.809
Yoshida-Moriguchi T, Yu L, Stalnaker SH, Davis S, Kunz S, Madson M, Oldstone MB, Schachter H, Wells L, Campbell KP: O-mannosyl phosphorylation of α-dystroglycan is required for laminin binding. Science 2010, 327: 88-92. 10.1126/science.1180512
Tisi D, Talts JF, Timpl R, Hohenester E: Structure of the C-terminal laminin G-like domain pair of the laminin α2 chain harbouring binding sites for α-dystroglycan and heparin. EMBO J 2000, 19: 1432-1440. 10.1093/emboj/19.7.1432
von der Mark H, Durr J, Sonnenberg A, von der Mark K, Deutzmann R, Goodman SL: Skeletal myoblasts utilize a novel β1-series integrin and not α6β1 for binding to the E8 and T8 fragments of laminin. J Biol Chem 1991, 266: 23593-23601.
Song WK, Wang W, Foster RF, Bielser DA, Kaufman SJ: H36-α7 is a novel integrin α chain that is developmentally regulated during skeletal myogenesis. J Cell Biol 1992, 117: 643-657. 10.1083/jcb.117.3.643
Vachon PH, Xu H, Liu L, Loechel F, Hayashi Y, Arahata K, Reed JC, Wewer UM, Engvall E: Integrins (α7β1) in muscle function and survival. Disrupted expression in merosin-deficient congenital muscular dystrophy. J Clin Invest 1997, 10: 1870-1881. 10.1172/JCI119716
Mayer U: Integrins: redundant or important players in skeletal muscle? J Biol Chem 2003, 278: 14587-14590. 10.1074/jbc.R200022200
Conti FJ, Monkley SJ, Wood MR, Critchley DR, Muller U: Talin 1 and 2 are required for myoblast fusion, sarcomere assembly and the maintenance of myotendinous junction. Development 2009, 136: 3597-3606. 10.1242/dev.035857
Wang HV, Chang LW, Brixius K, Wickström SA, Montanez E, Thievessen I, Schwander M, Muller U, Bloch W, Mayer U, Fässler R: Integrin-linked kinase stabilizes myotendinous junctions and protects muscle from stress-induced damage. J Cell Biol 2008, 180: 1037-1049. 10.1083/jcb.200707175
Häger M, Bigotti MG, Meszaros R, Carmignac V, Holmberg J, Allamand V, Åkerlund M, Kalamajski S, Brancaccio A, Mayer U, Durbeej M: Cib2 binds integrin α7Bβ1D and is reduced in laminin α2 chain-deficient muscular dystrophy. J Biol Chem 2008, 283: 24760-247695.
Lisi MT, Cohn RD: Congenital muscular dystrophies: new aspects of an expanding group of disorders. Biochim Biophys Acta 2007, 1772: 159-172.
Hayashi YK, Chou FL, Engvall E, Ogawa M, Matsuda C, Hirabayashi S, Yokochi K, Ziober BL, Kramer RH, Kaufman SJ, Ozawa E, Goto Y, Nonaka I, Tsukahara T, Wang JZ, Hoffman EP, Arahata K: Mutations in the integrin α7 gene cause congenital myopathy. Nat Genet 1998, 19: 94-97. 10.1038/ng0598-94
Guo C, Willem M, Werner A, Raivich G, Emerson M, Neyses L, Mayer U: Absence of α7 integrin in dystrophin-deficient mice causes a myopathy similar to Duchenne muscular dystrophy. Hum Mol Genet 2006, 15: 989-98. 10.1093/hmg/ddl018
Rooney JE, Welser JV, Dechert MA, Flintoff-Dye NL, Kaufman SJ, Burkin DJ: Severe muscular dystrophy in mice that lack dystrophin and α7 integrin. J Cell Sci 2006, 119: 2185-2195. 10.1242/jcs.02952
Han R, Kanagawa M, Yoshida-Moriguchi T, Rader EP, Ng RA, Michele DE, Muirhead DE, Kunz S, Moore SA, Iannaccone ST, Miyake K, McNeil PL, Mayer U, Oldstone MBA, Faulkner JA, Campbell KP: Basal lamina strengthens cell membrane integrity via the laminin G domain-binding motif of α-dystroglycan. Proc Natl Acad Sci USA 2009, 106: 12573-12579. 10.1073/pnas.0906545106
Gawlik KI, Åkerlund M, Carmignac V, Elamaa H, Durbeej M: Distinct roles for laminin globular domains in laminin α1 chain mediated rescue of murine laminin α2 chain deficiency. PLoS ONE 2010, 5: e11549. 10.1371/journal.pone.0011549
Yang B, Jung D, Motto D, Meyer J, Koretzky G, Campbell KP: SH3 domain-mediated interaction of dystroglycan and Grb2. J Biol Chem 1995, 270: 11711-11714. 10.1074/jbc.270.20.11711
Zhou YW, Oak SA, Senogles SE, Jarrett HW: Laminin-α1 globular domains 3 and 4 induce heterotrimeric G protein binding to α-syntrophin's PDZ domain and alter intracellular Ca2+in muscle. Am J Physiol Cell Physiol 2005, 288: C377-388. 10.1152/ajpcell.00279.2004
Zhou Y, Jiang D, Thomason DB, Jarrett HW: Laminin-induced activation of Rac1 and JNKp46 is initiated by Src family kinases and mimics the effects of skeletal muscle contraction. Biochemistry 2007, 46: 14907-14916. 10.1021/bi701384k
Langenbach KJ, Rando TA: Inhibition of dystroglycan binding to laminin disrupts the PI3K/AKT pathway and survival signaling in muscle cells. Muscle Nerve 2002, 26: 644-653. 10.1002/mus.10258
Xiong Y, Zhou Y, Jarrett HW: Dystrophin glycoprotein complex-associated Gβγ subunits activate phosphatidylinositol-3-kinase/Akt signaling in skeletal muscle in a laminin-dependent manner. J Cell Physiol 2009, 219: 402-414. 10.1002/jcp.21684
Laprise P, Poirier EM, Vezina A, Rivard N, Vachon PH: Merosin-integrin promotion of skeletal myofiber cell survival: Differentiation state-distinct involvement of p60Fyn tyrosine kinase and p38alpha stress-activated MAP kinase. J Cell Physiol 2002, 191: 69-81. 10.1002/jcp.10075
Suzuki N, Yokoyama F, Nomizu M: Functional sites in the laminin alpha chains. Conn Tiss Res 2005, 46: 142-152. 10.1080/03008200591008527
Allamand V, Guicheney P: Merosin-deficient muscular dystrophy, autosomal recessive (MDC1A, MIM#156225, LAMA2 gene coding for α2 chain of laminin). Eur J Hum Genet 2002, 10: 91-94. 10.1038/sj.ejhg.5200743
Voit T, Tomé FS: The congenital muscular dystrophies. In Myology. Volume 2. Edited by: Angel A, Franzini-Armstrong C. New York: McGraw-Hill; 2004:1203-1238.
Geranmayeh F, Clement E, Feng LH, Sewry C, Pagan J, Mein R, Abbs S, Brueton L, Childs AM, Jungbluth H, De Goede CG, Lynch B, Lin JP, Chow G, Sousa C, O'Mahony O, Majumdar A, Straub V, Bushby K, Muntoni F: Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord 2010, 4: 241-250. 10.1016/j.nmd.2010.02.001
Mostacciuolo ML, Miorin M, Martinello F, Angelini C, Perini P, Trevisan CP: Genetic epidemiology of congenital muscular dystrophy in a sample from north-east Italy. Hum Genet 1996, 97: 277-279. 10.1007/BF02185752
Wang CH, Bonnemann CG, Rutkowski A, Sejersen T, Bellini J, Battista V, Florence JM, Schara U, Schuler PM, Wahbi K, Aloysius A, Bash RO, Béroud C, Bertini E, Bushby K, Cohn RD, Connolly AM, Deconinck N, Desguerre I, Eagle M, Estournet-Mathiaud B, Ferreiro A, Fujak A, Goemans N, Iannaccone ST, Jouinot P, Main M, Melacini P, Mueller-Felber W, Muntoni F, Nelson LL, Rahbek J, Quijano-Roy S, Sewry C, Storhaug K, Simonds A, Tseng B, Vajsar J, Vianello A, Zeller R: Consensus statement on standard care for congenital muscular dystrophies. J Child Neurol 2010, 25: 1559-1581. 10.1177/0883073810381924
Guo LT, Zhang XU, Kuang W, Xu H, Liu LA, Vilquin JT, Miyagoe-Suzuki Y, Takeda S, Ruegg MA, Wewer UM, Engvall E: Laminin α2 deficiency and muscular dystrophy; genotype-phenotype correlation in mutant mice. Neuromusc Disord 2003, 3: 207-215. 10.1016/s0960-8966(02)00266-3
Chun SJ, Rasband MN, Sidman RL, Habib AA, Vartanian T: Integrin-linked kinase is required for laminin-2-induced oligodendrocyte cell spreading and CNS myelination. J Cell Biol 2003, 163: 397-408. 10.1083/jcb.200304154
Xu H, Wu XR, Wewer UM, Engvall E: Murine muscular dystrophy caused by a mutation in the laminin α2 (Lama2) gene. Nat Genet 1994, 8: 297-302. 10.1038/ng1194-297
Sunada Y, Bernier SM, Utani A, Yamada Y, Campbell KP: Identification of a novel mutant transcript of laminin α2 chain gene responsible for muscular dystrophy and dysmyelination in dy2J mice. Hum Mol Genet 1995, 4: 1055-1061. 10.1093/hmg/4.6.1055
Patton BL, Wang B, Tarumi YS, Seburn KL, Burgess RW: A single point mutation in the LN domain of LAMA2 causes muscular dystrophy and peripheral amyelination. J Cell Sci 2008, 121: 1593-1604. 10.1242/jcs.015354
Kuang W, Xu H, Vilquin JT, Engvall E: Activation of the lama2 gene in muscle regeneration: abortive regeneration in laminin α2-deficiency. Lab Invest 1999, 79: 1601-1613.
Hayashi YK, Tezak Z, Momoi T, Nonaka I, Garcia CA, Hoffman EP, Arahata K: Massive muscle cell degeneration in the early stage of merosin-deficient congenital muscular dystrophy. Neuromuscul Disord 2001, 11: 350-359. 10.1016/S0960-8966(00)00203-0
Ervasti JM, Campbell KP: Membrane organization of the dystrophin-glycoprotein complex. Cell 1993, 66: 1121-1131. 10.1016/0092-8674(91)90035-W
Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL: Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA 1993, 90: 3710-3714. 10.1073/pnas.90.8.3710
Hall TE, Bryson-Richardson RJ, Berger S, Jacoby AS, Cole NJ, Hollway GE, Berger J, Currie PD: The zebrafish candyfloss mutant implies extracellular matrix adhesion failure in laminin α2-deficient congenital muscular dystrophy. Proc Natl Acad Sci USA 2007, 104: 7093-7097.
Erb M, Meinen S, Barzaghi P, Sumanovski LT, Courdier-Fruh I, Ruegg MA, Meier T: Omigapil ameliorates the pathology of muscle dystrophy caused by laminin-α2 deficiency. J Pharmacol Exp Ther 2009, 331: 787-795. 10.1124/jpet.109.160754
Sandri M: Autophagy in skeletal muscle. FEBS Lett 2010, 584: 1411-1416. 10.1016/j.febslet.2010.01.056
Carmignac V, Quéré R, Durbeej M: Proteasome inhibition improves the muscle of laminin α2 chain-deficient mice. Hum Mol Genet 2011, 20: 541-552. 10.1093/hmg/ddq499
Bonuccelli G, Sotiga F, Schubert W, Park DS, Frank PG, Woodman SE, Insabato L, Cammer M, Minetti C, Lisanti MP: Proteasome inhibitor (MG-132) treatment of mdx mice rescues the expression and localization of dystrophin and dystrophin-associated proteins. Am J Path 2003, 163: 1663-1675. 10.1016/S0002-9440(10)63523-7
Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, Blaauw B, Urciuolo A, Tiepolo T, Merlini L, Maraldi NM, Bernardi P, Sandri M, Bonaldo P: Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med 2010, 16: 1313-1321. 10.1038/nm.2247
Ringelmann B, Roder C, Hallmann R, Maley M, Davies M, Grounds M, Sorokin L: Expression of laminin α1, α2, α4, and α5 chains, fibronectin, and tenascin-C in skeletal muscle of dystrophic 129ReJ dy/dy mice. Exp Cell Res 1999, 246: 165-182. 10.1006/excr.1998.4244
Talts JF, Sasaki T, Miosge N, Gohring W, Mann K, Mayne R, Timpl R: Structural and functional analyses of the recombinant G domain of the laminin α4 chain and its proteolytic processing in tissues. J Biol Chem 2000, 275: 35192-35199. 10.1074/jbc.M003261200
Porter JD, Karathanasis P: Extraocular muscle in merosin-deficient muscular dystrophy: cation homeostasis is maintained but is not mechanistic in muscle sparing. Cell Tissue Res 1998, 292: 495-501. 10.1007/s004410051078
Nyström A, Holmblad J, Pedrosa-Domellöf F, Sasaki T, Durbeej M: Extraocular muscle is spared upon complete laminin α2 chain deficiency: comparative expression of laminin and integrin isoforms. Matrix Biol 2006, 25: 382-385.
Hodges BS, Hayashi YK, Nonaka I, Wang A, Arahata K, Kaufman SJ: Altered expression of the α7β1 integrin in human and murine muscular dystrophies. J Cell Sci 1997, 110: 2873-2881.
Cohn RD, Mayer U, Saher G, Herrmann R, van der Flier A, Sonnenberg A, Sorokin L, Voit T: Secondary reduction of integrin α7B in laminin α2 deficient congenital muscular dystrophy supports an additional transmembrane link in skeletal muscle. J Neurol Sci 1999, 63: 140-152. 10.1016/S0022-510X(99)00012-X
Gawlik KI, Mayer U, Blomberg K, Sonnenberg A, Ekblom P, Durbeej M: Laminin α1 chain mediated reduction of laminin α2 chain deficient muscular dystrophy involves integrin α7β1 and dystroglycan. FEBS Lett 2006, 580: 1759-1765. 10.1016/j.febslet.2006.02.027
Moll J, Barzaghi P, Lin S, Bezakova G, Lochmuller H, Engvall E, Muller U, Ruegg MA: An agrin minigene rescues dystrophic symptoms in a mouse model for congenital muscular dystrophy. Nature 2001, 413: 302-307. 10.1038/35095054
Jimenez-Mallebrera C, Torelli S, Feng L, Kim J, Godfrey C, Clement E, Mein R, Abbs S, Brown SC, Campbell KP, Kröger S, Talim B, Topaloglu H, Quinlivan R, Roper H, Childs AM, Kinali M, Sewry CA, Muntoni F: A comparative study of α-dystroglycan glycosylation in dystroglycanopathies suggests that the hypoglycosylation of alpha-dystroglycan does not consistently correlate with clinical severity. Brain Pathol 2009, 19: 596-611. 10.1111/j.1750-3639.2008.00198.x
Bentzinger CF, Barzaghi P, Lin S, Ruegg MA: Overexpression of mini-agrin in skeletal muscle increases muscle integrity and regenerative capacity in laminin-α2-deficient mice. FASEB J 2005, 19: 934-942. 10.1096/fj.04-3376com
Gawlik K, Miyagoe-Suzuki Y, Ekblom P, Takeda S, Durbeej M: Laminin α1 chain reduces muscular dystrophy in laminin α2 chain deficient mice. Hum Mol Genet 2004, 13: 1775-1784. 10.1093/hmg/ddh190
Xu R, Chandrasekharan K, Yoon JH, Camboni M, Martin PT: Overexpression of the cytotoxic T cell (CT) carbohydrate inhibits muscular dystrophy in the dyWmouse model of congenital muscular dystrophy 1A. Am J Path 2007, 171: 181-199. 10.2353/ajpath.2007.060927
Meinen S, Barzaghi P, Lin S, Lochmuller H, Ruegg MA: Linker molecules between laminins and dystroglycan ameliorate laminin- α2-deficient muscular dystrophy at all disease stages. J Cell Biol 2007, 176: 979-993. 10.1083/jcb.200611152
Gawlik KI, Durbeej M: Transgenic overexpression of laminin α1 chain in laminin α2 chain-deficient mice rescues the disease throughout the lifespan. Muscle Nerve 2010, 42: 30-37. 10.1002/mus.21616
von der Mark H, Williams I, Wendler O, Sorokin L, von der Mark K, Pöschl E: Alternative splice variants of α7β1 integrin selectively recognize different laminin isoforms. J Biol Chem 2002, 277: 6012-6016. 10.1074/jbc.M102188200
Qiao C, Li J, Zhu T, Draviam R, Watkins S, Ye X, Chen C, Li J, Xiao X: Amelioration of laminin-α2-deficient congenital muscular dystrophy by somatic gene transfer of miniagrin. Proc Natl Acad Sci USA 2005, 102: 11999-12004. 10.1073/pnas.0502137102
Girgenrath M, Dominov JA, Kostek CA, Miller JB: Inhibition of apoptosis improves outcome in a model of congenital muscular dystrophy. J Clin Invest 2004, 114: 1635-1639.
Dominov JA, Kravetz AJ, Ardelt M, Kostek CA, Beermann ML, Miller JB: Muscle-specific BCL2 expression ameliorates muscle disease in laminin α2-deficient, but not in dystrophin-deficient, mice. Hum Mol Genet 2005, 14: 1029-1040. 10.1093/hmg/ddi095
Girgenrath M, Beermann ML, Vishnudas VK, Homma S, Miller JB: Pathology is alleviated by doxycycline in a laminin-α2-null model of congenital muscular dystrophy. Ann Neurol 2009, 65: 47-56. 10.1002/ana.21523
Millay DP, Sargent MA, Osinska H, Baines CP, Barton ER, Vaugniaux G, Sweeney HL, Robbins J, Molkentin JD: Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat Med 2008, 14: 442-447. 10.1038/nm1736
Vilquin JT, Guerette B, Puymirat J, Yaffe D, Tome FMS, Fardeau M, Fiszman M, Schwartz K, Tremblay JP: Myoblast transplantations lead to the expression of the laminin α2 chain in normal and dystrophic (dy/dy) mouse muscles. Gene Therapy 1999, 6: 792-800. 10.1038/sj.gt.3300889
Fukada S, Yamamoto Y, Segawa M, Sakamoto K, Nakajima M, Sato M, Morokawa D, Uezumi A, Miyagoe-Suzuki Y, Takeda S, Tsujikawa K, Yamamoto H: CD90-postive cells, an additional cell population, produce laminin α2 chain upon transplantation in dy3k/ dy3kmice. Exp Cell Res 2007, 314: 193-203. 10.1016/j.yexcr.2007.09.020
Hagiwara H, Ohsawa Y, Asakura S, Murakami T, Teshima T, Sunada Y: Bone marrow transplantation improves outcome in a mouse model of congenital muscular dystrophy. FEBS Lett 2006, 580: 4463-4468. 10.1016/j.febslet.2006.07.015
Pillers DA, Kempton JB, Duncan NM, Pang J, Dwinnel SJ, Trune DR: Hearing loss in the laminin-deficient dy mouse model of congenital muscular dystrophy. Mol Genet Metab 2002, 76: 217-224. 10.1016/S1096-7192(02)00039-2
Wagner WJ, Chang AC, Owens J, Hong MJ, Brooks A, Coligan JE: Aberrant development of thymocytes in mice lacking laminin-2. Dev Immunol 2000, 7: 179-193. 10.1155/2000/90943
Nakagawa M, Miyagoe-Suzuki Y, Ikezoe K, Miyata Y, Nonaka I, Harii K, Takeda S: Schwann cell myelination occurred without basal lamina formation in laminin α2 chain-null mutant (dy3K/dy3K) mice. Glia 2001, 35: 101-110. 10.1002/glia.1075
Häger M, Gawlik K, Nyström A, Sasaki T, Durbeej M: Laminin α1 chain corrects male infertility caused by absence of laminin α2 chain. Am J Path 2005, 167: 823-833.
Yuasa K, Fukumoto S, Kamasaki Y, Yamada A, Fukumoto E, Kanaoka K, Saito K, Harada H, Arikawa-Hirasawa E, Miyagoe-Suzuki Y, Takeda S, Okamoto K, Kato Y, Fujiwara T: Laminin α2 is essential for odontoblast differentiation regulating dentin sialoprotein expression. J Biol Chem 2004, 279: 10286-10292. 10.1074/jbc.M310013200
Besse S, Allamand V, Vilquin JT, Li Z, Poirier C, Vignier N, Hori H, Guenet JL, Guicheney P: Spontaneous muscular dystrophy caused by a retrotransposal insertion in the mouse laminin α2 chain gene. Neuromuscul Disord 2003, 13: 216-222. 10.1016/s0960-8966(02)00278-x
Acknowledgements
This work was supported by Muscular Dystrophy Association. We are very grateful to Professor Volker Straub and the patient and family for their permission to use the photographs.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
KIG And MD wrote the paper.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Gawlik, K.I., Durbeej, M. Skeletal muscle laminin and MDC1A: pathogenesis and treatment strategies. Skeletal Muscle 1, 9 (2011). https://doi.org/10.1186/2044-5040-1-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2044-5040-1-9